Canonical and Noncanonical Roles of Fanconi Anemia Proteins: Implications in Cancer Predisposition

 ,
,  ,
,
Simple Summary
Abstract
1. Introduction
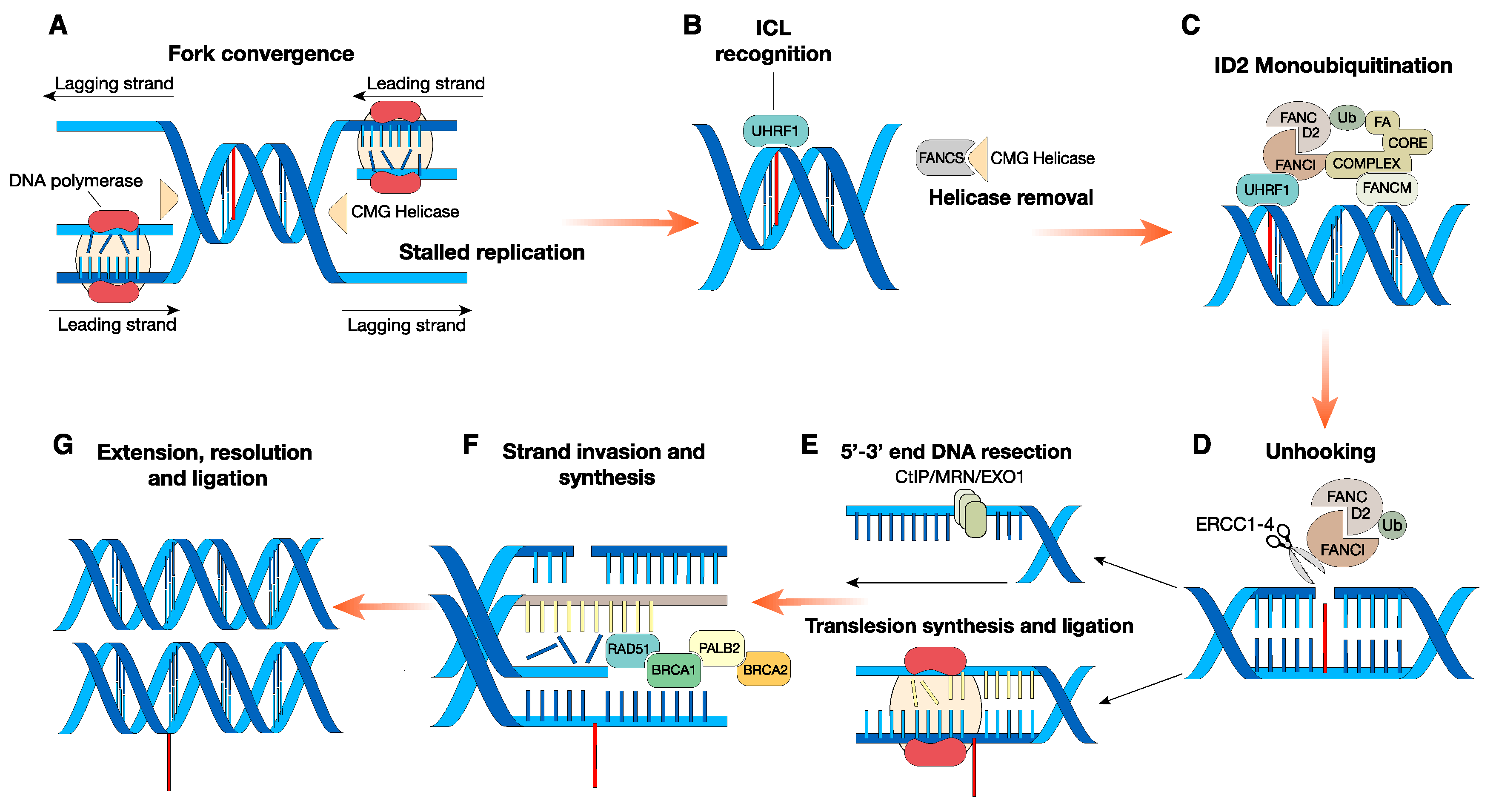
2. Canonical Role of FA Proteins: FA Signaling in the DNA ICL Repair Pathway
3. Noncanonical Role of FA Proteins
3.1. Expanded Nuclear Functions of FA Proteins outside ICLs Removal
3.1.1. FA and Replication Stress
3.1.2. FA in DSB Repair
3.1.3. FA and Transcription‒Replication Conflicts
3.2. Emerging Cytosolic Roles of FA Proteins
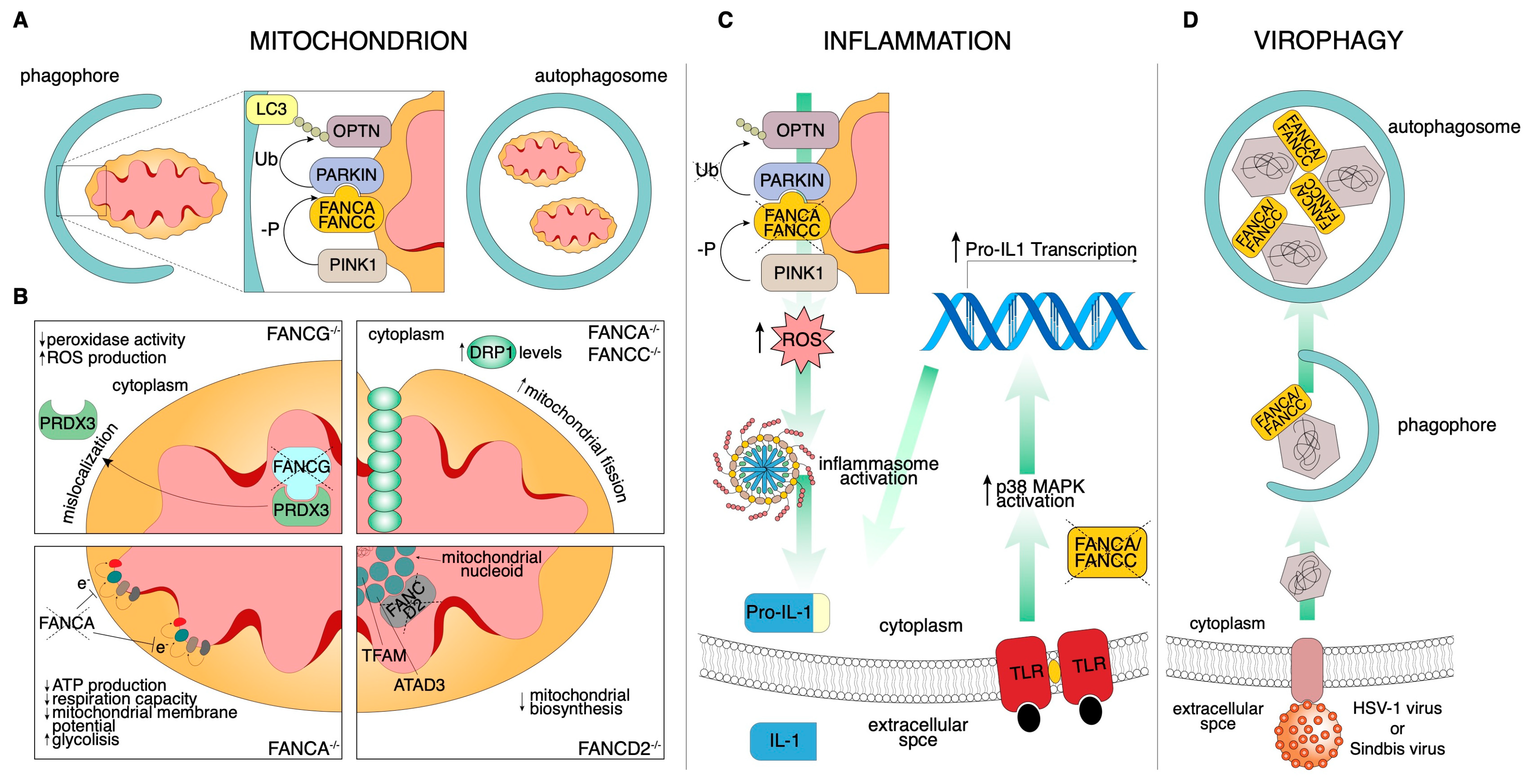
3.2.1. Mitochondria
3.2.2. Endocrinopathies
3.2.3. Inflammation
3.2.4. Virophagy
4. Hematopoietic Defects in FA Patients
5. FA Proteins and Cancer Predisposition
5.1. DNA Damage Response-Targeted Therapies in FA Patients
5.2. Clinical Implications for Emerging FA Noncanonical Roles in Cancer Predisposition
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rosenberg, P.S.; Tamary, H.; Alter, B.P. How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel. Am. J. Med. Genet. A 2011, 155, 1877–1883. [Google Scholar] [CrossRef]
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res. 2009, 668, 4–10. [Google Scholar] [CrossRef]
- Petryk, A.; Kanakatti Shankar, R.; Giri, N.; Hollenberg, A.N.; Rutter, M.M.; Nathan, B.; Lodish, M.; Alter, B.P.; Stratakis, C.A.; Rose, S.R. Endocrine Disorders in Fanconi Anemia: Recommendations for Screening and Treatment. J. Clin. Endocrinol. Metab. 2015, 100, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H.; Nathan, D.G.; Ginsburg, D.; Look, A.T.; Fisher, D.E.; Lux, S. Nathan and Oski’s Hematology of Infancy and Childhood E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2008; ISBN 978-1-4377-2130-0. [Google Scholar]
- Alter, B.P. Inherited bone marrow failure syndromes: Considerations pre- and posttransplant. Blood 2017, 130, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Giampietro, P.F.; Verlander, P.C.; Davis, J.G.; Auerbach, A.D. Diagnosis of Fanconi anemia in patients without congenital malformations: An international Fanconi Anemia Registry Study. Am. J. Med. Genet. 1997, 68, 58–61. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Zhang, J.; Dewar, J.M.; Budzowska, M.; Motnenko, A.; Cohn, M.A.; Walter, J.C. DNA interstrand cross-link repair requires replication fork convergence. Nat. Struct. Mol. Biol. 2015, 22, 242–247. [Google Scholar] [CrossRef]
- Singh, T.R.; Ali, A.M.; Paramasivam, M.; Pradhan, A.; Wahengbam, K.; Seidman, M.M.; Meetei, A.R. ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res. 2013, 73, 4300–4310. [Google Scholar] [CrossRef]
- Wang, Y.; Leung, J.W.; Jiang, Y.; Lowery, M.G.; Do, H.; Vasquez, K.M.; Chen, J.; Wang, W.; Li, L. FANCM and FAAP24 maintain genome stability via cooperative as well as unique functions. Mol. Cell 2013, 49, 997–1009. [Google Scholar] [CrossRef]
- Tian, Y.; Paramasivam, M.; Ghosal, G.; Chen, D.; Shen, X.; Huang, Y.; Akhter, S.; Legerski, R.; Chen, J.; Seidman, M.M.; et al. UHRF1 contributes to DNA damage repair as a lesion recognition factor and nuclease scaffold. Cell Rep. 2015, 10, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Zhan, B.; Yoshikawa, Y.; Haas, W.; Gygi, S.P.; Cohn, M.A. UHRF1 is a sensor for DNA interstrand crosslinks and recruits FANCD2 to initiate the Fanconi anemia pathway. Cell Rep. 2015, 10, 1947–1956. [Google Scholar] [CrossRef] [PubMed]
- Basbous, J.; Constantinou, A. A tumor suppressive DNA translocase named FANCM. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Long, D.T.; Joukov, V.; Budzowska, M.; Walter, J.C. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol. Cell 2014, 56, 174–185. [Google Scholar] [CrossRef]
- Huang, Y.; Leung, J.W.C.; Lowery, M.; Matsushita, N.; Wang, Y.; Shen, X.; Huong, D.; Takata, M.; Chen, J.; Li, L. Modularized functions of the Fanconi anemia core complex. Cell Rep. 2014, 7, 1849–1857. [Google Scholar] [CrossRef]
- Hodskinson, M.R.G.; Silhan, J.; Crossan, G.P.; Garaycoechea, J.I.; Mukherjee, S.; Johnson, C.M.; Schärer, O.D.; Patel, K.J. Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol. Cell 2014, 54, 472–484. [Google Scholar] [CrossRef]
- Klein Douwel, D.; Boonen, R.A.C.M.; Long, D.T.; Szypowska, A.A.; Räschle, M.; Walter, J.C.; Knipscheer, P. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell 2014, 54, 460–471. [Google Scholar] [CrossRef]
