Applications of Genome-Wide Screening and Systems Biology Approaches in Drug Repositioning

 ,
,  , and
, and
Simple Summary
Abstract
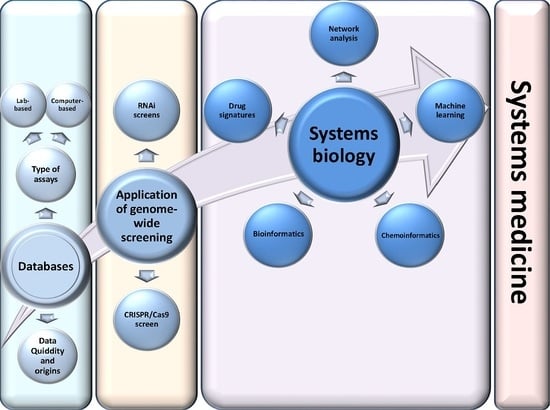
1. Introduction
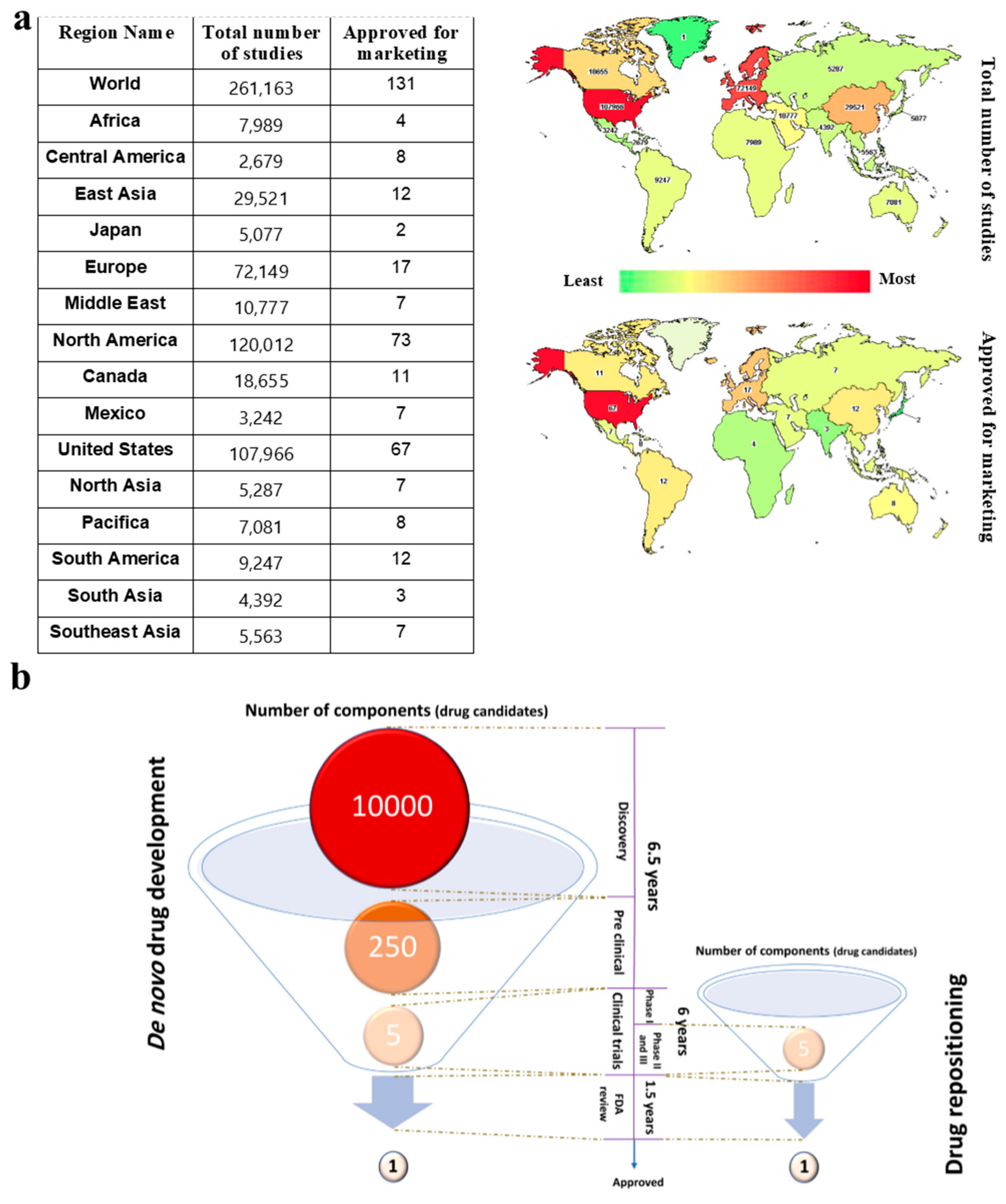
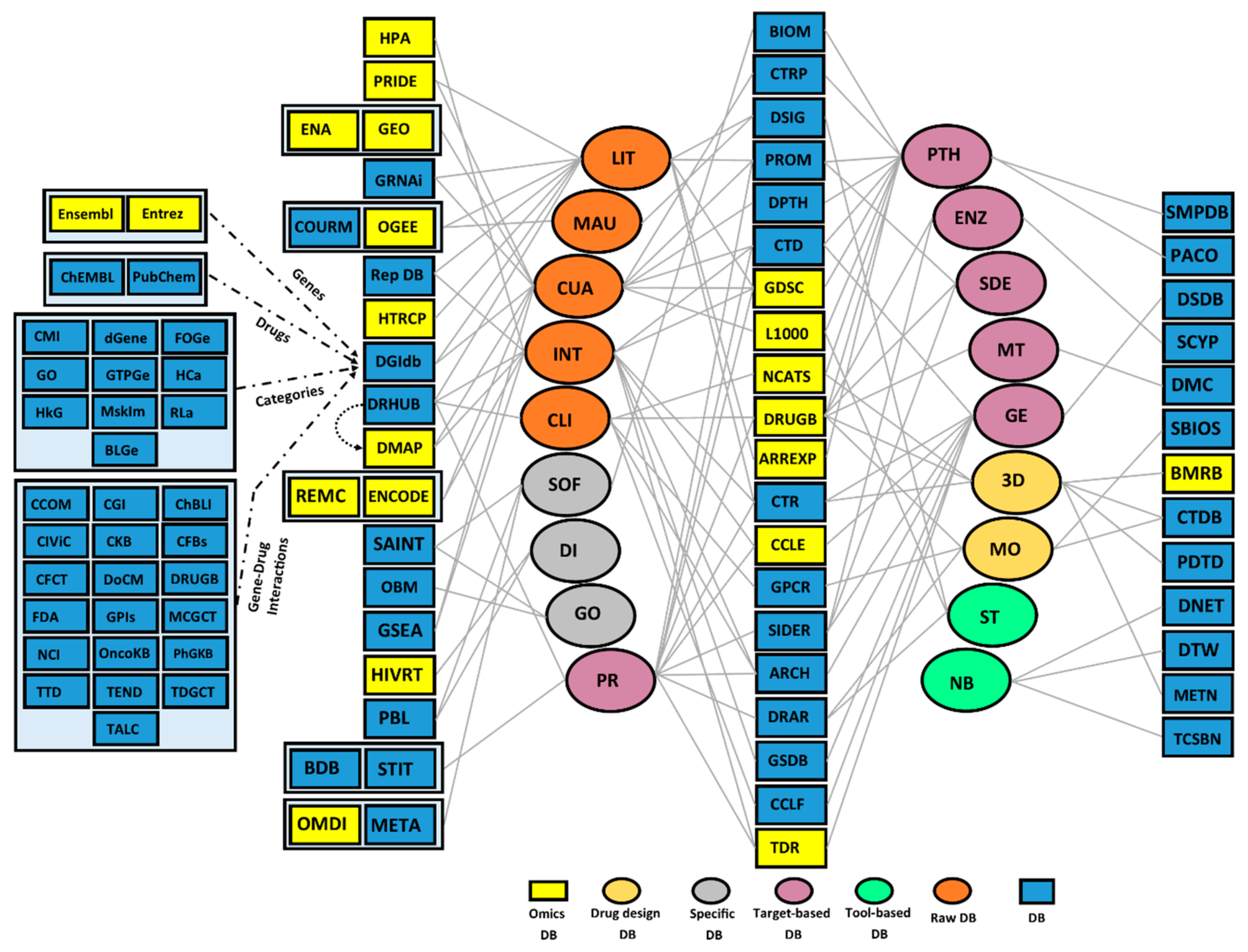
2. High-Throughput Resources for the Identification of Drug Targets and Repositioning of Drugs
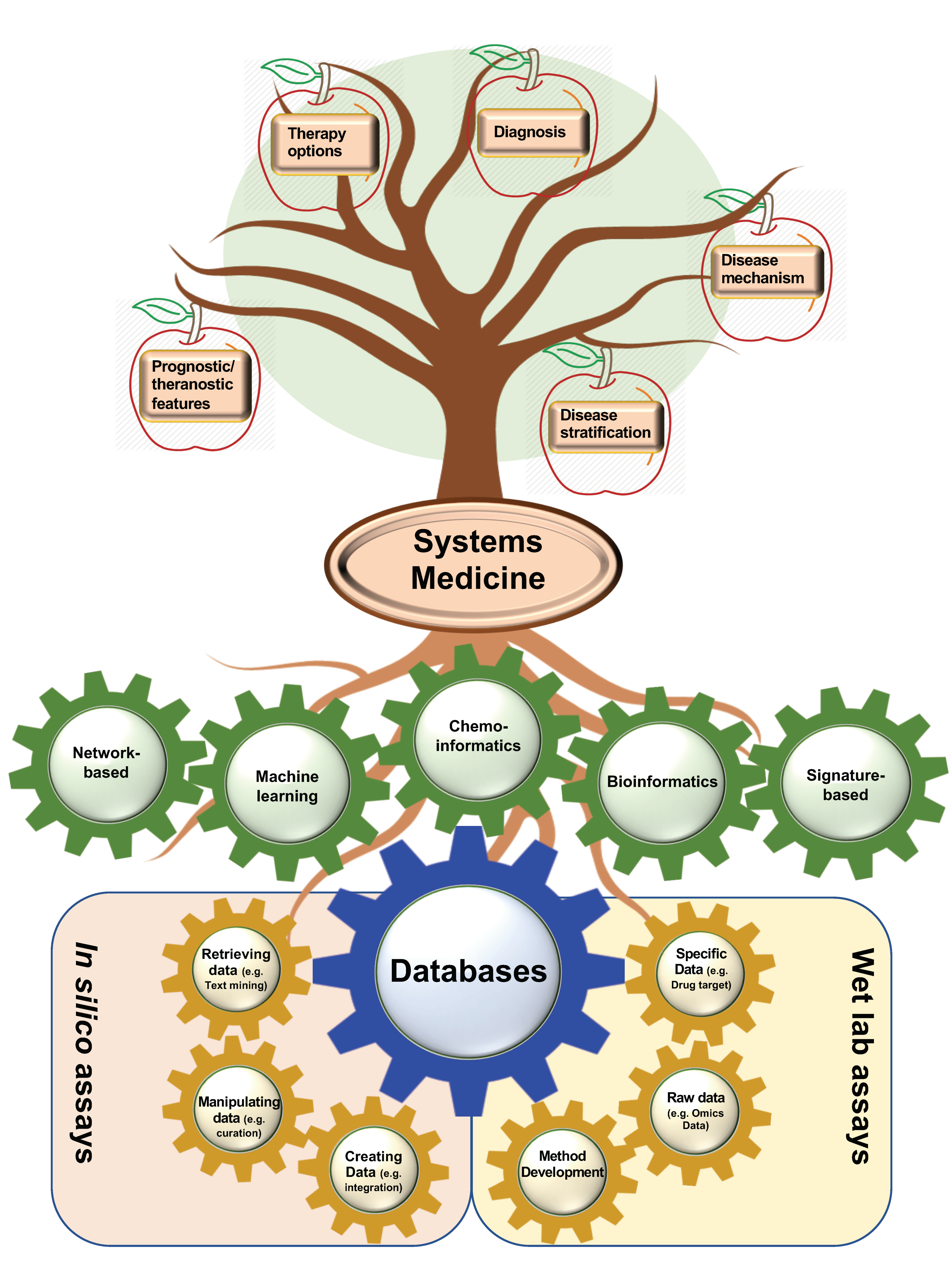
3. Computational- and Wet Lab-Based Assay DBs
3.1. Computational-Based Assay DBs
3.2. Wet Lab-Based Assay DBs
4. Application of Genome-Wide Screening in Drug Repositioning
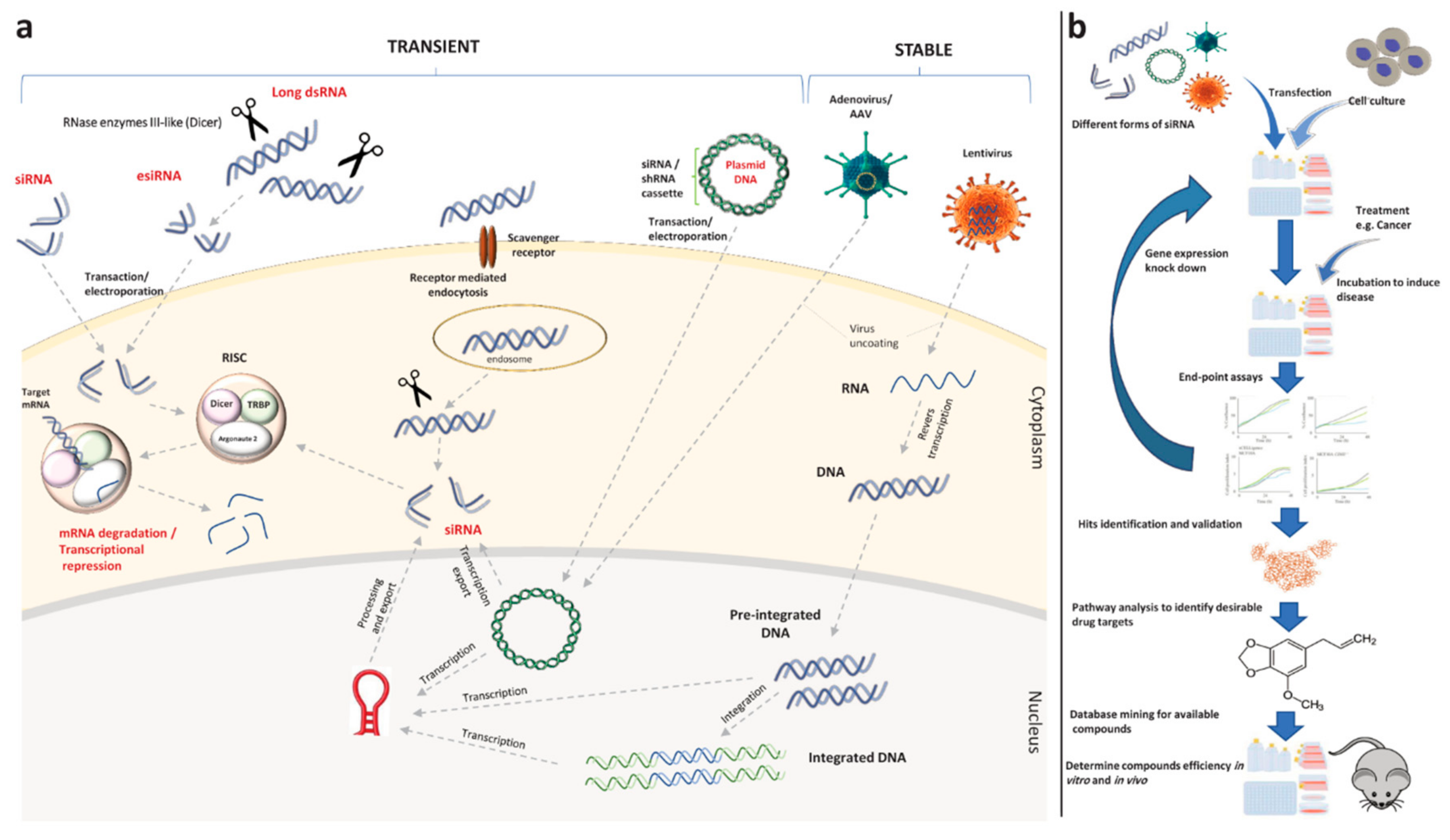
