Targeted Therapies for Epstein-Barr Virus-Associated Lymphomas



Simple Summary
Abstract
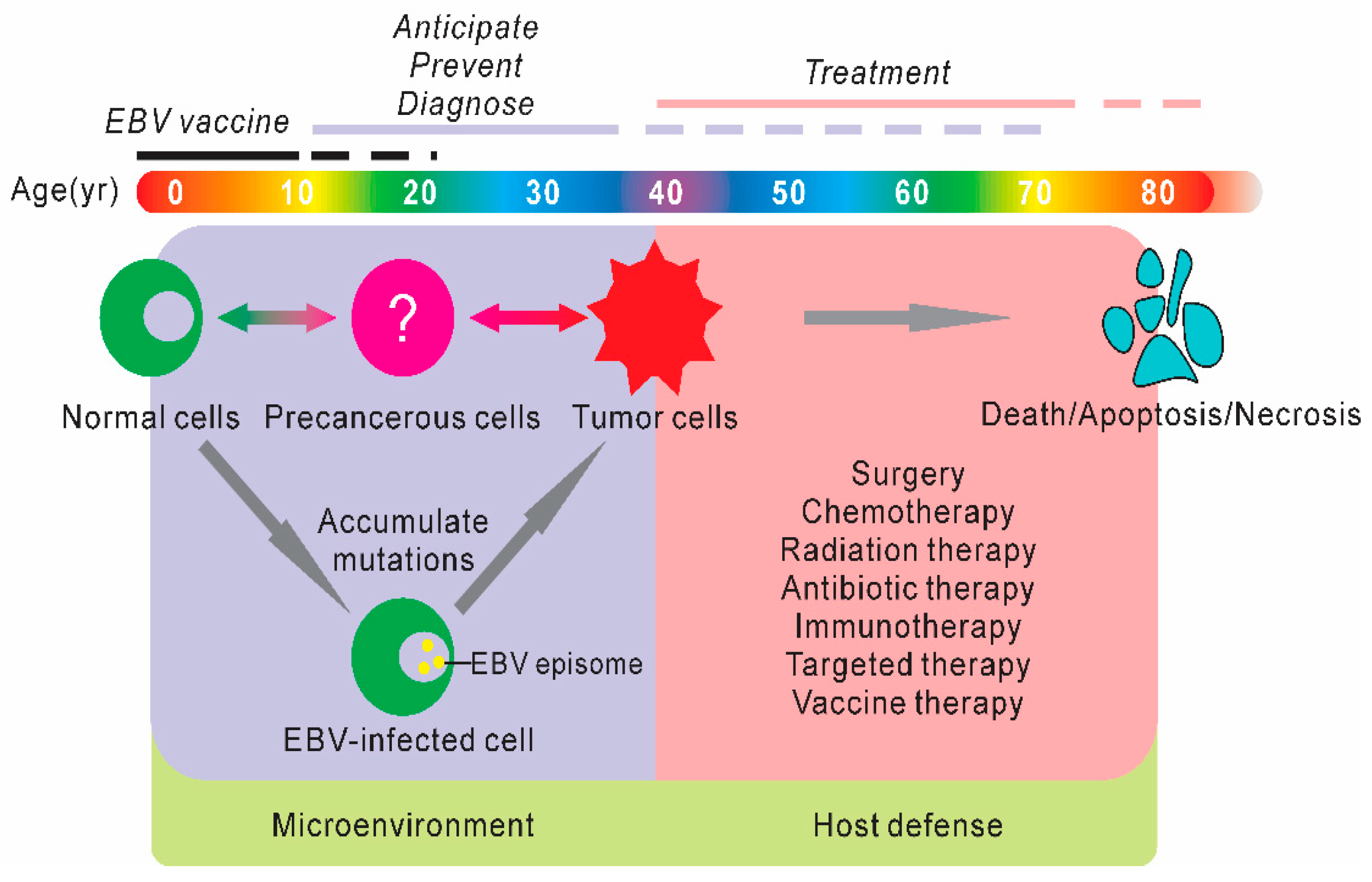
1. Introduction
2. Discovery of Small Molecule Inhibitors
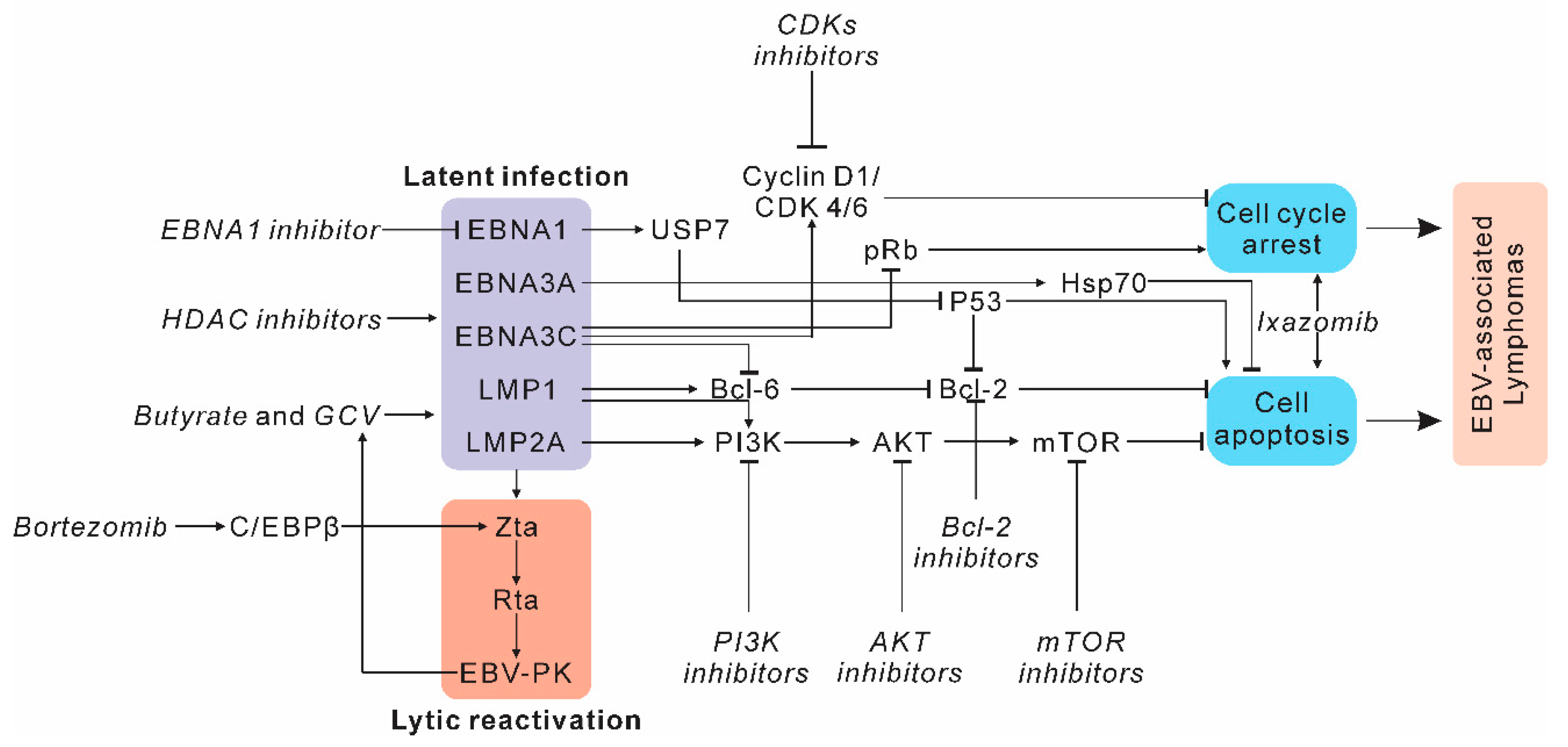
2.1. Targeting Host Factors and Signaling Pathways in EBV-Induced Lymphomas
2.2. Strategies of Specifically Targeting EBV Antigens in Lymphomas
2.3. Application of CRISPR Therapeutics for the Treatment of EBV-Associated Lymphomas
3. Immunotherapy and Cell Therapy in EBV-Associated Lymphomas
3.1. Immune Checkpoint Inhibitors (PD-1/PD-L1 Antibody)
3.2. Monoclonal Antibodies
3.3. Adoptive EBV-Specific T-Cell (EBVST) Therapy
3.4. T-Cell Receptor-Modified T-Cell Therapy
3.5. Chimeric Antigen Receptor T-Cell Therapy
4. Preventative and Therapeutic Vaccines for EBV Infection
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Mesri, E.A. Kaposi sarcoma-associated herpesvirus and other viruses in human lymphomagenesis. Curr. Top. Microbiol. Immunol. 2007, 312, 263–287. [Google Scholar] [CrossRef]
- Kutok, J.L.; Wang, F. Spectrum of Epstein-Barr virus-associated diseases. Annu. Rev. Pathol. 2006, 1, 375–404. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Thornburg, N.J.; Kulwichit, W.; Edwards, R.H.; Shair, K.H.; Bendt, K.M.; Raab-Traub, N. LMP1 signaling and activation of NF-kappaB in LMP1 transgenic mice. Oncogene 2006, 25, 288–297. [Google Scholar] [CrossRef]
- Izumi, K.M.; Kieff, E.D. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc. Natl. Acad. Sci. USA 1997, 94, 12592–12597. [Google Scholar] [CrossRef]
- Swart, R.; Ruf, I.K.; Sample, J.; Longnecker, R. Latent membrane protein 2A-mediated effects on the phosphatidylinositol 3-Kinase/Akt pathway. J. Virol. 2000, 74, 10838–10845. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Longnecker, R. Latent membrane protein 2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J. Virol. 2004, 78, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Hwang, N.; Lang, F.; Zhou, L.; Wong, J.H.-Y.; Singh, R.K.; Jha, H.C.; El-Deiry, W.S.; Du, Y.; Robertson, E.S. Quassinoid analogs with enhanced efficacy for treatment of hematologic malignancies target the PI3Kγ isoform. Commun. Biol. 2020, 3, 267. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.D.; Liu, J.; Fujimuro, M. Notch and Wnt signaling: Mimicry and manipulation by gamma herpesviruses. Sci. STKE 2006, 2006, re4. [Google Scholar] [CrossRef] [PubMed]
- Cerimele, F.; Battle, T.; Lynch, R.; Frank, D.A.; Murad, E.; Cohen, C.; Macaron, N.; Sixbey, J.; Smith, K.; Watnick, R.S.; et al. Reactive oxygen signaling and MAPK activation distinguish Epstein-Barr Virus (EBV)-positive versus EBV-negative Burkitt’s lymphoma. Proc. Natl. Acad. Sci. USA 2005, 102, 175–179. [Google Scholar] [CrossRef]
- Hausen, H.Z. The search for infectious causes of human cancers: Where and why (Nobel lecture). Angew. Chem. Int. Ed. Engl. 2009, 48, 5798–5808. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Saha, A.; Halder, S.; Upadhyay, S.K.; Lu, J.; Kumar, P.; Murakami, M.; Cai, Q.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C facilitates G1-S transition by stabilizing and enhancing the function of cyclin D1. PLoS Pathog. 2011, 7, e1001275. [Google Scholar] [CrossRef]
- Pei, Y.; Singh, R.K.; Shukla, S.K.; Lang, F.; Zhang, S.; Robertson, E.S. Epstein-Barr Virus nuclear antigen 3C facilitates cell proliferation by regulating Cyclin D2. J. Virol. 2018, 92, e00663-18. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, L.; Yaseen, N.; Sharma, S. Latent membrane protein-1 induces cyclin D2 expression, pRb hyperphosphorylation, and loss of TGF-beta 1-mediated growth inhibition in EBV-positive B cells. J. Immunol. 1995, 155, 1047–1056. [Google Scholar] [PubMed]
- Tsang, C.M.; Yip, Y.L.; Lo, K.W.; Deng, W.; To, K.F.; Hau, P.M.; Lau, V.M.; Takada, K.; Lui, V.W.; Lung, M.L.; et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3473–E3482. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef]
- Knight, J.S.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C regulates cyclin A/p27 complexes and enhances cyclin A-dependent kinase activity. J. Virol. 2004, 78, 1981–1991. [Google Scholar] [CrossRef]
