RAS Subcellular Localization Inversely Regulates Thyroid Tumor Growth and Dissemination


 and
and 
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
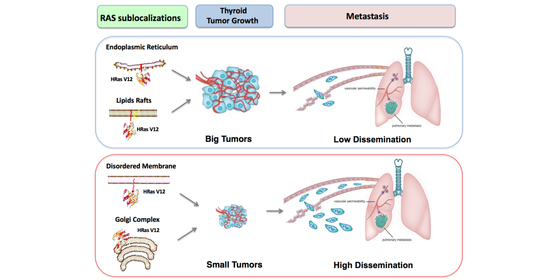
2.1. RAS Sublocalization Differentially Affects Transformed Thyroid Cells Proliferation and Dissemination
2.2. HRAS Regulates Tumor Behavior via VEGF-B Secretion
2.3. APT-1 Overexpression Correlates with Better Prognosis in Thyroid Tumors
2.4. APT-1 Levels Determine Thyroid Tumor Behavior
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids and siRNAs
4.3. Immunoblotting
4.4. Proliferation and Migration Analyses
4.5. Chick Embryo Spontaneous Metastasis Model
4.6. DNA/RNA Extraction from Chick Embryos and Quantification
4.7. Real Time qPCR
4.8. Plasma Membrane Fractionation in Sucrose Gradients
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The frequency of Ras mutations in cancer. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Calvo, F.; Crespo, P. Ras, an actor on many stages: Posttranslational modifications, localization, and site-specified events. Genes Cancer 2011, 2, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hancock, J.F. Compartmentalization of Ras proteins. J. Cell Sci. 2001, 114, 1603–1608. [Google Scholar]
- Prior, I.A.; Muncke, C.; Parton, R.G.; Hancock, J.F. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 2003, 160, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F.; Parton, R.G. Ras plasma membrane signalling platforms. Biochem. J. 2005, 389, 1–11. [Google Scholar] [CrossRef]
- Kranenburg, O.; Verlaan, I.; Moolenar, W.H. Regulation of c-Ras function: Cholesterol depletion affects caveolin association, GTP loading and signaling. Curr. Biol. 2001, 11, 1880–1884. [Google Scholar] [CrossRef]
- Matallanas, D.; Arozarena, I.; Berciano, M.T.; Aaronson, D.S.; Pellicer, A.; Lafarga, M.; Crespo, P. Differences in the inhibitory specificities of H-Ras, K-Ras and N-Ras (N17) dominant negative mutants are related to their membrane microlocalization. J. Biol. Chem. 2003, 278, 4572–4581. [Google Scholar] [CrossRef]
- Calvo, F.; Agudo-Ibáñez, L.; Crespo, P. The Ras-ERK pathway: Understanding site-specific signaling provides hope of new anti-tumor therapies. Bioessays 2010, 32, 412–421. [Google Scholar] [CrossRef]
- Matallanas, D.; Sanz-Moreno, V.; Arozarena, I.; Calvo, F.; Agudo-Ibanez, L.; Santos, E.; Berciano, M.T.; Crespo, P. Distinct utilization of effectors and biological outcomes resulting from site-specific Ras activation: Ras functions in lipid rafts and Golgi complex are dispensable for proliferation and transformation. Mol. Cell. Biol. 2006, 26, 100–116. [Google Scholar] [CrossRef]