4.1. Drug Target Identification through RNA Interference Screening
4.2. Drug Target Identification through Genome-Wide CRISPR-Cas9 Screening
5. Systems Biology Application in Drug Repositioning
5.1. Network-Based Methods in Drug Repositioning
5.2. The Application of Machine Learning in Drug Repositioning
5.3. Chemoinformatic-Based Methods in Drug Repositioning
5.4. Bioinformatics-Based Methods in Drug Repositioning
5.5. Signature-Based Drug Repositioning
6. Conclusions and Future Direction
Supplementary Materials
Funding
Conflicts of Interest
References
- Fischl, M.A.; Richman, D.D.; Grieco, M.H.; Gottlieb, M.S.; Volberding, P.A.; Laskin, O.L.; Leedom, J.M.; Groopman, J.E.; Mildvan, D.; Schooley, R.T. The efficacy of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. N. Engl. J. Med. 1987, 317, 185–191. [Google Scholar] [CrossRef]
- Vlasits, A.L.; Simon, J.A.; Raible, D.W.; Rubel, E.W.; Owens, K.N. Screen of FDA-approved drug library reveals compounds that protect hair cells from aminoglycosides and cisplatin. Hear. Res. 2012, 294, 153–165. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef]
- Perwitasari, O.; Tripp, R.A. RNAi Screening to Facilitate Drug Repurposing. Front. Rnai 2014, 19, 247–265. [Google Scholar]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov (accessed on 20 February 2020).