- Knight, J.S.; Sharma, N.; Kalman, D.E.; Robertson, E.S. A cyclin-binding motif within the amino-terminal homology domain of EBNA3C binds cyclin A and modulates cyclin A-dependent kinase activity in Epstein-Barr virus-infected cells. J. Virol. 2004, 78, 12857–12867. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Flemington, E.K. The Epstein-Barr virus bZIP transcription factor Zta causes G0/G1 cell cycle arrest through induction of cyclin-dependent kinase inhibitors. EMBO J. 1996, 15, 2748–2759. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. A BH3 mimetic for killing cancer cells. Cell 2016, 165, 1560. [Google Scholar] [CrossRef]
- Henderson, S.; Rowe, M.; Gregory, C.; Croom-Carter, D.; Wang, F.; Longnecker, R.; Kieff, E.; Rickinson, A. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell 1991, 65, 1107–1115. [Google Scholar] [CrossRef]
- Pei, Y.; Banerjee, S.; Jha, H.C.; Sun, Z.; Robertson, E.S. An essential EBV latent antigen 3C binds Bcl6 for targeted degradation and cell proliferation. PLoS Pathog. 2017, 13, e1006500. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Zhang, Y.; Teng, M.; Jiang, C.; Schineller, M.; Zhao, B.; Doench, J.G.; O’Reilly, R.J.; Cesarman, E.; Giulino-Roth, L.; et al. DNA methylation enzymes and PRC1 restrict B-cell Epstein-Barr virus oncoprotein expression. Nat. Microbiol. 2020, 5, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Lu, J.; Cai, Q.; Saha, A.; Jha, H.C.; Dzeng, R.K.; Robertson, E.S. The EBV latent antigen 3C inhibits apoptosis through targeted regulation of interferon regulatory factors 4 and 8. PLoS Pathog. 2013, 9, e1003314. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Price, A.M.; Dai, J.; Bazot, Q.; Patel, L.; Nikitin, P.A.; Djavadian, R.; Winter, P.S.; Salinas, C.A.; Barry, A.P.; Wood, K.C.; et al. Epstein-Barr virus ensures B cell survival by uniquely modulating apoptosis at early and late times after infection. eLife 2017, 6, e22509. [Google Scholar] [CrossRef] [PubMed]
- Desbien, A.L.; Kappler, J.W.; Marrack, P. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc. Natl. Acad. Sci. USA 2009, 106, 5663–5668. [Google Scholar] [CrossRef]
- Procko, E.; Berguig, G.Y.; Shen, B.W.; Song, Y.; Frayo, S.; Convertine, A.J.; Margineantu, D.; Booth, G.; Correia, B.E.; Cheng, Y.; et al. A computationally designed inhibitor of an Epstein-Barr viral Bcl-2 protein induces apoptosis in infected cells. Cell 2014, 157, 1644–1656. [Google Scholar] [CrossRef]
- Holowaty, M.N.; Zeghouf, M.; Wu, H.; Tellam, J.; Athanasopoulos, V.; Greenblatt, J.; Frappier, L. Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J. Biol. Chem. 2003, 278, 29987–29994. [Google Scholar] [CrossRef]
- Saridakis, V.; Sheng, Y.; Sarkari, F.; Holowaty, M.N.; Shire, K.; Nguyen, T.; Zhang, R.G.; Liao, J.; Lee, W.; Edwards, A.M.; et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol. Cell 2005, 18, 25–36. [Google Scholar] [CrossRef]
- Levitskaya, J.; Sharipo, A.; Leonchiks, A.; Ciechanover, A.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA 1997, 94, 12616–12621. [Google Scholar] [CrossRef]
- Knight, J.S.; Sharma, N.; Robertson, E.S. Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 18562–18566. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Bamidele, A.; Murakami, M.; Robertson, E.S. EBNA3C attenuates the function of p53 through interaction with inhibitor of growth family proteins 4 and 5. J. Virol. 2011, 85, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, S.; Kuravi, S.; Alleboina, S.; Mudduluru, G.; Jensen, R.A.; McGuirk, J.P.; Balusu, R. Targeted therapy for EBV-associated B-cell neoplasms. Mol. Cancer Res. 2019, 17, 839–844. [Google Scholar] [CrossRef]
- Shirley, C.M.; Chen, J.; Shamay, M.; Li, H.; Zahnow, C.A.; Hayward, S.D.; Ambinder, R.F. Bortezomib induction of C/EBPbeta mediates Epstein-Barr virus lytic activation in Burkitt lymphoma. Blood 2011, 117, 6297–6303. [Google Scholar] [CrossRef]
- Fu, D.X.; Tanhehco, Y.; Chen, J.; Foss, C.A.; Fox, J.J.; Chong, J.M.; Hobbs, R.F.; Fukayama, M.; Sgouros, G.; Kowalski, J.; et al. Bortezomib-induced enzyme-targeted radiation therapy in herpesvirus-associated tumors. Nat. Med. 2008, 14, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Lin, W.H.; Chen, S.Y.; Longnecker, R.; Tsai, S.C.; Chen, C.L.; Tsai, C.H. Syk tyrosine kinase mediates Epstein-Barr virus latent membrane protein 2A-induced cell migration in epithelial cells. J. Biol. Chem. 2006, 281, 8806–8814. [Google Scholar] [CrossRef]
