ATP Production Relies on Fatty Acid Oxidation Rather than Glycolysis in Pancreatic Ductal Adenocarcinoma

 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Glucose Is Not a Source for ATP Production in Cancer Cells
2.2. Identification of the Metabolic Pathway for ATP Production in PDAC Cells
2.3. The TCA Cycle Does Not Affect ATP Production in PDAC Cells
2.4. MAS Is an Important Pathway for ATP Production in PDAC Cells
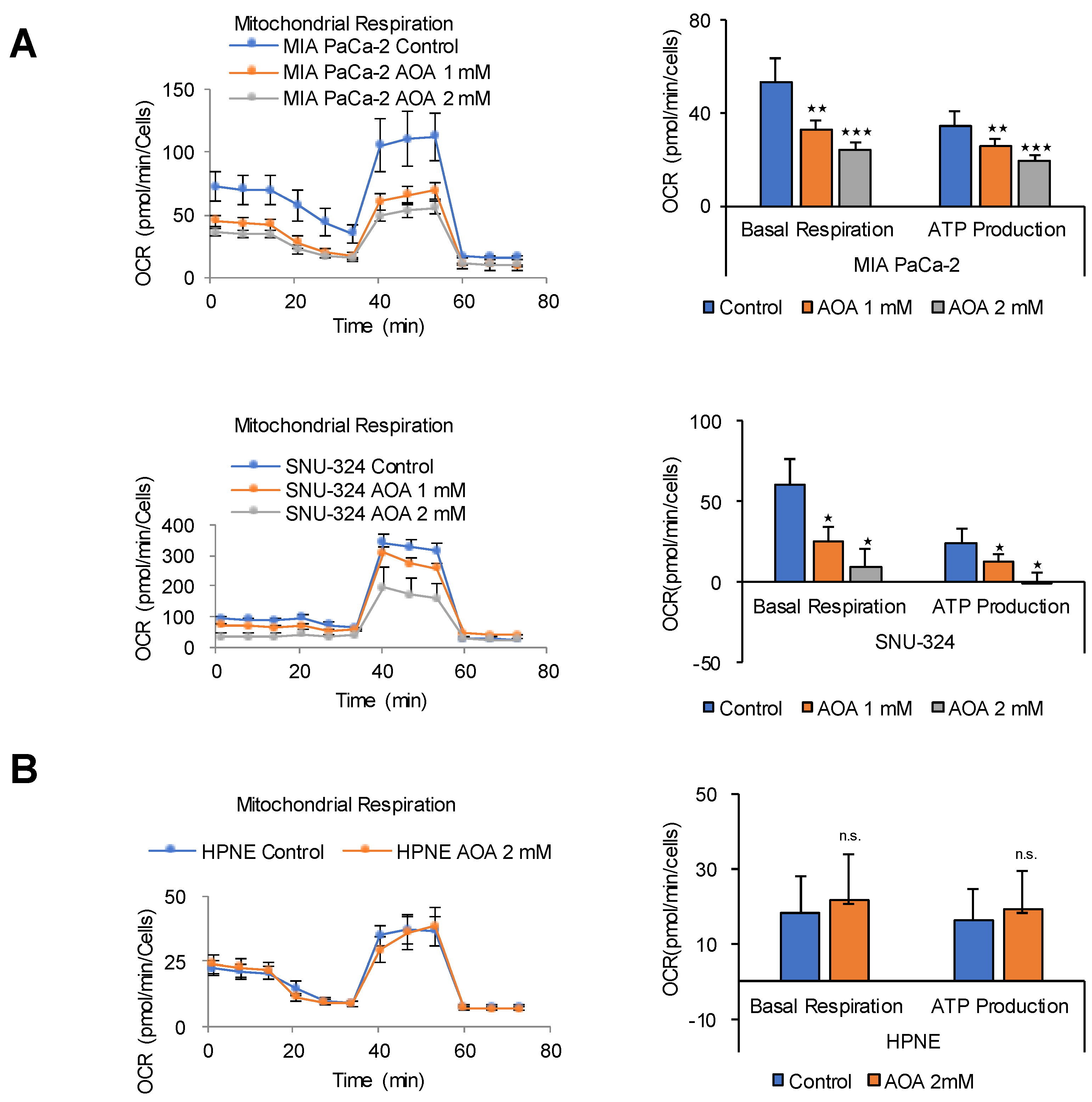
2.5. FAO Mediates ATP Production in PDAC Cells
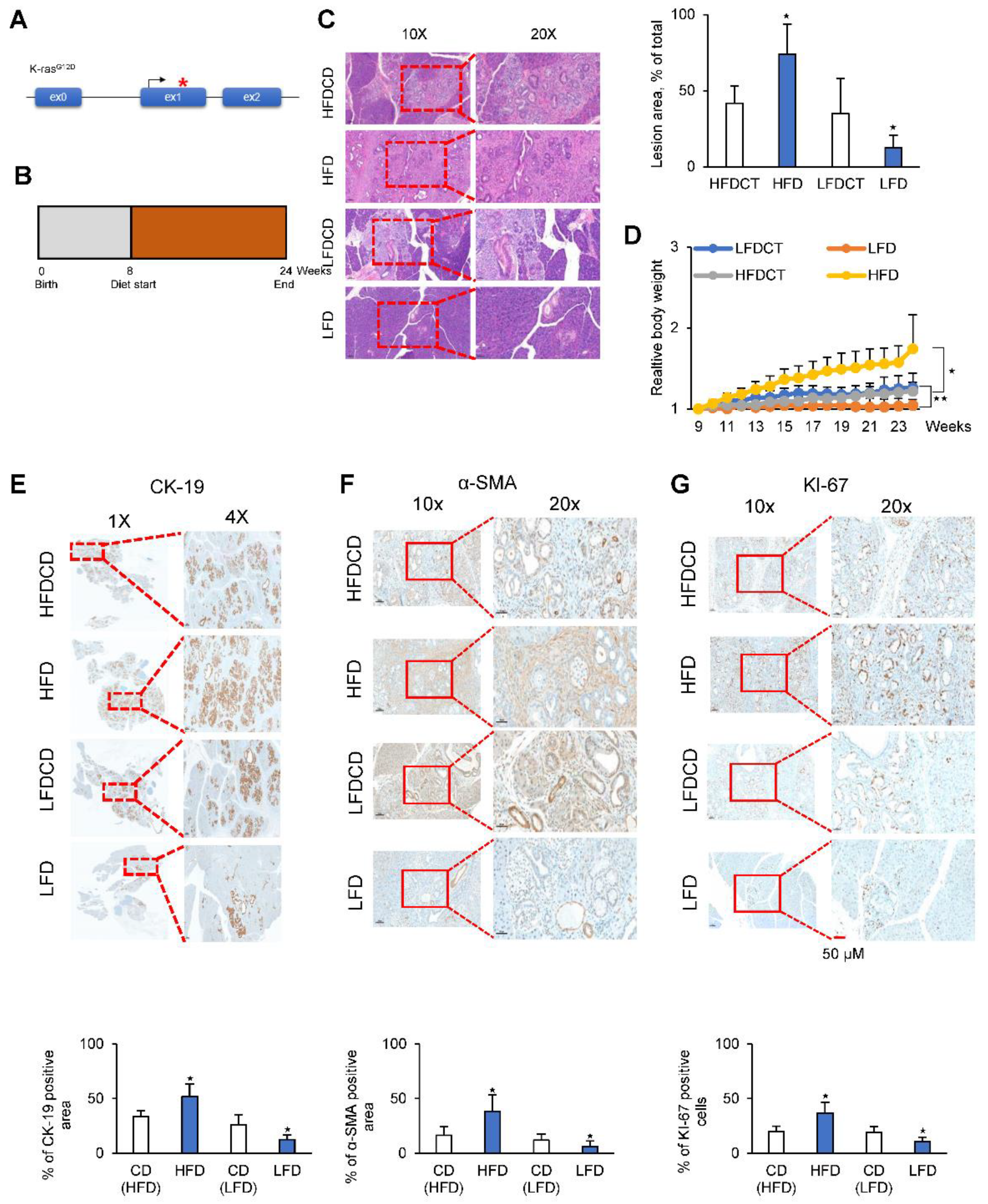
2.6. A High-Fat Diet Promotes PDAC Tumor Growth in an In Vivo Model
2.7. A Low-Fat Diet Decreases PDAC Tumor Growth in a KC Mouse Model
3. Discussion
4. Materials and Methods
4.1. Mouse Kras Model of Pancreatic Cancer
4.2. Preclinical Xenograft Tumor Models
4.3. Relative Quantification of Metabolites by Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
4.4. Relative Quantification of Energy Metabolites and Fatty Acyl CoA Using LC-MS/MS
Sample Preparation for LC-MS/MS
4.5. LC-MS/MS
4.6. Relative Quantification of Metabolites by Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
Sample Preparation for GC-MS
4.7. Fatty Acid Methyl Ester (FAME) Derivatization
4.8. GC-MS
4.9. XF Cell Mito Stress Analysis
4.10. Immunohistochemistry
4.11. Measurement of Mitochondrial Membrane Potential (∆ψm)
4.12. Cell Culture
4.13. FITC Annexin V and Propidium Iodide (PI) Cell Death Detection
4.14. Sulforhodamine B (SRB) Assay: Cell Proliferation Assay
4.15. Measurement of OAA and α-KG
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Pedersen, P.L. Tumor mitochondria and the bioenergetics of cancer cells. Prog. Exp. Tumor Res. 1978, 22, 190–274. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar] [PubMed]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Weinhouse, S. On respiratory impairment in cancer cells. Science 1956, 124, 267–269. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem Biophys Res. Commun. 2004, 313, 459–465. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Smolkova, K.; Bellance, N.; Scandurra, F.; Genot, E.; Gnaiger, E.; Plecita-Hlavata, L.; Jezek, P.; Rossignol, R. Mitochondrial bioenergetic adaptations of breast cancer cells to aglycemia and hypoxia. J. Bioenerg. Biomembr. 2010, 42, 55–67. [Google Scholar] [CrossRef]