- Chong, C.R.; Sullivan, D.J. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef]
- Altay, O.; Mohammadi, E.; Lam, S.; Turkez, H.; Boren, J.; Nielsen, J.; Uhlen, M.; Mardinoglu, A. Current status of COVID-19 therapies and drug repositioning applications. Iscience 2020, 23, 101303. [Google Scholar] [CrossRef]
- Ekins, S.; Williams, A.J.; Krasowski, M.D.; Freundlich, J.S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov. Today 2011, 16, 298–310. [Google Scholar] [CrossRef]
- Picco, G.; Chen, E.D.; Alonso, L.G.; Behan, F.M.; Gonçalves, E.; Bignell, G.; Matchan, A.; Fu, B.; Banerjee, R.; Anderson, E. Functional linkage of gene fusions to cancer cell fitness assessed by pharmacological and CRISPR-Cas9 screening. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Friedman, A.J. Spironolactone for adult female acne. Cutis 2015, 96, 216–217. [Google Scholar]
- Terrett, N.K.; Bell, A.S.; Brown, D.; Ellis, P. Sildenafil (Viagra), a potent and selective inhibitor of type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction. Bioorg. Med. Chem. Lett. 1996, 6, 1819–1824. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Safety and efficacy of sildenafil citrate in the treatment of male erectile dysfunction. Clin. Ther. 1998, 20, 1033–1048. [Google Scholar] [CrossRef]
- Rao, M.; Gupta, R.; Liguori, M.; Hu, M.; Huang, X.; Mantena, S.; Mittelstadt, S.; Blomme, E.; van Vleet, T. Novel computational approach to predict off-target interactions for small molecules. Front. Big Data 2019, 2, 25. [Google Scholar] [CrossRef]
- Huang, Y.; Furuno, M.; Arakawa, T.; Takizawa, S.; de Hoon, M.; Suzuki, H.; Arner, E. A framework for identification of on-and off-target transcriptional responses to drug treatment. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lyman, G.H.; Moses, H.L. Biomarker Tests for Molecularly Targeted Therapies—The Key to Unlocking Precision Medicine. N. Engl. J. Med. 2016, 375, 4. [Google Scholar] [CrossRef] [PubMed]
- Masoudi-Sobhanzadeh, Y.; Omidi, Y.; Amanlou, M.; Masoudi-Nejad, A. Drug databases and their contributions to drug repurposing. Genomics 2019, 112, 1087–1095. [Google Scholar] [CrossRef]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T. ArrayExpress update—Simplifying data submissions. Nucleic Acids Res. 2015, 43, CD1113–CD1116. [Google Scholar] [CrossRef]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z. BioMagResBank. Nucleic Acids Res. 2007, 36, CD402–CD408. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H. Next-generation characterization of the cancer cell line encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M. Defining a cancer dependency map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef]
- Harrison, P.W.; Alako, B.; Amid, C.; Cerdeño-Tárraga, A.; Cleland, I.; Holt, S.; Hussein, A.; Jayathilaka, S.; Kay, S.; Keane, T. The European nucleotide archive in 2018. Nucleic Acids Res. 2019, 47, D84–D88. [Google Scholar] [CrossRef] [PubMed]
- Dunham, I.; Birney, E.; Lajoie, B.R.; Sanyal, A.; Dong, X.; Greven, M.; Lin, X.; Wang, J.; Whitfield, T.W.; Zhuang, J. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res. 2010, 39, D52–D57. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; Ledoux, P.; Rudnev, D.; Evangelista, C.; Kim, I.F.; Soboleva, A.; Tomashevsky, M.; Edgar, R. NCBI GEO: Mining tens of millions of expression profiles—Database and tools update. Nucleic Acids Res. 2007, 35, D760–D765. [Google Scholar] [CrossRef]
- Tzou, P.L.; Descamps, D.; Rhee, S.-Y.; Raugi, D.N.; Charpentier, C.; Taveira, N.; Smith, R.A.; Soriano, V.; de Mendoza, C.; Holmes, S.P. Expanded Spectrum of Antiretroviral-Selected Mutations in Human Immunodeficiency Virus Type 2. J. Infect. Dis. 2020, 221, 1962–1972. [Google Scholar] [CrossRef]
- Han, L.; He, H.; Li, F.; Cui, X.; Xie, D.; Liu, Y.; Zheng, X.; Bai, H.; Wang, S.; Bo, X. Inferring infection patterns based on a connectivity map of host transcriptional responses. Sci. Rep. 2015, 5, 15820. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Robinson, J.L.; Kocabaş, P.; Wang, H.; Cholley, P.-E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An atlas of human metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, M.; da Veiga-Leprevost, F.; Squizzato, S.; Park, Y.M.; Haug, K.; Carroll, A.J.; Spalding, D.; Paschall, J.; Wang, M. Discovering and linking public omics data sets using the Omics Discovery Index. Nat. Biotechnol. 2017, 35, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Grishagin, I.; Wang, Y.; Zhao, T.; Greene, J.; Obenauer, J.C.; Ngan, D.; Nguyen, D.-T.; Guha, R.; Jadhav, A. The NCATS BioPlanet–an integrated platform for exploring the universe of cellular signaling pathways for toxicology, systems biology, and chemical genomics. Front. Pharmacol. 2019, 10, 445. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-H.; Lu, G.; Chen, X.; Zhao, X.-M.; Bork, P. OGEE v2: An update of the online gene essentiality database with special focus on differentially essential genes in human cancer cell lines. Nucleic Acids Res. 2016, gkw1013. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Karlić, R.; Koren, A.; Thurman, R.; Sandstrom, R.; Lawrence, M.S.; Reynolds, A.; Rynes, E.; Vlahoviček, K.; Stamatoyannopoulos, J.A. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015, 518, 360–364. [Google Scholar] [CrossRef]