- Cen, O.; Kannan, K.; Huck Sappal, J.; Yu, J.; Zhang, M.; Arikan, M.; Ucur, A.; Ustek, D.; Cen, Y.; Gordon, L.; et al. Spleen Tyrosine Kinase Inhibitor TAK-659 prevents splenomegaly and tumor development in a murine model of Epstein-Barr Virus-associated Lymphoma. mSphere 2018, 3, e00378-18. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Small-molecule modulation of protein homeostasis. Chem. Rev. 2017, 117, 11269–11301. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Crews, C.M. Chemical biology: Greasy tags for protein removal. Nature 2012, 487, 308–309. [Google Scholar] [CrossRef]
- Li, N.; Thompson, S.; Schultz, D.C.; Zhu, W.; Jiang, H.; Luo, C.; Lieberman, P.M. Discovery of selective inhibitors against EBNA1 via high throughput in silico virtual screening. PLoS ONE 2010, 5, e10126. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.; Messick, T.; Schultz, D.C.; Reichman, M.; Lieberman, P.M. Development of a high-throughput screen for inhibitors of Epstein-Barr virus EBNA1. J. Biomol. Screen. 2010, 15, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Bochkarev, A.; Barwell, J.A.; Pfuetzner, R.A.; Furey, W., Jr.; Edwards, A.M.; Frappier, L. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein EBNA 1. Cell 1995, 83, 39–46. [Google Scholar] [CrossRef]
- Bochkarev, A.; Barwell, J.A.; Pfuetzner, R.A.; Bochkareva, E.; Frappier, L.; Edwards, A.M. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein, EBNA1, bound to DNA. Cell 1996, 84, 791–800. [Google Scholar] [CrossRef]
- Kirchmaier, A.L.; Sugden, B. Dominant-negative inhibitors of EBNA-1 of Epstein-Barr virus. J. Virol. 1997, 71, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Messick, T.E.; Smith, G.R.; Soldan, S.S.; McDonnell, M.E.; Deakyne, J.S.; Malecka, K.A.; Tolvinski, L.; van den Heuvel, A.P.J.; Gu, B.W.; Cassel, J.A.; et al. Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth. Sci. Transl. Med. 2019, 11, eaau5612. [Google Scholar] [CrossRef] [PubMed]
- Chodosh, J.; Holder, V.P.; Gan, Y.J.; Belgaumi, A.; Sample, J.; Sixbey, J.W. Eradication of latent Epstein-Barr virus by hydroxyurea alters the growth-transformed cell phenotype. J. Infect. Dis. 1998, 177, 1194–1201. [Google Scholar] [CrossRef]
- Slobod, K.S.; Taylor, G.H.; Sandlund, J.T.; Furth, P.; Helton, K.J.; Sixbey, J.W. Epstein-Barr virus-targeted therapy for AIDS-related primary lymphoma of the central nervous system. Lancet 2000, 356, 1493–1494. [Google Scholar] [CrossRef]
- Feederle, R.; Kost, M.; Baumann, M.; Janz, A.; Drouet, E.; Hammerschmidt, W.; Delecluse, H.J. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000, 19, 3080–3089. [Google Scholar] [CrossRef]
- McKenzie, J.; El-Guindy, A. Epstein-Barr virus lytic cycle reactivation. Curr. Top. Microbiol. Immunol. 2015, 391, 237–261. [Google Scholar] [CrossRef]
- Chang, L.K.; Liu, S.T. Activation of the BRLF1 promoter and lytic cycle of Epstein-Barr virus by histone acetylation. Nucleic Acids Res. 2000, 28, 3918–3925. [Google Scholar] [CrossRef] [PubMed]
- Hausen, H.Z.; O’Neill, F.J.; Freese, U.K.; Hecker, E. Persisting oncogenic herpesvirus induced by the tumour promotor TPA. Nature 1978, 272, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Luka, J.; Kallin, B.; Klein, G. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology 1979, 94, 228–231. [Google Scholar] [CrossRef]
- Westphal, E.M.; Blackstock, W.; Feng, W.; Israel, B.; Kenney, S.C. Activation of lytic Epstein-Barr virus (EBV) infection by radiation and sodium butyrate in vitro and in vivo: A potential method for treating EBV-positive malignancies. Cancer Res. 2000, 60, 5781–5788. [Google Scholar] [PubMed]
- Shimizu, N.; Takada, K. Analysis of the BZLF1 promoter of Epstein-Barr virus: Identification of an anti-immunoglobulin response sequence. J. Virol. 1993, 67, 3240–3245. [Google Scholar] [CrossRef]
- Tovey, M.G.; Lenoir, G.; Begon-Lours, J. Activation of latent Epstein-Barr virus by antibody to human IgM. Nature 1978, 276, 270–272. [Google Scholar] [CrossRef]