- Murina, O.; von Aesch, C.; Karakus, U.; Ferretti, L.P.; Bolck, H.A.; Hänggi, K.; Sartori, A.A. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep. 2014, 7, 1030–1038. [Google Scholar] [CrossRef]
- Unno, J.; Itaya, A.; Taoka, M.; Sato, K.; Tomida, J.; Sakai, W.; Sugasawa, K.; Ishiai, M.; Ikura, T.; Isobe, T.; et al. FANCD2 binds CtIP and regulates DNA-end resection during DNA interstrand crosslink repair. Cell Rep. 2014, 7, 1039–1047. [Google Scholar] [CrossRef]
- Clauson, C.; Schärer, O.D.; Niedernhofer, L. Advances in Understanding the Complex Mechanisms of DNA Interstrand Cross-Link Repair. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Zimmer, J.; Tarsounas, M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016, 35, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callén, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; López-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef]
- Howlett, N.G.; Taniguchi, T.; Durkin, S.G.; D’Andrea, A.D.; Glover, T.W. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet. 2005, 14, 693–701. [Google Scholar] [CrossRef]
- Chaudhury, I.; Sareen, A.; Raghunandan, M.; Sobeck, A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res. 2013, 41, 6444–6459. [Google Scholar] [CrossRef]
- Lossaint, G.; Larroque, M.; Ribeyre, C.; Bec, N.; Larroque, C.; Décaillet, C.; Gari, K.; Constantinou, A. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol. Cell 2013, 51, 678–690. [Google Scholar] [CrossRef]
- Michl, J.; Zimmer, J.; Buffa, F.M.; McDermott, U.; Tarsounas, M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat. Struct. Mol. Biol. 2016, 23, 755–757. [Google Scholar] [CrossRef]
- Johnson, R.D.; Jasin, M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 2000, 19, 3398–3407. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell 2009, 36, 943–953. [Google Scholar] [CrossRef]
- Rosado, I.V.; Niedzwiedz, W.; Alpi, A.F.; Patel, K.J. The Walker B motif in avian FANCM is required to limit sister chromatid exchanges but is dispensable for DNA crosslink repair. Nucleic Acids Res. 2009, 37, 4360–4370. [Google Scholar] [CrossRef] [PubMed]
- Gari, K.; Décaillet, C.; Stasiak, A.Z.; Stasiak, A.; Constantinou, A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell 2008, 29, 141–148. [Google Scholar] [CrossRef] [PubMed]
- FANCM, BRCA1, and BLM Cooperatively Resolve the Replication Stress at the ALT Telomeres | PNAS. Available online: https://www.pnas.org/content/114/29/E5940 (accessed on 26 August 2020).
- Lu, R.; O’Rourke, J.J.; Sobinoff, A.P.; Allen, J.A.M.; Nelson, C.B.; Tomlinson, C.G.; Lee, M.; Reddel, R.R.; Deans, A.J.; Pickett, H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019, 10, 2252. [Google Scholar] [CrossRef]
- Maizels, N. Genomic Stability: FANCJ-Dependent G4 DNA Repair. Curr. Biol. 2008, 18, R613–R614. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shin-ya, K.; Brosh, R.M. FANCJ Helicase Defective in Fanconia Anemia and Breast Cancer Unwinds G-Quadruplex DNA to Defend Genomic Stability. Mol. Cell. Biol. 2008, 28, 4116–4128. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.G.; Spies, M. G-quadruplex recognition and remodeling by the FANCJ helicase. Nucleic Acids Res. 2016, 44, 8742–8753. [Google Scholar] [CrossRef] [PubMed]
- Benitez, A.; Liu, W.; Palovcak, A.; Wang, G.; Moon, J.; An, K.; Kim, A.; Zheng, K.; Zhang, Y.; Bai, F.; et al. FANCA Promotes DNA Double-Strand Break Repair by Catalyzing Single-Strand Annealing and Strand Exchange. Mol. Cell 2018, 71, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.D.; Kazane, K.R.; Feng, S.J.; Zelin, E.; Bray, N.L.; Schäfer, A.J.; Floor, S.N.; Corn, J.E. CRISPR-Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat. Genet. 2018, 50, 1132–1139. [Google Scholar] [CrossRef]
- García-Muse, T.; Aguilera, A. Transcription-replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 Recruitment to Transcriptional Pause Sites Is Required for R-Loop-Driven DNA Damage Repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.Y.-C.; Tsai, S.; Aristizabal, M.J.; Wells, J.P.; Coulombe, Y.; Busatto, F.F.; Chan, Y.A.; Kumar, A.; Dan Zhu, Y.; Wang, A.Y.-H.; et al. MRE11-RAD50-NBS1 promotes Fanconi Anemia R-loop suppression at transcription-replication conflicts. Nat. Commun. 2019, 10, 4265. [Google Scholar] [CrossRef]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef]
- Nalepa, G.; Enzor, R.; Sun, Z.; Marchal, C.; Park, S.J.; Yang, Y.; Tedeschi, L.; Kelich, S.; Hanenberg, H.; Clapp, D.W. Fanconi anemia signaling network regulates the spindle assembly checkpoint. J. Clin. Investig. 2013, 123, 3839–3847. [Google Scholar] [CrossRef]