- Urán-Landaburu, L.; Berenstein, A.J.; Videla, S.; Maru, P.; Shanmugam, D.; Chernomoretz, A.; Agüero, F. TDR Targets 6: Driving drug discovery for human pathogens through intensive chemogenomic data integration. Nucleic Acids Res. 2020, 48, D992–D1005. [Google Scholar] [CrossRef]
- Allahyari, M.; Pouriyeh, S.; Assefi, M.; Safaei, S.; Trippe, E.D.; Gutierrez, J.B.; Kochut, K. A brief survey of text mining: Classification, clustering and extraction techniques. arXiv 2017, arXiv:1707.02919. [Google Scholar]
- Odell, S.G.; Lazo, G.R.; Woodhouse, M.R.; Hane, D.L.; Sen, T.Z. The art of curation at a biological database: Principles and application. Curr. Plant Biol. 2017, 11, 2–11. [Google Scholar] [CrossRef]
- Gligorijević, V.; Pržulj, N. Methods for biological data integration: Perspectives and challenges. J. R. Soc. Interface 2015, 12, 20150571. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.E.; Pelz, O.; Buhlmann, S.; Kerr, G.; Horn, T.; Boutros, M. GenomeRNAi: A database for cell-based and in vivo RNAi phenotypes, 2013 update. Nucleic Acids Res. 2013, 41, D1021–D1026. [Google Scholar] [CrossRef] [PubMed]
- Pawson, A.J.; Sharman, J.L.; Benson, H.E.; Faccenda, E.; Alexander, S.P.; Buneman, O.P.; Davenport, A.P.; McGrath, J.C.; Peters, J.A.; Southan, C. The IUPHAR/BPS Guide to PHARMACOLOGY: An expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014, 42, D1098–D1106. [Google Scholar] [CrossRef] [PubMed]
- Araki, H.; Knapp, C.; Tsai, P.; Print, C. GeneSetDB: A comprehensive meta-database, statistical and visualisation framework for gene set analysis. FEBS Open Bio 2012, 2, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, S.-J.; Yang, S.-Y.; Peng, J.-W.; Wang, S.-N.; Wang, F.-Y.; Song, Y.-X.; Qi, T.; Li, Y.-X.; Li, Y.-Y. DNetDB: The human disease network database based on dysfunctional regulation mechanism. BMC Syst. Biol. 2016, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Zhang, C.; Arif, M.; Liu, Z.; Benfeitas, R.; Bidkhori, G.; Deshmukh, S.; Al-Shobky, M.; Lovric, A.; Boren, J. TCSBN: A database of tissue and cancer specific biological networks. Nucleic Acids Res. 2018, 46, D595–D600. [Google Scholar] [CrossRef]
- von Eichborn, J.; Murgueitio, M.S.; Dunkel, M.; Koerner, S.; Bourne, P.E.; Preissner, R. PROMISCUOUS: A database for network-based drug-repositioning. Nucleic Acids Res. 2010, 39, D1060–D1066. [Google Scholar] [CrossRef]
- Davis, A.P.; Murphy, C.G.; Johnson, R.; Lay, J.M.; Lennon-Hopkins, K.; Saraceni-Richards, C.; Sciaky, D.; King, B.L.; Rosenstein, M.C.; Wiegers, T.C. The comparative toxicogenomics database: Update 2013. Nucleic Acids Res. 2013, 41, D1104–D1114. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef]
- Cotto, K.C.; Wagner, A.H.; Feng, Y.-Y.; Kiwala, S.; Coffman, A.C.; Spies, G.; Wollam, A.; Spies, N.C.; Griffith, O.L.; Griffith, M. DGIdb 3.0: A redesign and expansion of the drug–gene interaction database. Nucleic Acids Res. 2018, 46, D1068–D1073. [Google Scholar] [CrossRef]
- Frolkis, A.; Knox, C.; Lim, E.; Jewison, T.; Law, V.; Hau, D.D.; Liu, P.; Gautam, B.; Ly, S.; Guo, A.C. SMPDB: The small molecule pathway database. Nucleic Acids Res. 2010, 38, D480–D487. [Google Scholar] [CrossRef] [PubMed]
- Shameer, K.; Glicksberg, B.S.; Hodos, R.; Johnson, K.W.; Badgeley, M.A.; Readhead, B.; Tomlinson, M.S.; O’Connor, T.; Miotto, R.; Kidd, B.A. Systematic analyses of drugs and disease indications in RepurposeDB reveal pharmacological, biological and epidemiological factors influencing drug repositioning. Brief. Bioinform. 2018, 19, 656–678. [Google Scholar] [CrossRef] [PubMed]
- Chelliah, V.; Juty, N.; Ajmera, I.; Ali, R.; Dumousseau, M.; Glont, M.; Hucka, M.; Jalowicki, G.; Keating, S.; Knight-Schrijver, V.; et al. BioModels: Ten-year anniversary. Nucleic Acids Res. 2014, 43, D542–D548. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, E.M.; Whirl-Carrillo, M.; Garten, Y.; Altman, R.B.; Klein, T.E. From pharmacogenomic knowledge acquisition to clinical applications: The PharmGKB as a clinical pharmacogenomic biomarker resource. Biomark. Med. 2011, 5, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2017, 45, D12. [Google Scholar]
- Duan, Q.; Flynn, C.; Niepel, M.; Hafner, M.; Muhlich, J.L.; Fernandez, N.F.; Rouillard, A.D.; Tan, C.M.; Chen, E.Y.; Golub, T.R. LINCS Canvas Browser: Interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucleic Acids Res. 2014, 42, W449–W460. [Google Scholar] [CrossRef]