- Tikhmyanova, N.; Schultz, D.C.; Lee, T.; Salvino, J.M.; Lieberman, P.M. Identification of a new class of small molecules that efficiently reactivate latent Epstein-Barr Virus. ACS Chem. Biol. 2014, 9, 785–795. [Google Scholar] [CrossRef]
- Perrine, S.P.; Hermine, O.; Small, T.; Suarez, F.; O’Reilly, R.; Boulad, F.; Fingeroth, J.; Askin, M.; Levy, A.; Mentzer, S.J.; et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 2007, 109, 2571–2578. [Google Scholar] [CrossRef]
- Faller, D.V.; Mentzer, S.J.; Perrine, S.P. Induction of the Epstein-Barr virus thymidine kinase gene with concomitant nucleoside antivirals as a therapeutic strategy for Epstein-Barr virus-associated malignancies. Curr. Opin. Oncol. 2001, 13, 360–367. [Google Scholar] [CrossRef]
- Meng, Q.; Hagemeier, S.R.; Fingeroth, J.D.; Gershburg, E.; Pagano, J.S.; Kenney, S.C. The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J. Virol. 2010, 84, 4534–4542. [Google Scholar] [CrossRef]
- Van Diemen, F.R.; Kruse, E.M.; Hooykaas, M.J.; Bruggeling, C.E.; Schurch, A.C.; van Ham, P.M.; Imhof, S.M.; Nijhuis, M.; Wiertz, E.J.; Lebbink, R.J. CRISPR/Cas9-mediated genome editing of herpesviruses limits productive and latent infections. PLoS Pathog. 2016, 12, e1005701. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Jiang, C.; Zhang, Y.; Govande, A.; Trudeau, S.J.; Chen, F.; Fry, C.J.; Puri, R.; Wolinsky, E.; Schineller, M.; et al. MYC controls the Epstein-Barr virus lytic switch. Mol. Cell. 2020, 78, 653–669.e8. [Google Scholar] [CrossRef] [PubMed]
- McKeown, M.R.; Bradner, J.E. Therapeutic strategies to inhibit MYC. Cold Spring Harb. Perspect. Med. 2014, 4, a014266. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-molecule MYC inhibitors suppress Tumor growth and enhance immunotherapy. Cancer cell 2019, 36, 483–497.e15. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.R.; Murray, J.; Cheung, B.B.; Gamble, L.; Koach, J.; Tsang, J.; Sutton, S.; Kalla, H.; Syed, S.; Gifford, A.J.; et al. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci. Transl. Med. 2015, 7, 312ra176. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Stroopinsky, D.; Alimperti, S.; Jiao, A.L.; Pyzer, A.R.; Cippitelli, C.; Pepe, G.; Severa, M.; Rosenblatt, J.; Etna, M.P.; et al. Epstein−Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia 2019, 33, 132–147. [Google Scholar] [CrossRef]
- Bi, X.-W.; Wang, H.; Zhang, W.-W.; Wang, J.-H.; Liu, W.-J.; Xia, Z.-J.; Huang, H.-Q.; Jiang, W.-Q.; Zhang, Y.-J.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef]
- Xue, T.; Wang, W.-G.; Zhou, X.-Y.; Li, X.-Q. EBV-positive diffuse large B-cell lymphoma features PD-L1 protein but not mRNA overexpression. Pathology 2018, 50, 725–729. [Google Scholar] [CrossRef]
- Laurent, C.; Fabiani, B.; Do, C.; Tchernonog, E.; Cartron, G.; Gravelle, P.; Amara, N.; Malot, S.; Palisoc, M.M.; Copie-Bergman, C.; et al. Immune-checkpoint expression in Epstein-Barr virus positive and negative plasmablastic lymphoma: A clinical and pathological study in 82 patients. Haematologica 2016, 101, 976. [Google Scholar] [CrossRef]
- Granai, M.; Mundo, L.; Akarca, A.U.; Siciliano, M.C.; Rizvi, H.; Mancini, V.; Onyango, N.; Nyagol, J.; Abinya, N.O.; Maha, I.; et al. Immune landscape in Burkitt lymphoma reveals M2-macrophage polarization and correlation between PD-L1 expression and non-canonical EBV latency program. Infect. Agents Cancer 2020, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.; Chen, X.; Liu, A.; Zhang, Y.; Guo, X.; Yan, S.; Liu, Y. PD-1 blockade can restore functions of T-cells in Epstein-Barr virus-positive diffuse large B-cell lymphoma in vitro. PLoS ONE 2015, 10, e0136476. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Hyeon, J.; Cho, I.; Ko, Y.H.; Kim, W.S. Comparison of efficacy of pembrolizumab between Epstein-Barr viruspositive and negative relapsed or refractory non-hodgkin lymphomas. Cancer Res. Treat. 2019, 51, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Kwong, Y.-L.; Chan, T.S.Y.; Tan, D.; Kim, S.J.; Poon, L.-M.; Mow, B.; Khong, P.-L.; Loong, F.; Au-Yeung, R.; Iqbal, J.; et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 2017, 129, 2437–2442. [Google Scholar] [CrossRef]