- Casar, B.; Arozarena, I.; Sanz-Moreno, V.; Pinto, A.; Agudo-Ibáñez, L.; Marais, R.; Lewis, R.E.; Berciano, M.T.; Crespo, P. Ras subcellular localization defines extracellular signal-regulated kinase 1 and 2 substrate specificity through distinct utilization of scaffold proteins. Mol. Cell. Biol. 2009, 29, 1338–1353. [Google Scholar] [CrossRef]
- Agudo-Ibanez, L.; Nunez, F.; Calvo, F.; Berenjeno, I.M.; Bustelo, X.R.; Crespo, P. Transcriptomal profiling of site-specific Ras signals. Cell. Signal. 2007, 19, 2264–2276. [Google Scholar] [CrossRef] [PubMed]
- Santra, T.; Herrero, A.; Rodriguez, J.; von Kriegsheim, A.; Iglesias-Martinez, L.F.; Schwarzl, T.; Higgins, D.; Aye, T.T.; Heck, A.J.R.; Calvo, F.; et al. An Integrated Global Analysis of Compartmentalized HRAS Signaling. Cell Rep. 2019, 26, 3100–3115. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.; Casar, B.; Colon-Bolea, P.; Agudo-Ibanez, L.; Crespo, P. Defined spatiotemporal features of RAS-ERK signals dictate cell fate in MCF-7 mammary epithelial cells. Mol. Biol Cell 2016, 27, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Casar, B.; Badrock, A.P.; Jimenez, I.; Arozarena, I.; Colon-Bolea, P.; Lorenzo-Martin, L.F.; Barinaga-Rementeria, I.; Barriuso, J.; Cappitelli, V.; Donoghue, D.J.; et al. RAS at the Golgi antagonizes malignant transformation through PTPRkappa-mediated inhibition of ERK activation. Nat. Commun. 2018, 9, 3595. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, P.; Wang, G.; Zhang, H.; Zhang, Y.; Yu, Y.; Zhang, M.; Xiao, J.; Crespo, P.; Hell, J.W.; et al. Ras and Rap Signal Bidirectional Synaptic Plasticity via Distinct Subcellular Microdomains. Neuron 2018, 98, 783–800. [Google Scholar] [CrossRef]
- Agudo-Ibanez, L.; Herrero, A.; Barbacid, M.; Crespo, P. H-ras distribution and signaling in plasma membrane microdomains are regulated by acylation and deacylation events. Mol. Cell. Biol. 2015, 35, 1898–1914. [Google Scholar] [CrossRef]
- Goodwin, J.S.; Drake, K.R.; Rogers, C.; Wright, L.; Lippincott-Schwartz, J.; Philips, M.R.; Kenworthy, A.K. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell Biol. 2005, 170, 261–272. [Google Scholar] [CrossRef]
- Rocks, O.; Peyker, A.; Kahms, M.; Verveer, P.J.; Koerner, C.; Lumbierres, M.; Kuhlmann, J.; Waldmann, H.; Wittinghofer, A.; Bastiaens, P.I. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 2005, 307, 1746–1752. [Google Scholar] [CrossRef]
- Camp, L.A.; Hofmann, S.L. Purification and properties of a palmitoyl-protein thioesterase that cleaves palmitate from H-Ras. J. Biol. Chem. 1993, 268, 22566–22574. [Google Scholar]
- Duncan, J.A.; Gilman, A.G. A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein alpha subunits and p21(RAS). J. Biol. Chem. 1998, 273, 15830–15837. [Google Scholar] [CrossRef]
- Design Better Experiments. Gain More Insights. Prepare to Publish Faster. Available online: www.oncomine.org (accessed on 1 May 2019).