- Mondal, G.; Rowley, M.; Guidugli, L.; Wu, J.; Pankratz, V.S.; Couch, F.J. BRCA2 localization to the midbody by filamin A regulates cep55 signaling and completion of cytokinesis. Dev. Cell 2012, 23, 137–152. [Google Scholar] [CrossRef]
- Schindler, D.; Hoehn, H. Fanconi anemia mutation causes cellular susceptibility to ambient oxygen. Am. J. Hum. Genet. 1988, 43, 429–435. [Google Scholar]
- Sumpter, R.; Levine, B. Emerging functions of the Fanconi anemia pathway at a glance. J. Cell Sci. 2017, 130, 2657–2662. [Google Scholar] [CrossRef]
- Garbati, M.R.; Hays, L.E.; Keeble, W.; Yates, J.E.; Rathbun, R.K.; Bagby, G.C. FANCA and FANCC modulate TLR and p38 MAPK-dependent expression of IL-1β in macrophages. Blood 2013, 122, 3197–3205. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Vaccaro, D.; Cuccarolo, P.; Columbaro, M.; Capanni, C.; Bartolucci, M.; Panfoli, I.; Morelli, A.; Dufour, C.; Cappelli, E.; et al. Mitochondrial respiratory chain Complex I defects in Fanconi anemia complementation group A. Biochimie 2013, 95, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, E.; Ravera, S.; Vaccaro, D.; Cuccarolo, P.; Bartolucci, M.; Panfoli, I.; Dufour, C.; Degan, P. Mitochondrial respiratory complex I defects in Fanconi anemia. Trends Mol. Med. 2013, 19, 513–514. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Shyamsunder, P.; Verma, R.S.; Lyakhovich, A. Damaged mitochondria in Fanconi anemia–an isolated event or a general phenomenon? Oncoscience 2014, 1, 287–295. [Google Scholar] [CrossRef]
- Kumari, U.; Ya Jun, W.; Huat Bay, B.; Lyakhovich, A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi anemia cells. Oncogene 2014, 33, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Kruyt, F.A.; Hoshino, T.; Liu, J.M.; Joseph, P.; Jaiswal, A.K.; Youssoufian, H. Abnormal microsomal detoxification implicated in Fanconi anemia group C by interaction of the FAC protein with NADPH cytochrome P450 reductase. Blood 1998, 92, 3050–3056. [Google Scholar] [CrossRef]
- Futaki, M.; Igarashi, T.; Watanabe, S.; Kajigaya, S.; Tatsuguchi, A.; Wang, J.; Liu, J.M. The FANCG Fanconi anemia protein interacts with CYP2E1: Possible role in protection against oxidative DNA damage. Carcinogenesis 2002, 23, 67–72. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.S.; Leung, K.S.; Hicks, M.J.; Hastings, P.J.; Youssoufian, H.; Plon, S.E. Defective mitochondrial peroxiredoxin-3 results in sensitivity to oxidative stress in Fanconi anemia. J. Cell Biol. 2006, 175, 225–235. [Google Scholar] [CrossRef]
- Bose, K.C.J.; Kapoor, S.B.; Mondal, K.; Ghosh, S.; Mokhamatam, B.R.; Manna, K.S.; Mukhopadhyay, S.S. Despite of DNA repair ability the Fanconi anemia mutant protein FANCGR22P destabilizes mitochondria and leads to genomic instability via FANCJ helicase. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cappelli, E.; Cuccarolo, P.; Stroppiana, G.; Miano, M.; Bottega, R.; Cossu, V.; Degan, P.; Ravera, S. Defects in mitochondrial energetic function compels Fanconi Anaemia cells to glycolytic metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1214–1221. [Google Scholar] [CrossRef]
- Rousset, S.; Nocentini, S.; Rouillard, D.; Baroche, C.; Moustacchi, E. Mitochondrial alterations in fanconi anemia fibroblasts following ultraviolet A or psoralen photoactivation. Photochem. Photobiol. 2002, 75, 159–166. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Bleazard, W.; McCaffery, J.M.; King, E.J.; Bale, S.; Mozdy, A.; Tieu, Q.; Nunnari, J.; Shaw, J.M. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1999, 1, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Shyamsunder, P.; Esner, M.; Barvalia, M.; Wu, Y.J.; Loja, T.; Boon, H.B.; Lleonart, M.E.; Verma, R.S.; Krejci, L.; Lyakhovich, A. Impaired mitophagy in Fanconi anemia is dependent on mitochondrial fission. Oncotarget 2016, 7, 58065–58074. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R.; Sirasanagandla, S.; Fernández, Á.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi anemia proteins function in mitophagy and immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef]
- Bottega, R.; Nicchia, E.; Cappelli, E.; Ravera, S.; Rocco, D.D.; Faleschini, M.; Corsolini, F.; Pierri, F.; Calvillo, M.; Russo, G.; et al. Hypomorphic FANCA mutations correlate with mild mitochondrial and clinical phenotype in Fanconi anemia. Haematologica 2018, 103, 417–426. [Google Scholar] [CrossRef]