- Ma, S.-D.; Xu, X.; Jones, R.; Delecluse, H.-J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 blockade inhibits Epstein-Barr virus-induced lymphoma growth in a cord blood humanized-mouse model. PLoS Pathog. 2016, 12, e1005642. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Zheng, Z.; Sun, R.; Zhao, H.-J.; Fu, D.; Zhong, H.-J.; Weng, X.-Q.; Qu, B.; Zhao, Y.; Wang, L.; Zhao, W.-L. MiR155 sensitized B-lymphoma cells to anti-PD-L1 antibody via PD-1/PD-L1-mediated lymphoma cell interaction with CD8+T cells. Mol. Cancer 2019, 18, 54. [Google Scholar] [CrossRef]
- Zimmermann, H.; Trappe, R.U. Therapeutic options in post-transplant lymphoproliferative disorders. Ther. Adv. Hematol. 2011, 2, 393–407. [Google Scholar] [CrossRef]
- Feugier, P.; Van Hoof, A.; Sebban, C.; Solal-Celigny, P.; Bouabdallah, R.; Fermé, C.; Christian, B.; Lepage, E.; Tilly, H.; Morschhauser, F.; et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: A study by the Groupe d’Etude des Lymphomes de l’Adulte. J. Clin. Oncol. 2005, 23, 4117–4126. [Google Scholar] [CrossRef]
- Comoli, P.; Basso, S.; Zecca, M.; Pagliara, D.; Baldanti, F.; Bernardo, M.E.; Barberi, W.; Moretta, A.; Labirio, M.; Paulli, M.; et al. Preemptive therapy of EBV-related lymphoproliferative disease after pediatric haploidentical stem cell transplantation. Am. J. Transplant. 2007, 7, 1648–1655. [Google Scholar] [CrossRef]
- Kobayashi, S.; Sano, H.; Mochizuki, K.; Ohara, Y.; Takahashi, N.; Ohto, H.; Kikuta, A. Pre-emptive rituximab for Epstein–Barr virus reactivation after haplo-hematopoietic stem cell transplantation. Pediatr. Int. 2017, 59, 973–978. [Google Scholar] [CrossRef]
- Van Esser, J.W.J.; Niesters, H.G.M.; van der Holt, B.; Meijer, E.; Osterhaus, A.D.M.E.; Gratama, J.W.; Verdonck, L.F.; Löwenberg, B.; Cornelissen, J.J. Prevention of Epstein-Barr virus–lymphoproliferative disease by molecular monitoring and preemptive rituximab in high-risk patients after allogeneic stem cell transplantation. Blood 2002, 99, 4364–4369. [Google Scholar] [CrossRef]
- Israel, B.F.; Gulley, M.; Elmore, S.; Ferrini, S.; Feng, W.H.; Kenney, S.C. Anti-CD70 antibodies: A potential treatment for EBV+ CD70-expressing lymphomas. Mol. Cancer Ther. 2005, 4, 2037–2044. [Google Scholar] [CrossRef]
- Papadopoulos, E.B.; Ladanyi, M.; Emanuel, D.; Mackinnon, S.; Boulad, F.; Carabasi, M.H.; Castro-Malaspina, H.; Childs, B.H.; Gillio, A.P.; Small, T.N.; et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N. Engl. J. Med. 1994, 330, 1185–1191. [Google Scholar] [CrossRef]
- Heslop, H.E.; Slobod, K.S.; Pule, M.A.; Hale, G.A.; Rousseau, A.; Smith, C.A.; Bollard, C.M.; Liu, H.; Wu, M.-F.; Rochester, R.J.; et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010, 115, 925–935. [Google Scholar] [CrossRef]
- Rooney, C.M.; Ng, C.Y.C.; Loftin, S.; Smith, C.A.; Li, C.; Krance, R.A.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet 1995, 345, 9–13. [Google Scholar] [CrossRef]
- Heslop, H.E.; Ng, C.Y.C.; Li, C.; Smith, C.A.; Loftin, S.K.; Krance, R.A.; Brenner, M.K.; Rooney, C.M. Long-term restoration of immunity against Epstein–Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat. Med. 1996, 2, 551–555. [Google Scholar] [CrossRef]
- Bollard, C.M.; Gottschalk, S.; Torrano, V.; Diouf, O.; Ku, S.; Hazrat, Y.; Carrum, G.; Ramos, C.; Fayad, L.; Shpall, E.J.; et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J. Clin. Oncol. 2014, 32, 798–808. [Google Scholar] [CrossRef]
- Icheva, V.; Kayser, S.; Wolff, D.; Tuve, S.; Kyzirakos, C.; Bethge, W.; Greil, J.; Albert, M.H.; Schwinger, W.; Nathrath, M.; et al. Adoptive transfer of Epstein-Barr virus (EBV) nuclear antigen 1-specific T cells as treatment for EBV reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J. Clin. Oncol. 2012, 31, 39–48. [Google Scholar] [CrossRef]
- Gallot, G.; Vollant, S.; Saïagh, S.; Clémenceau, B.; Vivien, R.; Cerato, E.; Bignon, J.-D.; Ferrand, C.; Jaccard, A.; Vigouroux, S.; et al. T-cell therapy using a bank of EBV-specific cytotoxic T cells: Lessons from a phase I/II feasibility and safety study. J. Immunother. 2014, 37, 170–179. [Google Scholar] [CrossRef]