- Wagner, B.A.; Venkataraman, S.; Buettner, G.R. The rate of oxygen utilization by cells. Free Radical Biol. Med. 2011, 51, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Lee, S.H.; Lee, J.S.; Nam, B.; Seong, T.W.; Son, J.; Jang, H.; Hong, K.M.; Lee, C.; Kim, S.Y. Aldehyde dehydrogenase inhibition combined with phenformin treatment reversed NSCLC through ATP depletion. Oncotarget 2016, 7, 49397–49410. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.S.; Seo, J.; Lee, S.H.; Kang, J.H.; Song, J.; Kim, S.Y. Targeting Mitochondrial Oxidative Phosphorylation Abrogated Irinotecan Resistance in NSCLC. Sci. Rep. 2018, 8, 15707. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kim, S.H.; Lee, S.; Kang, J.H.; Lee, S.H.; Cheong, J.H.; Kim, S.Y. Gastric cancer depends on aldehyde dehydrogenase 3A1 for fatty acid oxidation. Sci. Rep. 2019, 9, 16313. [Google Scholar] [CrossRef]
- Guppy, M.; Leedman, P.; Zu, X.; Russell, V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem. J. 2002, 364, 309–315. [Google Scholar] [CrossRef]
- Elwood, J.C.; Lin, Y.C.; Cristofalo, V.J.; Weinhouse, S.; Morris, H.P. Glucose Utilization in Homogenates of the Morris Hepatoma 5123 and Related Tumors. Cancer Res. 1963, 23, 906–913. [Google Scholar]
- Kallinowski, F.; Schlenger, K.H.; Runkel, S.; Kloes, M.; Stohrer, M.; Okunieff, P.; Vaupel, P. Blood flow, metabolism, cellular microenvironment, and growth rate of human tumor xenografts. Cancer Res. 1989, 49, 3759–3764. [Google Scholar]
- Kim, S.Y. Targeting cancer energy metabolism: A potential systemic cure for cancer. Arch. Pharmacal Res. 2019, 42, 140–149. [Google Scholar] [CrossRef]
- Kim, S.Y. Cancer Energy Metabolism: Shutting Power off Cancer Factory. Biomolecules ther. 2018, 26, 39–44. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, S.H.; Hong, D.; Lee, J.S.; Ahn, H.S.; Ahn, J.H.; Seong, T.W.; Lee, C.H.; Jang, H.; Hong, K.M.; et al. Aldehyde dehydrogenase is used by cancer cells for energy metabolism. Exp. Mol. Med. 2016, 48, e272. [Google Scholar] [CrossRef]