- He, J.; Mao, C.C.; Reyes, A.; Sembongi, H.; Di Re, M.; Granycome, C.; Clippingdale, A.B.; Fearnley, I.M.; Harbour, M.; Robinson, A.J.; et al. The AAA+ protein ATAD3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J. Cell Biol. 2007, 176, 141–146. [Google Scholar] [CrossRef]
- Zhang, T.; Du, W.; Wilson, A.F.; Namekawa, S.H.; Andreassen, P.R.; Meetei, A.R.; Pang, Q. Fancd2 in vivo interaction network reveals a non-canonical role in mitochondrial function. Sci. Rep. 2017, 7, 45626. [Google Scholar] [CrossRef]
- Dillon, B.; Feben, C.; Segal, D.; Plessis, J.; Reynders, D.; Wainwright, R.; Poole, J.; Krause, A. Endocrine profiling in patients with Fanconi anemia, homozygous for a FANCG founder mutation. Mol. Genet. Genom. Med. 2020, e1351. [Google Scholar] [CrossRef]
- Ravera, S.; Degan, P.; Sabatini, F.; Columbaro, M.; Dufour, C.; Cappelli, E. Altered lipid metabolism could drive the bone marrow failure in fanconi anaemia. Br. J. Haematol. 2019, 184, 693–696. [Google Scholar] [CrossRef]
- Li, J.; Sipple, J.; Maynard, S.; Mehta, P.A.; Rose, S.R.; Davies, S.M.; Pang, Q. Fanconi Anemia Links Reactive Oxygen Species to Insulin Resistance and Obesity. Antioxid. Redox Signal. 2012, 17, 1083–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Brusadelli, M.G.; Sauter, S.; Butsch Kovacic, M.; Zhang, W.; Romick-Rosendale, L.E.; Lambert, P.F.; Setchell, K.D.R.; Wells, S.I. Lipidomic Profiling Links the Fanconi Anemia Pathway to Glycosphingolipid Metabolism in Head and Neck Cancer Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2700–2709. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.C.; Shahidi, N.T. Tumor necrosis factor-α overproduction in Fanconi’s anemia. Am. J. Hematol. 1993, 42, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Rosselli, F.; Sanceau, J.; Gluckman, E.; Wietzerbin, J.; Moustacchi, E. Abnormal lymphokine production: A novel feature of the genetic disease Fanconi anemia. II. In vitro and in vivo spontaneous overproduction of tumor necrosis factor alpha. Blood 1994, 83, 1216–1225. [Google Scholar] [CrossRef]
- Du, W.; Adam, Z.; Rani, R.; Zhang, X.; Pang, Q. Oxidative stress in Fanconi anemia hematopoiesis and disease progression. Antioxid. Redox Signal. 2008, 10, 1909–1921. [Google Scholar] [CrossRef]
- Briot, D.; Macé-Aimé, G.; Subra, F.; Rosselli, F. Aberrant activation of stress-response pathways leads to TNF-alpha oversecretion in Fanconi anemia. Blood 2008, 111, 1913–1923. [Google Scholar] [CrossRef]
- Korthof, E.T.; Svahn, J.; Peffault de Latour, R.; Terranova, P.; Moins-Teisserenc, H.; Socié, G.; Soulier, J.; Kok, M.; Bredius, R.G.M.; van Tol, M.; et al. Immunological profile of Fanconi anemia: A multicentric retrospective analysis of 61 patients. Am. J. Hematol. 2013, 88, 472–476. [Google Scholar] [CrossRef]
- Ibáñez, A.; Río, P.; Casado, J.A.; Bueren, J.A.; Fernández-Luna, J.L.; Pipaón, C. Elevated levels of IL-1β in Fanconi anaemia group A patients due to a constitutively active phosphoinositide 3-kinase-Akt pathway are capable of promoting tumour cell proliferation. Biochem. J. 2009, 422, 161–170. [Google Scholar] [CrossRef]
- Chatzinikolaou, G.; Karakasilioti, I.; Garinis, G.A. DNA damage and innate immunity: Links and trade-offs. Trends Immunol. 2014, 35, 429–435. [Google Scholar] [CrossRef]
- Pálmai-Pallag, T.; Bachrati, C.Z. Inflammation-induced DNA damage and damage-induced inflammation: A vicious cycle. Microbes Infect. 2014, 16, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Huang, W.; Hjort, E.; Eklund, E.A. Increased Fanconi C expression contributes to the emergency granulopoiesis response. J. Clin. Investig. 2013, 123, 3952–3966. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Fagerlie, S.; Christianson, T.A.; Keeble, W.; Faulkner, G.; Diaz, J.; Rathbun, R.K.; Bagby, G.C. The Fanconi Anemia Protein FANCC Binds to and Facilitates the Activation of STAT1 by Gamma Interferon and Hematopoietic Growth Factors. Mol. Cell. Biol. 2000, 20, 4724–4735. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Keeble, W.; Christianson, T.A.; Faulkner, G.R.; Bagby, G.C. FANCC interacts with Hsp70 to protect hematopoietic cells from IFN-γ/TNF-α-mediated cytotoxicity. EMBO J. 2001, 20, 4478–4489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Sejas, D.P.; Rathbun, K.R.; Bagby, G.C.; Pang, Q. The Fanconi anemia proteins functionally interact with the protein kinase regulated by RNA (PKR). J. Biol. Chem. 2004, 279, 43910–43919. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Sumpter, R.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.F.; Radtke, S.; von Kalle, C.; Balcik, B.; Bohn, K.; Mueller, R.; Schuesler, T.; Haren, M.; Reeves, L.; Cancelas, J.A.; et al. Stem cell collection and gene transfer in Fanconi anemia. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 211–219. [Google Scholar] [CrossRef]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegué, E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.S.; Pondarre, C.; et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012, 11, 36–49. [Google Scholar] [CrossRef]