- Han, H.-W.; Hahn, S.; Jeong, H.Y.; Jee, J.-H.; Nam, M.-O.; Kim, H.K.; Lee, D.H.; Lee, S.-Y.; Choi, D.K.; Yu, J.H. LINCS L1000 dataset-based repositioning of CGP-60474 as a highly potent anti-endotoxemic agent. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Aksoy, B.A.; Dančík, V.; Smith, K.; Mazerik, J.N.; Ji, Z.; Gross, B.; Nikolova, O.; Jaber, N.; Califano, A.; Schreiber, S.L. CTD2 Dashboard: A searchable web interface to connect validated results from the Cancer Target Discovery and Development Network. Database 2017, 2017, bax054. [Google Scholar] [CrossRef]
- Cancer Therapeutics Response Portal: A CTD² Network Resource for Mining Candidate Cancer Dependencies. Available online: https://ocg.cancer.gov/e-newsletter-issue/issue-11/cancer-therapeutics-response-portal-ctd%C2%B2-network (accessed on 21 September 2020).
- Coussens, N.P.; Sittampalam, G.S.; Guha, R.; Brimacombe, K.; Grossman, A.; Chung, T.D.; Weidner, J.R.; Riss, T.; Trask, O.J.; Auld, D. Assay guidance manual: Quantitative biology and pharmacology in preclinical drug discovery. Clin. Transl. Sci. 2018, 11, 461–470. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhu, H.; Shinn, P.; Ngan, D.; Ye, L.; Thakur, A.; Grewal, G.; Zhao, T.; Southall, N.; Hall, M.D. The NCATS Pharmaceutical Collection: A 10-year update. Drug Discov. Today 2019, 24, 2341–2349. [Google Scholar] [CrossRef] [PubMed]
- Probe Reports from the NIH Molecular Libraries Program, National Center for Biotechnology Information (US). Available online: https://www.ncbi.nlm.nih.gov/books/NBK47352/ (accessed on 21 September 2020).
- Zhang, R.; Xie, X. Tools for GPCR drug discovery. Acta Pharmacol. Sin. 2012, 33, 372–384. [Google Scholar] [CrossRef] [PubMed]
- So, R.W.; Chung, S.W.; Lau, H.H.; Watts, J.J.; Gaudette, E.; Al-Azzawi, Z.A.; Bishay, J.; Lin, L.T.-W.; Joung, J.; Wang, X. Application of CRISPR genetic screens to investigate neurological diseases. Mol. Neurodegener. 2019, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Hijmans, E.M.; Mullenders, J.; Brummelkamp, T.R.; Velds, A.; Heimerikx, M.; Kerkhoven, R.M.; Madiredjo, M.; Nijkamp, W.; Weigelt, B. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004, 428, 431–437. [Google Scholar] [CrossRef]
- Turanli, B.; Altay, O.; Borén, J.; Turkez, H.; Nielsen, J.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Systems biology based drug repositioning for development of cancer therapy. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Mohr, S.E.; Perrimon, N. RNAi screening: New approaches, understandings, and organisms. Wiley Interdiscip. Rev. Rna 2012, 3, 145–158. [Google Scholar] [CrossRef]
- Perwitasari, O.; Bakre, A.; Tompkins, S.M.; Tripp, R.A. siRNA genome screening approaches to therapeutic drug repositioning. Pharmaceuticals 2013, 6, 124–160. [Google Scholar] [CrossRef]
- Fareh, M.; Yeom, K.-H.; Haagsma, A.C.; Chauhan, S.; Heo, I.; Joo, C. TRBP ensures efficient Dicer processing of precursor microRNA in RNA-crowded environments. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.-K.; Elledge, S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Takai, A.; Dang, H.; Oishi, N.; Khatib, S.; Martin, S.P.; Dominguez, D.A.; Luo, J.; Bagni, R.; Wu, X.; Powell, K. Genome-wide RNAi Screen identifies PMPCB as a therapeutic vulnerability in EpCAM+ hepatocellular carcinoma. Cancer Res. 2019, 79, 2379–2391. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, E.; Milazzo, J.P.; Wang, Z.; Kinney, J.B.; Vakoc, C.R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015, 33, 661. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262. [Google Scholar] [CrossRef]
- Rouet, P.; Smih, F.; Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 1994, 14, 8096–8106. [Google Scholar] [CrossRef]
- Kurata, M.; Yamamoto, K.; Moriarity, B.S.; Kitagawa, M.; Largaespada, D.A. CRISPR/Cas9 library screening for drug target discovery. J. Hum. Genet. 2018, 63, 179–186. [Google Scholar] [CrossRef]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Zhang, F. High-throughput functional genomics using CRISPR–Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Koike-Yusa, H.; Li, Y.; Tan, E.-P.; Velasco-Herrera, M.D.C.; Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014, 32, 267. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Sinha, C.; Kalyana-Sundaram, S.; Chinnaiyan, A.M. Landscape of gene fusions in epithelial cancers: Seq and ye shall find. Genome Med. 2015, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef]
- Talevi, A. Drug repositioning: Current approaches and their implications in the precision medicine era. Expert Rev. Precis. Med. Drug Dev. 2018, 3, 49–61. [Google Scholar] [CrossRef]
- John-Harris, C.; Hill, R.; Sheppard, D.; Slater, M.; Stouten, P. The design and application of target-focused compound libraries. Comb. Chem. High Throughput Screen. 2011, 14, 521–531. [Google Scholar] [CrossRef]