- Lee, J.S.; Kang, J.H.; Lee, S.H.; Lee, C.H.; Son, J.; Kim, S.Y. Glutaminase 1 inhibition reduces thymidine synthesis in NSCLC. Biochem. Biophys. Res. Commun. 2016, 477, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lee, H.; Lee, S.; Kang, J.H.; Lee, S.H.; Kim, S.G.; Cho, E.S.; Kim, N.H.; Yook, J.I.; Kim, S.Y. Loss of SLC25A11 causes suppression of NSCLC and melanoma tumor formation. EBioMedicine 2019, 40, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Wick, A.N.; Drury, D.R.; Nakada, H.I.; Wolfe, J.B. Localization of the primary metabolic block produced by 2-deoxyglucose. J. Biol. Chem. 1957, 224, 963–969. [Google Scholar]
- Proudfoot, A.T.; Bradberry, S.M.; Vale, J.A. Sodium fluoroacetate poisoning. Toxicol. Rev. 2006, 25, 213–219. [Google Scholar] [CrossRef]
- McKenna, M.C.; Waagepetersen, H.S.; Schousboe, A.; Sonnewald, U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: Current evidence and pharmacological tools. Biochem. Pharmacol. 2006, 71, 399–407. [Google Scholar] [CrossRef]
- Dezsi, C.A. Trimetazidine in Practice: Review of the Clinical and Experimental Evidence. Am. J. Ther. 2016, 23, e871–e879. [Google Scholar] [CrossRef]
- Lee, S.H.; Jeon, Y.; Kang, J.H.; Jang, H.; Lee, H.; Kim, S.Y. The Combination of Loss of ALDH1L1 Function and Phenformin Treatment Decreases Tumor Growth in KRAS-Driven Lung Cancer. Cancers 2020, 12, 1382. [Google Scholar] [CrossRef]
- Greenhouse, W.V.; Lehninger, A.L. Occurrence of the malate-aspartate shuttle in various tumor types. Cancer Res. 1976, 36, 1392–1396. [Google Scholar]
- Egnatchik, R.A.; Leamy, A.K.; Sacco, S.A.; Cheah, Y.E.; Shiota, M.; Young, J.D. Glutamate-oxaloacetate transaminase activity promotes palmitate lipotoxicity in rat hepatocytes by enhancing anaplerosis and citric acid cycle flux. J. Biol. Chem. 2019, 294, 3081–3090. [Google Scholar] [CrossRef]
- Lee, K.M.; Yasuda, H.; Hollingsworth, M.A.; Ouellette, M.M. Notch 2-positive progenitors with the intrinsic ability to give rise to pancreatic ductal cells. Lab. Investig. 2005, 85, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Porter, N.A.; Brash, A.R. Routes to 4-hydroxynonenal: Fundamental issues in the mechanisms of lipid peroxidation. J. Biol. Chem. 2008, 283, 15539–15543. [Google Scholar] [CrossRef] [PubMed]
- Welsch, C.W. Enhancement of mammary tumorigenesis by dietary fat: Review of potential mechanisms. Am. J. Clin. Nutr. 1987, 45, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Birt, D.F.; Kris, E.S.; Choe, M.; Pelling, J.C. Dietary energy and fat effects on tumor promotion. Cancer Res. 1992, 52, 2035s–2039s. [Google Scholar] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef]