- Haque, T.; Wilkie, G.M.; Jones, M.M.; Higgins, C.D.; Urquhart, G.; Wingate, P.; Burns, D.; McAulay, K.; Turner, M.; Bellamy, C.; et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: Results of a phase 2 multicenter clinical trial. Blood 2007, 110, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N.; Lankiewicz, B.; Drexhage, J.; Berrevoets, C.; Moss, D.J.; Levitsky, V.; Bonneville, M.; Lee, S.P.; McMichael, A.J.; Gratama, J.-W.; et al. T cell re-targeting to EBV antigens following TCR gene transfer: CD28-containing receptors mediate enhanced antigen-specific IFNγ production. Int. Immunol. 2006, 18, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-I.; Kim, U.-H.; Shin, A.R.; Won, J.-N.; Lee, H.-J.; Sohn, H.-J.; Kim, T.-G. A novel Epstein–Barr virus-latent membrane protein-1-specific T-cell receptor for TCR gene therapy. Br. J. Cancer 2018, 118, 534–545. [Google Scholar] [CrossRef]
- Xue, S.-A.; Gao, L.; Ahmadi, M.; Ghorashian, S.; Barros, R.D.; Pospori, C.; Holler, A.; Wright, G.; Thomas, S.; Topp, M.; et al. Human MHC class I-restricted high avidity CD4(+) T cells generated by co-transfer of TCR and CD8 mediate efficient tumor rejection in vivo. Oncoimmunology 2013, 2, e22590. [Google Scholar] [CrossRef]
- Yang, D.; Shao, Q.; Sun, H.; Mu, X.; Gao, Y.; Jiang, R.; Hou, J.; Yao, K.; Chen, Y.; Sun, B. Evaluation of Epstein-Barr virus latent membrane protein 2 specific T-cell receptors driven by T-cell specific promoters using lentiviral vector. Clin. Dev. Immunol. 2011, 2011, 716926. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef]
- Maus, M.V.; Grupp, S.A.; Porter, D.L.; June, C.H. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014, 123, 2625–2635. [Google Scholar] [CrossRef]
- Cruz, C.R.Y.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef]
- Shen, R.R.; Pham, C.D.; Wu, M.; Munson, D.J.; Aftab, B.T. CD19 chimeric antigen receptor (CAR) engineered epstein-barr virus (EBV) specific T cells—An off-the-shelf, allogeneic CAR T-cell immunotherapy platform. Cytotherapy 2019, 21, S11. [Google Scholar] [CrossRef]
- Tang, X.; Zhou, Y.; Li, W.; Tang, Q.; Chen, R.; Zhu, J.; Feng, Z. T cells expressing a LMP1-specific chimeric antigen receptor mediate antitumor effects against LMP1-positive nasopharyngeal carcinoma cells in vitro and in vivo. J. Biomed. Res. 2014, 28, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A.; Morgan, A.J.; Finerty, S.; Randle, B.J.; Kirkwood, J.K. Protection of cottontop tamarins against Epstein-Barr virus-induced malignant lymphoma by a prototype subunit vaccine. Nature 1985, 318, 287–289. [Google Scholar] [CrossRef]
- Morgan, A.J.; Mackett, M.; Finerty, S.; Arrand, J.R.; Scullion, F.T.; Epstein, M.A. Recombinant vaccinia virus expressing Epstein-Barr virus glycoprotein gp340 protects cottontop tamarins against EB virus-induced malignant lymphomas. J. Med. Virol. 1988, 25, 189–195. [Google Scholar] [CrossRef]
- Wilson, A.D.; Lovgren-Bengtsson, K.; Villacres-Ericsson, M.; Morein, B.; Morgan, A.J. The major Epstein-Barr virus (EBV) envelope glycoprotein gp340 when incorporated into Iscoms primes cytotoxic T-cell responses directed against EBV lymphoblastoid cell lines. Vaccine 1999, 17, 1282–1290. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Poodry, C.A. Identification and isolation of the main component (gp350–gp220) of Epstein-Barr virus responsible for generating neutralizing antibodies in vivo. J. Virol. 1982, 43, 730–736. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Geilinger, K. Monoclonal antibodies against the major glycoprotein (gp350/220) of Epstein-Barr virus neutralize infectivity. Proc. Natl. Acad. Sci. USA 1980, 77, 5307–5311. [Google Scholar] [CrossRef]
- Cohen, J.I. Epstein-barr virus vaccines. Clin. Transl. Immunol. 2015, 4, e32. [Google Scholar] [CrossRef]
- Jackman, W.T.; Mann, K.A.; Hoffmann, H.J.; Spaete, R.R. Expression of Epstein-Barr virus gp350 as a single chain glycoprotein for an EBV subunit vaccine. Vaccine 1999, 17, 660–668. [Google Scholar] [CrossRef]
- Kanekiyo, M.; Bu, W.; Joyce, M.G.; Meng, G.; Whittle, J.R.; Baxa, U.; Yamamoto, T.; Narpala, S.; Todd, J.P.; Rao, S.S.; et al. Rational design of an Epstein-Barr virus vaccine targeting the receptor-binding site. Cell 2015, 162, 1090–1100. [Google Scholar] [CrossRef]
- Sokal, E.M.; Hoppenbrouwers, K.; Vandermeulen, C.; Moutschen, M.; Leonard, P.; Moreels, A.; Haumont, M.; Bollen, A.; Smets, F.; Denis, M. Recombinant gp350 vaccine for infectious mononucleosis: A phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J. Infect. Dis. 2007, 196, 1749–1753. [Google Scholar] [CrossRef]