- Goh, K.-I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.-L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, 45, gkw943. [Google Scholar] [CrossRef]
- Zhu, F.; Han, B.; Kumar, P.; Liu, X.; Ma, X.; Wei, X.; Huang, L.; Guo, Y.; Han, L.; Zheng, C. Update of TTD: Therapeutic target database. Nucleic Acids Res. 2010, 38, D787–D791. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Desai, R.J.; Handy, D.E.; Wang, R.; Schneeweiss, S.; Barabási, A.-L.; Loscalzo, J. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Cheng, F.; Kovács, I.A.; Barabási, A.-L. Network-based prediction of drug combinations. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Conte, F.; Fiscon, G.; Licursi, V.; Bizzarri, D.; D’Antò, T.; Farina, L.; Paci, P. A paradigm shift in medicine: A comprehensive review of network-based approaches. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194416. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.; Ester, M.; Kriegel, H.-P.; Xu, X. Density-based clustering in spatial databases: The algorithm gdbscan and its applications. Data Min. Knowl. Discov. 1998, 2, 169–194. [Google Scholar] [CrossRef]
- Wang, W.; Yang, J.; Muntz, R. STING: A statistical information grid approach to spatial data mining. In Proceedings of the VLDB, Athens, Greece, 25–29 August 1997; pp. 186–195. [Google Scholar]
- Hodgkin, J. Seven types of pleiotropy. Int. J. Dev. Biol. 2002, 42, 501–505. [Google Scholar]
- Lu, J.; Chen, L.; Yin, J.; Huang, T.; Bi, Y.; Kong, X.; Zheng, M.; Cai, Y.-D. Identification of new candidate drugs for lung cancer using chemical–chemical interactions, chemical–protein interactions and a K-means clustering algorithm. J. Biomol. Struct. Dyn. 2016, 34, 906–917. [Google Scholar] [CrossRef]
- Mei, J.-P.; Kwoh, C.-K.; Yang, P.; Li, X.-L.; Zheng, J. Drug–target interaction prediction by learning from local information and neighbors. Bioinformatics 2013, 29, 238–245. [Google Scholar] [CrossRef]
- Köhler, S.; Bauer, S.; Horn, D.; Robinson, P.N. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 2008, 82, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Aittokallio, T.; Schwikowski, B. Graph-based methods for analysing networks in cell biology. Brief. Bioinform. 2006, 7, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Emig, D.; Ivliev, A.; Pustovalova, O.; Lancashire, L.; Bureeva, S.; Nikolsky, Y.; Bessarabova, M. Drug target prediction and repositioning using an integrated network-based approach. PLoS ONE 2013, 8, e60618. [Google Scholar] [CrossRef]
- Vanunu, O.; Magger, O.; Ruppin, E.; Shlomi, T.; Sharan, R. Associating genes and protein complexes with disease via network propagation. PLoS Comput. Biol. 2010, 6, e1000641. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.; Navarro, C.; Cano, C.; Fajardo, W.; Blanco, A. DrugNet: Network-based drug–disease prioritization by integrating heterogeneous data. Artif. Intell. Med. 2015, 63, 41–49. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef]
- Turanli, B.; Zhang, C.; Kim, W.; Benfeitas, R.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Discovery of therapeutic agents for prostate cancer using genome-scale metabolic modeling and drug repositioning. EBioMedicine 2019, 42, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Raškevičius, V.; Mikalayeva, V.; Antanavičiūtė, I.; Ceslevičienė, I.; Skeberdis, V.A.; Kairys, V.; Bordel, S. Genome scale metabolic models as tools for drug design and personalized medicine. PLoS ONE 2018, 13, e0190636. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Nookaew, I.; Jacobson, P.; Walley, A.J.; Froguel, P.; Carlsson, L.M.; Uhlen, M. Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol. Syst. Biol. 2013, 9, 649. [Google Scholar] [CrossRef] [PubMed]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Réda, C.; Kaufmann, E.; Delahaye-Duriez, A. Machine learning applications in drug development. Comput. Struct. Biotechnol. J. 2020, 18, 241–252. [Google Scholar] [CrossRef]
- Gottlieb, A.; Stein, G.Y.; Ruppin, E.; Sharan, R. PREDICT: A method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 2011, 7, 496. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Choi, A.-s.; Nam, H. Drug repositioning of herbal compounds via a machine-learning approach. BMC Bioinform. 2019, 20, 247. [Google Scholar] [CrossRef] [PubMed]
- Bijalwan, V.; Kumar, V.; Kumari, P.; Pascual, J. KNN based machine learning approach for text and document mining. Int. J. Database Theory Appl. 2014, 7, 61–70. [Google Scholar] [CrossRef]
- Cohen, T.; Widdows, D.; Schvaneveldt, R.W.; Davies, P.; Rindflesch, T.C. Discovering discovery patterns with predication-based semantic indexing. J. Biomed. Inform. 2012, 45, 1049–1065. [Google Scholar] [CrossRef]