- Tulpule, A.; Lensch, M.W.; Miller, J.D.; Austin, K.; D’Andrea, A.; Schlaeger, T.M.; Shimamura, A.; Daley, G.Q. Knockdown of Fanconi anemia genes in human embryonic stem cells reveals early developmental defects in the hematopoietic lineage. Blood 2010, 115, 3453–3462. [Google Scholar] [CrossRef]
- Zhang, H.; Kozono, D.E.; O’Connor, K.W.; Vidal-Cardenas, S.; Rousseau, A.; Hamilton, A.; Moreau, L.; Gaudiano, E.F.; Greenberger, J.; Bagby, G.; et al. TGF-β Inhibition Rescues Hematopoietic Stem Cell Defects and Bone Marrow Failure in Fanconi Anemia. Cell Stem Cell 2016, 18, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.U.W.; Milsom, M.D.; Harris, C.E.; Vyas, R.; Brumme, K.M.; Parmar, K.; Moreau, L.A.; Schambach, A.; Park, I.H.; London, W.B.; et al. Overcoming reprogramming resistance of Fanconi anemia cells. Blood 2012, 119, 5449–5457. [Google Scholar] [CrossRef] [PubMed]
- Hadjur, S.; Ung, K.; Wadsworth, L.; Dimmick, J.; Rajcan-Separovic, E.; Scott, R.W.; Buchwald, M.; Jirik, F.R. Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding Fancc and Cu/Zn superoxide dismutase. Blood 2001, 98, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Yabe, M.; Yabe, H.; Morimoto, T.; Fukumura, A.; Ohtsubo, K.; Koike, T.; Yoshida, K.; Ogawa, S.; Ito, E.; Okuno, Y.; et al. The phenotype and clinical course of Japanese Fanconi Anaemia infants is influenced by patient, but not maternal ALDH2 genotype. Br. J. Haematol. 2016, 175, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Hira, A.; Yoshida, K.; Muramatsu, H.; Okuno, Y.; Shiraishi, Y.; Anmae, M.; Yasuda, J.; Tadaka, S.; Kinoshita, K.; et al. Pathogenic mutations identified by a multimodality approach in 117 Japanese Fanconi anemia patients. Haematologica 2019, 104, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Hira, A.; Yabe, H.; Yoshida, K.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Miyano, S.; Nakamura, J.; Kojima, S.; et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood 2013, 122, 3206–3209. [Google Scholar] [CrossRef] [PubMed]
- Bakker, S.T.; de Winter, J.P.; te Riele, H. Learning from a paradox: Recent insights into Fanconi anaemia through studying mouse models. Dis. Model. Mech. 2013, 6, 40–47. [Google Scholar] [CrossRef]
- Easton, D.F.; Bishop, D.T.; Ford, D.; Crockford, G.P. Genetic linkage analysis in familial breast and ovarian cancer: Results from 214 families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1993, 52, 678–701. [Google Scholar]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.C. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Higuera, I.; Taniguchi, T.; Ganesan, S.; Meyn, M.S.; Timmers, C.; Hejna, J.; Grompe, M.; D’Andrea, A.D. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell 2001, 7, 249–262. [Google Scholar] [CrossRef]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Rafnar, T.; Gudbjartsson, D.F.; Sulem, P.; Jonasdottir, A.; Sigurdsson, A.; Jonasdottir, A.; Besenbacher, S.; Lundin, P.; Stacey, S.N.; Gudmundsson, J.; et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 2011, 43, 1104–1107. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ruark, E.; Xicola, R.M.M.; Ramsay, E.; Hughes, D.; Warren-Perry, M.; Snape, K.; Eccles, D.; Evans, D.G.; et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat. Genet. 2012, 44, 475–476. [Google Scholar] [CrossRef]
- Easton, D.F.; Lesueur, F.; Decker, B.; Michailidou, K.; Li, J.; Allen, J.; Luccarini, C.; Pooley, K.A.; Shah, M.; Bolla, M.K.; et al. No evidence that protein truncating variants in BRIP1 are associated with breast cancer risk: Implications for gene panel testing. J. Med. Genet. 2016, 53, 298–309. [Google Scholar] [CrossRef]