- Moutschen, M.; Leonard, P.; Sokal, E.M.; Smets, F.; Haumont, M.; Mazzu, P.; Bollen, A.; Denamur, F.; Peeters, P.; Dubin, G.; et al. Phase I/II studies to evaluate safety and immunogenicity of a recombinant gp350 Epstein-Barr virus vaccine in healthy adults. Vaccine 2007, 25, 4697–4705. [Google Scholar] [CrossRef]
- Weiss, E.R.; Alter, G.; Ogembo, J.G.; Henderson, J.L.; Tabak, B.; Bakis, Y.; Somasundaran, M.; Garber, M.; Selin, L.; Luzuriaga, K. High Epstein-Barr virus load and genomic diversity are associated with generation of gp350-Specific neutralizing antibodies following acute infectious mononucleosis. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Bu, W.; Joyce, M.G.; Nguyen, H.; Banh, D.V.; Aguilar, F.; Tariq, Z.; Yap, M.L.; Tsujimura, Y.; Gillespie, R.A.; Tsybovsky, Y.; et al. Immunization with components of the viral fusion apparatus elicits antibodies that neutralize Epstein-Barr virus in B cells and epithelial cells. Immunity 2019, 50, 1305–1316.e16. [Google Scholar] [CrossRef]
- Snijder, J.; Ortego, M.S.; Weidle, C.; Stuart, A.B.; Gray, M.D.; McElrath, M.J.; Pancera, M.; Veesler, D.; McGuire, A.T. An antibody targeting the fusion machinery neutralizes dual-tropic infection and defines a site of vulnerability on Epstein-Barr virus. Immunity 2018, 48, 799–811.e799. [Google Scholar] [CrossRef]
- Mundo, L.; Del Porro, L.; Granai, M.; Siciliano, M.C.; Mancini, V.; Santi, R.; Marcar, L.; Vrzalikova, K.; Vergoni, F.; Di Stefano, G.; et al. Frequent traces of EBV infection in Hodgkin and non-Hodgkin lymphomas classified as EBV-negative by routine methods: Expanding the landscape of EBV-related lymphomas. Mod. Pathol. 2020. [Google Scholar] [CrossRef]
- Ruiss, R.; Jochum, S.; Wanner, G.; Reisbach, G.; Hammerschmidt, W.; Zeidler, R. A virus-like particle-based Epstein-Barr virus vaccine. J. Virol. 2011, 85, 13105–13113. [Google Scholar] [CrossRef]
- Pavlova, S.; Feederle, R.; Gartner, K.; Fuchs, W.; Granzow, H.; Delecluse, H.J. An Epstein-Barr virus mutant produces immunogenic defective particles devoid of viral DNA. J. Virol. 2013, 87, 2011–2022. [Google Scholar] [CrossRef]
- Van Zyl, D.G.; Mautner, J.; Delecluse, H.J. Progress in EBV vaccines. Front. Oncol. 2019, 9, 104. [Google Scholar] [CrossRef]
- Shumilov, A.; Tsai, M.H.; Schlosser, Y.T.; Kratz, A.S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein-Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef]
- Van Zyl, D.G.; Tsai, M.H.; Shumilov, A.; Schneidt, V.; Poirey, R.; Schlehe, B.; Fluhr, H.; Mautner, J.; Delecluse, H.J. Immunogenic particles with a broad antigenic spectrum stimulate cytolytic T cells and offer increased protection against EBV infection ex vivo and in mice. PLoS Pathog. 2018, 14, e1007464. [Google Scholar] [CrossRef]
- Hui, E.P.; Taylor, G.S.; Jia, H.; Ma, B.B.; Chan, S.L.; Ho, R.; Wong, W.L.; Wilson, S.; Johnson, B.F.; Edwards, C.; et al. Phase I trial of recombinant modified vaccinia ankara encoding Epstein-Barr viral tumor antigens in nasopharyngeal carcinoma patients. Cancer Res. 2013, 73, 1676–1688. [Google Scholar] [CrossRef]
- Taylor, G.S.; Jia, H.; Harrington, K.; Lee, L.W.; Turner, J.; Ladell, K.; Price, D.A.; Tanday, M.; Matthews, J.; Roberts, C.; et al. A recombinant modified vaccinia ankara vaccine encoding Epstein-Barr Virus (EBV) target antigens: A phase I trial in UK patients with EBV-positive cancer. Clin. Cancer Res. 2014, 20, 5009–5022. [Google Scholar] [CrossRef]
- Taylor, G.S.; Haigh, T.A.; Gudgeon, N.H.; Phelps, R.J.; Lee, S.P.; Steven, N.M.; Rickinson, A.B. Dual stimulation of Epstein-Barr Virus (EBV)-specific CD4+- and CD8+-T-cell responses by a chimeric antigen construct: Potential therapeutic vaccine for EBV-positive nasopharyngeal carcinoma. J. Virol. 2004, 78, 768–778. [Google Scholar] [CrossRef]
- Roskrow, M.A.; Suzuki, N.; Gan, Y.; Sixbey, J.W.; Ng, C.Y.; Kimbrough, S.; Hudson, M.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for the treatment of patients with EBV-positive relapsed Hodgkin’s disease. Blood 1998, 91, 2925–2934. [Google Scholar] [CrossRef]