- Zhu, Y.; Jung, W.; Wang, F.; Che, C. Drug repurposing against Parkinson’s disease by text mining the scientific literature. Libr. Hi Tech 2020. [Google Scholar] [CrossRef]
- Li, J.; Zhu, X.; Chen, J.Y. Building disease-specific drug-protein connectivity maps from molecular interaction networks and PubMed abstracts. PLoS Comput. Biol. 2009, 5, e1000450. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019, 35, 5191–5198. [Google Scholar] [CrossRef]
- Wu, L.; Ai, N.; Liu, Y.; Wang, Y.; Fan, X. Relating anatomical therapeutic indications by the ensemble similarity of drug sets. J. Chem. Inf. Model. 2013, 53, 2154–2160. [Google Scholar] [CrossRef] [PubMed]
- Sawada, R.; Iwata, H.; Mizutani, S.; Yamanishi, Y. Target-based drug repositioning using large-scale chemical–protein interactome data. J. Chem. Inf. Model. 2015, 55, 2717–2730. [Google Scholar] [CrossRef]
- Ehrt, C.; Brinkjost, T.; Koch, O. Impact of binding site comparisons on medicinal chemistry and rational molecular design. J. Med. Chem. 2016, 59, 4121–4151. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Ma, A.; Qin, Z.S. An integrated system biology approach yields drug repositioning candidates for the treatment of heart failure. Front. Genet. 2019, 10, 916. [Google Scholar] [CrossRef]
- Vargesson, N. Thalidomide-induced teratogenesis: History and mechanisms. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Tanhaian, A.; Mohammadi, E.; Vakili-Ghartavol, R.; Saberi, M.R.; Mirzayi, M.; Jaafari, M.R. In silico and In vitro investigation of a likely pathway for anti-cancerous effect of Thrombocidin-1 as a novel anticancer peptide. Protein Pept. Lett. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, S. Molecular Docking: A Structure-Based Approach for Drug Repurposing. In Silico Drug Design; Elsevier: Amsterdam, The Netherlands, 2019; pp. 161–189. [Google Scholar]
- Haupt, V.J.; Daminelli, S.; Schroeder, M. Drug promiscuity in PDB: Protein binding site similarity is key. PLoS ONE 2013, 8, e65894. [Google Scholar] [CrossRef]
- Peyvandipour, A.; Saberian, N.; Shafi, A.; Donato, M.; Draghici, S. A novel computational approach for drug repurposing using systems biology. Bioinformatics 2018, 34, 2817–2825. [Google Scholar] [CrossRef]
- Akhoon, B.A.; Tiwari, H.; Nargotra, A. In Silico Drug Design Methods for Drug Repurposing. In Silico Drug Design; Elsevier: Amsterdam, The Netherlands, 2019; pp. 47–84. [Google Scholar]
- Shigemizu, D.; Hu, Z.; Hung, J.-H.; Huang, C.-L.; Wang, Y.; DeLisi, C. Using functional signatures to identify repositioned drugs for breast, myelogenous leukemia and prostate cancer. PLoS Comput. Biol. 2012, 8, e1002347. [Google Scholar] [CrossRef]
- Siavelis, J.C.; Bourdakou, M.M.; Athanasiadis, E.I.; Spyrou, G.M.; Nikita, K.S. Bioinformatics methods in drug repurposing for Alzheimer’s disease. Brief. Bioinform. 2016, 17, 322–335. [Google Scholar] [CrossRef]
- Dudley, J.T.; Sirota, M.; Shenoy, M.; Pai, R.K.; Roedder, S.; Chiang, A.P.; Morgan, A.A.; Sarwal, M.M.; Pasricha, P.J.; Butte, A.J. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med. 2011, 3, 96ra76. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Musa, A.; Ghoraie, L.S.; Zhang, S.-D.; Glazko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A review of connectivity map and computational approaches in pharmacogenomics. Brief. Bioinform. 2018, 19, 506–523. [Google Scholar] [PubMed]
- Liu, C.; Su, J.; Yang, F.; Wei, K.; Ma, J.; Zhou, X. Compound signature detection on LINCS L1000 big data. Mol. Biosyst. 2015, 11, 714–722. [Google Scholar] [CrossRef]
- Liu, T.-P.; Hsieh, Y.-Y.; Chou, C.-J.; Yang, P.-M. Systematic polypharmacology and drug repurposing via an integrated L1000-based Connectivity Map database mining. R. Soc. Open Sci. 2018, 5, 181321. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammadi, E.; Benfeitas, R.; Turkez, H.; Boren, J.; Nielsen, J.; Uhlen, M.; Mardinoglu, A. Applications of Genome-Wide Screening and Systems Biology Approaches in Drug Repositioning. Cancers 2020, 12, 2694. https://doi.org/10.3390/cancers12092694
Mohammadi E, Benfeitas R, Turkez H, Boren J, Nielsen J, Uhlen M, Mardinoglu A. Applications of Genome-Wide Screening and Systems Biology Approaches in Drug Repositioning. Cancers. 2020; 12(9):2694. https://doi.org/10.3390/cancers12092694
Chicago/Turabian StyleMohammadi, Elyas, Rui Benfeitas, Hasan Turkez, Jan Boren, Jens Nielsen, Mathias Uhlen, and Adil Mardinoglu. 2020. "Applications of Genome-Wide Screening and Systems Biology Approaches in Drug Repositioning" Cancers 12, no. 9: 2694. https://doi.org/10.3390/cancers12092694
APA StyleMohammadi, E., Benfeitas, R., Turkez, H., Boren, J., Nielsen, J., Uhlen, M., & Mardinoglu, A. (2020). Applications of Genome-Wide Screening and Systems Biology Approaches in Drug Repositioning. Cancers, 12(9), 2694. https://doi.org/10.3390/cancers12092694