- Thompson, E.R.; Doyle, M.A.; Ryland, G.L.; Rowley, S.M.; Choong, D.Y.H.; Tothill, R.W.; Thorne, H.; Barnes, D.R.; Li, J.; Ellul, J.; et al. Exome Sequencing Identifies Rare Deleterious Mutations in DNA Repair Genes FANCC and BLM as Potential Breast Cancer Susceptibility Alleles. PLOS Genet. 2012, 8, e1002894. [Google Scholar] [CrossRef]
- Kiiski, J.I.; Pelttari, L.M.; Khan, S.; Freysteinsdottir, E.S.; Reynisdottir, I.; Hart, S.N.; Shimelis, H.; Vilske, S.; Kallioniemi, A.; Schleutker, J.; et al. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 15172–15177. [Google Scholar] [CrossRef]
- Easton, D.F.; Pharoah, P.D.P.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Bluteau, D.; Lespinasse, J.; Pujol, R.; Vasquez, N.; d’Enghien, C.D.; Stoppa-Lyonnet, D.; Leblanc, T.; Soulier, J.; Surrallés, J. Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Bakker, S.T.; van de Vrugt, H.J.; Rooimans, M.A.; Oostra, A.B.; Steltenpool, J.; Delzenne-Goette, E.; van der Wal, A.; van der Valk, M.; Joenje, H.; te Riele, H.; et al. Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum. Mol. Genet. 2009, 18, 3484–3495. [Google Scholar] [CrossRef] [PubMed]
- Catucci, I.; Osorio, A.; Arver, B.; Neidhardt, G.; Bogliolo, M.; Zanardi, F.; Riboni, M.; Minardi, S.; Pujol, R.; Azzollini, J.; et al. Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer, chemotherapy toxicity and may display chromosome fragility. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 452–457. [Google Scholar] [CrossRef]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef]
- Steinberg-Shemer, O.; Goldberg, T.A.; Yacobovich, J.; Levin, C.; Koren, A.; Revel-Vilk, S.; Ben-Ami, T.; Kuperman, A.A.; Shkalim Zemer, V.; Toren, A.; et al. Characterization and genotype-phenotype correlation of patients with Fanconi anemia in a multi-ethnic population. Haematologica 2019. [Google Scholar] [CrossRef]
- Romick-Rosendale, L.E.; Lui, V.W.Y.; Grandis, J.R.; Wells, S.I. The Fanconi Anemia Pathway: Repairing the Link Between DNA Damage and Squamous Cell Carcinoma. Mutat. Res. 2013, 0, 78–88. [Google Scholar] [CrossRef]
- Rochowski, A.; Olson, S.B.; Alonzo, T.A.; Gerbing, R.B.; Lange, B.J.; Alter, B.P. Patients with Fanconi Anemia and AML have Different Cytogenetic Clones than de novo Cases of AML. Pediatr. Blood Cancer 2012, 59, 922–924. [Google Scholar] [CrossRef]
- Maung, K.Z.Y.; Leo, P.J.; Bassal, M.; Casolari, D.A.; Gray, J.X.; Bray, S.C.; Pederson, S.; Singhal, D.; Samaraweera, S.E.; Nguyen, T.; et al. Rare variants in Fanconi anemia genes are enriched in acute myeloid leukemia. Blood Cancer J. 2018, 8, 50. [Google Scholar] [CrossRef]
- Myers, K.C.; Sauter, S.; Zhang, X.; Bleesing, J.J.; Davies, S.M.; Wells, S.I.; Mehta, P.A.; Kumar, A.; Marmer, D.; Marsh, R.; et al. Impaired immune function in children and adults with Fanconi anemia. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef]
- Beddok, A.; Krieger, S.; Castera, L.; Stoppa-Lyonnet, D.; Thariat, J. Management of Fanconi Anemia patients with head and neck carcinoma: Diagnosis and treatment adaptation. Oral Oncol. 2020, 108, 104816. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun. Signal. 2017, 15, 41. [Google Scholar] [CrossRef] [PubMed]
- Rageul, J.; Kim, H. Fanconi anemia and the underlying causes of genomic instability. Environ. Mol. Mutagen. 2020, 61, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Renaudin, X.; Rosselli, F. The FANC/BRCA Pathway Releases Replication Blockades by Eliminating DNA Interstrand Cross-Links. Genes 2020, 11, 585. [Google Scholar] [CrossRef]
- Domchek, S.M.; Tang, J.; Stopfer, J.; Lilli, D.R.; Hamel, N.; Tischkowitz, M.; Monteiro, A.N.A.; Messick, T.E.; Powers, J.; Yonker, A.; et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013, 3, 399–405. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Tian, L.; Kahkonen, M.; Schwartzentruber, J.; Kircher, M.; Majewski, J.; Dyment, D.A.; Innes, A.M.; Boycott, K.M.; Moreau, L.A.; et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015, 5, 135–142. [Google Scholar] [CrossRef]
- Gluckman, E.; Devergie, A.; Schaison, G.; Bussel, A.; Berger, R.; Sohier, J.; Bernard, J. Bone Marrow Transplantation in Fanconi Anaemia. Br. J. Haematol. 1980, 45, 557–564. [Google Scholar] [CrossRef]
- Gluckman, E.; Devergie, A.; Dutreix, J. Radiosensitivity in Fanconi anaemia: Application to the conditioning regimen for bone marrow transplantation. Br. J. Haematol. 1983, 54, 431–440. [Google Scholar] [CrossRef]
- Schifferli, A.; Kühne, T. Fanconi Anemia: Overview of the Disease and the Role of Hematopoietic Transplantation. J. Pediatr. Hematol. Oncol. 2015, 37, 335–343. [Google Scholar] [CrossRef]