- Zhang, Y.; Zhu, Y.; Risch, H.A. Changing incidence of thyroid cancer. JAMA 2006, 296, 1350. [Google Scholar] [CrossRef] [PubMed]
- Wesola, M.; Jelen, M. Bethesda System in the evaluation of thyroid nodules: Review. Adv. Clin. Exp. Med. 2017, 26, 177–182. [Google Scholar] [CrossRef]
- Gharib, H.; Papini, E. Thyroid nodules: Clinical importance, assessment, and treatment. Endocrinol. Metab. Clin. North. Am. 2007, 36, 707–735. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef]
- Howell, G.M.; Hodak, S.P.; Yip, L. RAS mutations in thyroid cancer. Oncologist 2013, 18, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Karga, H.; Lee, J.K.; Vickery, A.L., Jr.; Thor, A.; Gaz, R.D.; Jameson, J.L. Ras oncogene mutations in benign and malignant thyroid neoplasms. J. Clin. Endocrinol. Metab. 1991, 73, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Basolo, F.; Pisaturo, F.; Pollina, L.E.; Fontanini, G.; Elisei, R.; Molinaro, E.; Iacconi, P.; Miccoli, P.; Pacini, F. N-ras mutation in poorly differentiated thyroid carcinomas: Correlation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid 2000, 10, 19–23. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Zhao, H.; Camp, R.L.; Pollan, M.; Herrero, A.; Pardo, J.; Wu, R.; Carcangiu, M.L.; Costa, J.; Tallini, G. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J. Clin. Oncol. 2003, 21, 3226–3235. [Google Scholar] [CrossRef]
- Volante, M.; Rapa, I.; Gandhi, M.; Bussolati, G.; Giachino, D.; Papotti, M.; Nikiforov, Y.E. RAS mutations are the predominant molecular alteration in poorly differentiated thyroid carcinomas and bear prognostic impact. J. Clin. Endocrinol. Metab. 2009, 94, 4735–4741. [Google Scholar] [CrossRef]
- Patel, S.G.; Carty, S.E.; McCoy, K.L.; Ohori, N.P.; LeBeau, S.O.; Seethala, R.R.; Nikiforova, M.N.; Nikiforov, Y.E.; Yip, L. Preoperative detection of RAS mutation may guide extent of thyroidectomy. Surgery 2017, 161, 168–175. [Google Scholar] [CrossRef]
- Censi, S.; Cavedon, E.; Bertazza, L.; Galuppini, F.; Watutantrige-Fernando, S.; De Lazzari, P.; Nacamulli, D.; Pennelli, G.; Fassina, A.; Iacobone, M.; et al. Frequency and Significance of Ras, Tert Promoter, and Braf Mutations in Cytologically Indeterminate Thyroid Nodules: A Monocentric Case Series at a Tertiary-Level Endocrinology Unit. Front. Endocrinol. 2017, 8, 273. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Ohori, N.P.; Hodak, S.P.; Carty, S.E.; LeBeau, S.O.; Ferris, R.L.; Yip, L.; Seethala, R.R.; Tublin, M.E.; Stang, M.T.; et al. Impact of mutational testing on the diagnosis and management of patients with cytologically indeterminate thyroid nodules: A prospective analysis of 1056 FNA samples. J. Clin. Endocrinol. Metab. 2011, 96, 3390–3397. [Google Scholar] [CrossRef]
- Nabhan, F.; Porter, K.; Lupo, M.A.; Randolph, G.W.; Patel, K.N.; Kloos, R.T. Heterogeneity in Positive Predictive Value of RAS Mutations in Cytologically Indeterminate Thyroid Nodules. Thyroid 2018, 28, 729–738. [Google Scholar] [CrossRef]
- Saavedra, H.I.; Knauf, J.A.; Shirokawa, J.M.; Wang, J.; Ouyang, B.; Elisei, R.; Stambrook, P.J.; Fagin, J.A. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene 2000, 19, 3948–3954. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Chick embryo chorioallantoic membrane model systems to study and visualize human tumor cell metastasis. Histochem. Cell Biol. 2008, 130, 1119–1130. [Google Scholar] [CrossRef]
- Herrero, A.; Pinto, A.; Colon-Bolea, P.; Casar, B.; Jones, M.; Agudo-Ibanez, L.; Vidal, R.; Tenbaum, S.P.; Nuciforo, P.; Valdizan, E.M.; et al. Small Molecule Inhibition of ERK Dimerization Prevents Tumorigenesis by RAS-ERK Pathway Oncogenes. Cancer Cell 2015, 28, 170–182. [Google Scholar] [CrossRef]