- Kuppers, R.; Engert, A.; Hansmann, M.L. Hodgkin lymphoma. J. Clin. Investig. 2012, 122, 3439–3447. [Google Scholar] [CrossRef]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Watterson, D.; Robinson, J.; Chappell, K.J.; Butler, M.S.; Edwards, D.J.; Fry, S.R.; Bermingham, I.M.; Cooper, M.A.; Young, P.R. A generic screening platform for inhibitors of virus induced cell fusion using cellular electrical impedance. Sci. Rep. 2016, 6, 22791. [Google Scholar] [CrossRef]
- Pei, Y.; Lewis, A.E.; Robertson, E.S. Current progress in EBV-associated B-Cell lymphomas. Adv. Exp. Med. Biol. 2017, 1018, 57–74. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Romero, D. HDAC inhibitors tested in phase III trial. Nat. Rev. Clin. Oncol. 2019, 16, 465. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Identifier | Year Started | Study Title | Phase |
|---|---|---|---|
| PD-1/PD-L1 | |||
| NCT04058470 | 2019 | Toripalimab in Combination with R-CHOP for Elderly Patients with Untreated Diffused B-Cell Lymphoma | I/II |
| NCT04181489 | 2019 | Sintilimab in Combination with R-CHOP in Patients with Treatment-naive EBV-positive Diffuse Large B-cell Lymphoma (DLBCL), NOS | II |
| NCT04084626 | 2019 | PD1 Antibody and Lenalidomide as a Treatment for EBV-associated Hemophagocytic Lymphohistiocytosis (HLH) or Chronic Active EBV Infection (CAEBV) | III |
| NCT03586024 | 2018 | Pembrolizumab in Relapsed/Refractory Extranodal NK/T- Cell Lymphoma, Nasal Type and EBV-associated DLBCL | I/II |
| NCT03258567 | 2017 | Nivolumab in EBV-Positive Lymphoproliferative Disorders and EBV-Positive Non-Hodgkin Lymphomas | II |
| NCT03038672 | 2017 | Nivolumab with or without Varlilumab in Treating Patients with Relapsed/Refractory Aggressive B-cell Lymphomas | II |
| T-cell Therapy | |||
| NCT04156217 | 2019 | EBV-TCR-T Cells for EBV Infection and EBV-Associated Post-Transplant Lymphoproliferative Disease After HSCT | I |
| NCT03789617 | 2018 | Evaluate the Efficacy and Safety of EBV Induced Natural T Lymphocyte (EBViNT) Cell in Patients with Progressive EBV Positive Extranodal NK/T-cell Lymphoma Where Standard Treatments Have Failed | I/II |
| NCT03671850 | 2018 | VT-EBV-N for Treatment of Severe in EBV Positive Extranodal NK/T Cell Lymphoma Patients | II |
| NCT03394365 | 2018 | Tabelecleucel for Solid Organ or Allogeneic Hematopoietic Cell Transplant Participants with EBV-Associated Post-Transplant Lymphoproliferative Disease (EBV+ PTLD) After Failure of Rituximab or Rituximab and Chemotherapy | III |
| NCT03392142 | 2018 | Tabelecleucel for Allogeneic Hematopoietic Cell Transplant Subjects with EBV-Associated Post-Transplant Lymphoproliferative Disease (EBV + PTLD) After Failure of Rituximab (MATCH) | III |
| NCT03044743 | 2017 | PD-1 Knockout EBV-CTLs for Advanced Stage EBV Associated Malignancies | I/II |
| Modified-TCR | |||
| NCT01956084 | 2013 | Cytotoxic T Cells to Treat Relapsed EBV-positive Lymphoma (ALCI2) | I |
| CAR-T Therapy | |||
| NCT03233854 | 2017 | CD19/CD22 Chimeric Antigen Receptor (CAR) T Cells in Adults with Recurrent/Refractory B-Cell Malignancies | I |
| HDAC Inhibitors | |||
| NCT03397706 | 2018 | Dose Escalation & Expansion Study of Oral VRx-3996 & Valganciclovir in Subjects with Relapsed/Refractory EBV-Associated Lymphoid Malignancies | I/II |
| Monoclonal Antibodies | |||
| NCT02924402 | 2016 | Study to Evaluate Safety and Tolerability of XmAb13676 in Patients with CD20-expressing Hematologic Malignancies | I |
| NCT02670616 | 2016 | Study of Ibrutinib in Combination with Rituximab-CHOP in EBV-positive Diffuse Large B-cell Lymphoma | II |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, Y.; Wong, J.H.Y.; Robertson, E.S. Targeted Therapies for Epstein-Barr Virus-Associated Lymphomas. Cancers 2020, 12, 2565. https://doi.org/10.3390/cancers12092565
Pei Y, Wong JHY, Robertson ES. Targeted Therapies for Epstein-Barr Virus-Associated Lymphomas. Cancers. 2020; 12(9):2565. https://doi.org/10.3390/cancers12092565
Chicago/Turabian StylePei, Yonggang, Josiah H. Y. Wong, and Erle S. Robertson. 2020. "Targeted Therapies for Epstein-Barr Virus-Associated Lymphomas" Cancers 12, no. 9: 2565. https://doi.org/10.3390/cancers12092565
APA StylePei, Y., Wong, J. H. Y., & Robertson, E. S. (2020). Targeted Therapies for Epstein-Barr Virus-Associated Lymphomas. Cancers, 12(9), 2565. https://doi.org/10.3390/cancers12092565