- Gluckman, E.; Rocha, V.; Ionescu, I.; Bierings, M.; Harris, E.R.; Wagner, J.; Kurtzberg, J.; Champagne, M.A.; Bonfim, C.; Bittencourt, M.; et al. Results of unrelated cord blood transplant in Fanconi anemia patients: Risk factor analysis for engraftment and survival. Biol Blood Marrow Transplant. 2007, 13(9), 1073–1082. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet Lond. Engl. 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Montanuy, H.; Martínez-Barriocanal, A.; Casado, J.A.; Rovirosa, L.; Ramírez, M.J.; Nieto, R.; Carrascoso-Rubio, C.; Riera, P.; Gonzalez, A.; Lerma, E.; et al. Gefitinib and afatinib show potential efficacy for Fanconi anemia-related head and neck cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Bordi, M.; Nazio, F.; Campello, S. The Close Interconnection between Mitochondrial Dynamics and Mitophagy in Cancer. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Vernucci, E.; Tomino, C.; Molinari, F.; Limongi, D.; Aventaggiato, M.; Sansone, L.; Tafani, M.; Russo, M.A. Mitophagy and Oxidative Stress in Cancer and Aging: Focus on Sirtuins and Nanomaterials. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Macleod, K.F. Mitophagy and Mitochondrial Dysfunction in Cancer. Annu. Rev. Cancer Biol. 2020, 4, 41–60. [Google Scholar] [CrossRef]
- Chen, Q.; Lei, J.H.; Bao, J.; Wang, H.; Hao, W.; Li, L.; Peng, C.; Masuda, T.; Miao, K.; Xu, J.; et al. BRCA1 Deficiency Impairs Mitophagy and Promotes Inflammasome Activation and Mammary Tumor Metastasis. Adv. Sci. 2020, 7. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD+/SIRT1 Reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef]
- Zhang, Q.S.; Eaton, L.; Snyder, E.R.; Houghtaling, S.; Mitchell, J.B.; Finegold, M.; Van Waes, C.; Grompe, M. Tempol protects against oxidative damage and delays epithelial tumor onset in Fanconi anemia mice. Cancer Res. 2008, 68, 1601–1608. [Google Scholar] [CrossRef]
- Zhang, Q.S.; Marquez-Loza, L.; Eaton, L.; Duncan, A.W.; Goldman, D.C.; Anur, P.; Watanabe-Smith, K.; Rathbun, R.K.; Fleming, W.H.; Bagby, G.C.; et al. Fancd2-/- mice have hematopoietic defects that can be partially corrected by resveratrol. Blood 2010, 116, 5140–5148. [Google Scholar] [CrossRef]
- Kuno, A.; Hosoda, R.; Sebori, R.; Hayashi, T.; Sakuragi, H.; Tanabe, M.; Horio, Y. Resveratrol Ameliorates Mitophagy Disturbance and Improves Cardiac Pathophysiology of Dystrophin-deficient mdx Mice. Sci. Rep. 2018, 8, 15555. [Google Scholar] [CrossRef]
- Panneerselvam, J.; Xie, G.; Che, R.; Su, M.; Zhang, J.; Jia, W.; Fei, P. Distinct Metabolic Signature of Human Bladder Cancer Cells Carrying an Impaired Fanconi Anemia Tumor Suppressor Signaling Pathway. J. Proteome Res. 2016, 15, 1333–1341. [Google Scholar] [CrossRef]
- Nepal, M.; Ma, C.; Xie, G.; Jia, W.; Fei, P. Fanconi Anemia complementation group C protein in metabolic disorders. Aging 2018, 10, 1506–1522. [Google Scholar] [CrossRef]
- Van Zeeburg, H.J.T.; Snijders, P.J.F.; Wu, T.; Gluckman, E.; Soulier, J.; Surralles, J.; Castella, M.; van der Wal, J.E.; Wennerberg, J.; Califano, J.; et al. Clinical and molecular characteristics of squamous cell carcinomas from Fanconi anemia patients. J. Natl. Cancer Inst. 2008, 100, 1649–1653. [Google Scholar] [CrossRef] [PubMed]
- Sauter, S.L.; Wells, S.I.; Zhang, X.; Hoskins, E.E.; Davies, S.M.; Myers, K.C.; Mueller, R.; Panicker, G.; Unger, E.R.; Sivaprasad, U.; et al. Oral human papillomavirus is common in individuals with Fanconi anemia. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2015, 24, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Khoury, R.; Sauter, S.; Butsch Kovacic, M.; Nelson, A.S.; Myers, K.C.; Mehta, P.A.; Davies, S.M.; Wells, S.I. Risk of Human Papillomavirus Infection in Cancer-Prone Individuals: What We Know. Viruses 2018, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Vescovo, T.; Pagni, B.; Piacentini, M.; Fimia, G.M.; Antonioli, M. Regulation of Autophagy in Cells Infected with Oncogenic Human Viruses and Its Impact on Cancer Development. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Mattoscio, D.; Medda, A.; Chiocca, S. Human Papilloma Virus and Autophagy. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| FA Gene | Alternative Name and Location | Key Features | Canonical Role | Cytosolic Noncanonical Role | Cancer Type Predisposition | |
|---|---|---|---|---|---|---|