- Herrero, A.; Reis-Cardoso, M.; Jimenez-Gomez, I.; Doherty, C.; Agudo-Ibanez, L.; Pinto, A.; Calvo, F.; Kolch, W.; Crespo, P.; Matallanas, D. Characterisation of HRas local signal transduction networks using engineered site-specific exchange factors. Small GTPases 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Okamoto, R.; Nagata, Y.; Kanojia, D.; Venkatesan, S.; MT, A.; Braunstein, G.D.; Said, J.W.; Doan, N.B.; Ho, Q.; et al. Establishment and characterization of novel human primary and metastatic anaplastic thyroid cancer cell lines and their genomic evolution over a year as a primagraft. J. Clin. Endocrinol. Metab. 2015, 100, 725–735. [Google Scholar] [CrossRef]
- Morrison, J.A.; Pike, L.A.; Lund, G.; Zhou, Q.; Kessler, B.E.; Bauerle, K.T.; Sams, S.B.; Haugen, B.R.; Schweppe, R.E. Characterization of thyroid cancer cell lines in murine orthotopic and intracardiac metastasis models. Horm. Cancer 2015, 6, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Li, X. VEGF-B: A thing of beauty. Cell Res. 2010, 20, 741–744. [Google Scholar] [CrossRef]
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 2010, 464, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. Clinical utility of RAS mutations in thyroid cancer: A blurred picture now emerging clearer. BMC Med. 2016, 14, 12. [Google Scholar] [CrossRef] [PubMed]
- Untch, B.R.; Dos Anjos, V.; Garcia-Rendueles, M.E.R.; Knauf, J.A.; Krishnamoorthy, G.P.; Saqcena, M.; Bhanot, U.K.; Socci, N.D.; Ho, A.L.; Ghossein, R.; et al. Tipifarnib Inhibits HRAS-Driven Dedifferentiated Thyroid Cancers. Cancer Res. 2018, 78, 4642–4657. [Google Scholar] [CrossRef] [PubMed]
- Maae, E.; Nielsen, M.; Steffensen, K.D.; Jakobsen, E.H.; Jakobsen, A.; Sørensen, F.B. Estimation of immunohistochemical expression of VEGF in ductal carcinomas of the breast. J. Histochem. Cytochem. 2011, 59, 750–760. [Google Scholar] [CrossRef]
- Zhang, F.; Tang, Z.; Hou, X.; Lennartsson, J.; Li, Y.; Koch, A.W.; Scotney, P.; Lee, C.; Arjunan, P.; Dong, L.; et al. VEGF-B is dispensable for blood vessel growth but critical for their survival, and VEGF-B targeting inhibits pathological angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 6152–6157. [Google Scholar] [CrossRef]
- Frampton, J.E. Lenvatinib: A Review in Refractory Thyroid Cancer. Target. Oncol. 2016, 11, 115–122. [Google Scholar] [CrossRef]
- Casar, B.; Sanz-Moreno, V.; Yazicioglu, M.N.; Rodriguez, J.; Berciano, M.T.; Lafarga, M.; Cobb, M.H.; Crespo, P. Mxi2 promotes stimulus-independent ERK nuclear translocation. EMBO J. 2007, 26, 635–646. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Ibáñez, Y.; Riesco-Eizaguirre, G.; Santisteban, P.; Casar, B.; Crespo, P. RAS Subcellular Localization Inversely Regulates Thyroid Tumor Growth and Dissemination. Cancers 2020, 12, 2588. https://doi.org/10.3390/cancers12092588
García-Ibáñez Y, Riesco-Eizaguirre G, Santisteban P, Casar B, Crespo P. RAS Subcellular Localization Inversely Regulates Thyroid Tumor Growth and Dissemination. Cancers. 2020; 12(9):2588. https://doi.org/10.3390/cancers12092588
Chicago/Turabian StyleGarcía-Ibáñez, Yaiza, Garcilaso Riesco-Eizaguirre, Pilar Santisteban, Berta Casar, and Piero Crespo. 2020. "RAS Subcellular Localization Inversely Regulates Thyroid Tumor Growth and Dissemination" Cancers 12, no. 9: 2588. https://doi.org/10.3390/cancers12092588
APA StyleGarcía-Ibáñez, Y., Riesco-Eizaguirre, G., Santisteban, P., Casar, B., & Crespo, P. (2020). RAS Subcellular Localization Inversely Regulates Thyroid Tumor Growth and Dissemination. Cancers, 12(9), 2588. https://doi.org/10.3390/cancers12092588