| FANCA | - | 16q24.3 | Fanconi anemia core complex | Promotes FANCD2/I heterodimer monoubiquitylation | Interact with cytochrome P450 to respond oxidative damage; mitophagy effector | - |
| FANCB | - | Xp22.2 | Fanconi anemia core complex | Promotes FANCD2/I heterodimer monoubiquitylation | - | - |
| FANCC | - | 9q22.32 | Fanconi anemia core complex | Promotes FANCD2/I heterodimer monoubiquitylation | Interact with cytochrome P450 to respond oxidative damage; mitophagy effector through PARKIN interaction; virophagy mediator | Breast cancer |
| FANCD1 | BRCA2 | 13q12. 3 | RAD51 activator | Fork stabilization; homologous recombination | Involved in mitophagy | AML; T-ALL; ALL; brain tumor; medulloblastoma; squamous cell carcinoma; breast cancer |
| FANCD2 | - | 3p25.3 | FANCI binding partner | Monoubiquitylated by the core complex; promote the unhooking | Regulation of mitochondrial nucleoid complex components Atad3 and Tufm; sustain stem cells functions through SF3Bq spliceosomal protein interaction; Involved in mitophagy | - |
| FANCE | - | 6p21.31 | Fanconi anemia core complex | Promotes FANCD2/I heterodimer monoubiquitylation | - | - |
| FANCF | - | 11p14.3 | Fanconi anemia core complex | Promotes FANCD2/I heterodimer monoubiquitylation | Involved in mitophagy; virophagy mediator | - |
| FANCG | - | 9p13.3 | Fanconi anemia core complex | Promotes FANCD2/I heterodimer monoubiquitylation | Interact with cytochrome P450 to respond oxidative damage; involved in mitochondria morphology regulation | - |
| FANCI | - | 15q26.1 | FANCD2 binding partner | Several functions in the ICL repair | Sustain stem cells functions through SF3Bq spliceosomal protein interaction | - |
| FANCJ | BRIP1 | 17q23.2 | BRCA1-interacting protein helicase | Promotes HR through BRCA1 binding | DNA-dependent ATPase activity | Ovarian cancer |
| FANCL | - | 2p16.1 | E3 ubiquitin ligase of the Fanconi anemia core complex | Promotes FANCD2/I heterodimer monoubiquitylation | β-Catenin ubiquitination; involved in mitophagy; virophagy mediator | - |
| FANCM | - | 14q21. 2 | ATR-activated DNA helicase | Recruits the FA core complex | - | Breast cancer |
| FANCN | PALB2 | 16p12.2 | Partner and localizer of BRCA2 | Promotes HR and RAD51 activity | - | AML; medulloblastoma; neuroblastoma; Wilms Tumor; breast cancer |
| FANCO | RAD51C | 17q23 | RAD51 paralog | HR | - | Ovarian cancer |
| FANCP | SLX4 | 16p13.3 | Multidomain scaffold protein | Nuclease scaffold for ICL unhooking | - | |
| FANCQ | ERCC4 | 16p13.12 | DNA repair endonuclease | DNA incision for ICL unhooking | - | |
| FANCR | RAD51 | 15q15.1 | DNA recombinase | Fork stabilization and HR | Regulation of axonal branching through Netrin-1 modulation | |
| FANCS | BRCA1 | 17q21.31 | E3 ubiquitin ligase | CMG complex unloading; Homologous recombination | Involved in mitophagy | Breast, ovarian cancer and leukemia |
| FANCT | UBE2T | 1q32.1 | E2 ubiquitin ligase of the Fanconi anemia core complex | Promotes FANCD2/I heterodimer monoubiquitylation | - | |
| FANCU | XRCC2 | 7q36.1 | DNA repair protein | HR | - | |
| FANCV | REV7 | 1p36.22 | DNA polymerase | Translesion DNA synthesis | - | |
| FANCW | - | 16q23.1 | E3 ubiquitin ligase | Essential for RAD51 turnover | - | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milletti, G.; Strocchio, L.; Pagliara, D.; Girardi, K.; Carta, R.; Mastronuzzi, A.; Locatelli, F.; Nazio, F. Canonical and Noncanonical Roles of Fanconi Anemia Proteins: Implications in Cancer Predisposition. Cancers 2020, 12, 2684. https://doi.org/10.3390/cancers12092684
Milletti G, Strocchio L, Pagliara D, Girardi K, Carta R, Mastronuzzi A, Locatelli F, Nazio F. Canonical and Noncanonical Roles of Fanconi Anemia Proteins: Implications in Cancer Predisposition. Cancers. 2020; 12(9):2684. https://doi.org/10.3390/cancers12092684
Chicago/Turabian StyleMilletti, Giacomo, Luisa Strocchio, Daria Pagliara, Katia Girardi, Roberto Carta, Angela Mastronuzzi, Franco Locatelli, and Francesca Nazio. 2020. "Canonical and Noncanonical Roles of Fanconi Anemia Proteins: Implications in Cancer Predisposition" Cancers 12, no. 9: 2684. https://doi.org/10.3390/cancers12092684
APA StyleMilletti, G., Strocchio, L., Pagliara, D., Girardi, K., Carta, R., Mastronuzzi, A., Locatelli, F., & Nazio, F. (2020). Canonical and Noncanonical Roles of Fanconi Anemia Proteins: Implications in Cancer Predisposition. Cancers, 12(9), 2684. https://doi.org/10.3390/cancers12092684