- Moreno-Sanchez, R.; Marin-Hernandez, A.; Saavedra, E.; Pardo, J.P.; Ralph, S.J.; Rodriguez-Enriquez, S. Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int. J. Biochem. Cell Biol. 2014, 50, 10–23. [Google Scholar] [CrossRef]
- Wice, B.M.; Reitzer, L.J.; Kennell, D. The continuous growth of vertebrate cells in the absence of sugar. J. Biol. Chem. 1981, 256, 7812–7819. [Google Scholar]
- Portais, J.C.; Schuster, R.; Merle, M.; Canioni, P. Metabolic flux determination in C6 glioma cells using carbon-13 distribution upon [1-13C]glucose incubation. Eur. J. Biochem. 1993, 217, 457–468. [Google Scholar] [CrossRef]
- Baggetto, L.G. Deviant energetic metabolism of glycolytic cancer cells. Biochimie 1992, 74, 959–974. [Google Scholar] [CrossRef]
- Israelsen, W.J.; Dayton, T.L.; Davidson, S.M.; Fiske, B.P.; Hosios, A.M.; Bellinger, G.; Li, J.; Yu, Y.; Sasaki, M.; Horner, J.W.; et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 2013, 155, 397–409. [Google Scholar] [CrossRef]
- Chandel, N.S.; Budinger, G.R.; Choe, S.H.; Schumacker, P.T. Cellular respiration during hypoxia. Role of cytochrome oxidase as the oxygen sensor in hepatocytes. J. Biol. Chem. 1997, 272, 18808–18816. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Khasawneh, J.; Schulz, M.D.; Walch, A.; Rozman, J.; Hrabe de Angelis, M.; Klingenspor, M.; Buck, A.; Schwaiger, M.; Saur, D.; Schmid, R.M.; et al. Inflammation and mitochondrial fatty acid beta-oxidation link obesity to early tumor promotion. Proc. Natl. Acad. Sci. USA 2009, 106, 3354–3359. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Michaud, D.S.; Giovannucci, E.; Willett, W.C.; Colditz, G.A.; Stampfer, M.J.; Fuchs, C.S. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001, 286, 921–929. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-S.; Oh, S.-J.; Choi, H.-J.; Kang, J.H.; Lee, S.-H.; Ha, J.S.; Woo, S.M.; Jang, H.; Lee, H.; Kim, S.-Y. ATP Production Relies on Fatty Acid Oxidation Rather than Glycolysis in Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 2477. https://doi.org/10.3390/cancers12092477
Lee J-S, Oh S-J, Choi H-J, Kang JH, Lee S-H, Ha JS, Woo SM, Jang H, Lee H, Kim S-Y. ATP Production Relies on Fatty Acid Oxidation Rather than Glycolysis in Pancreatic Ductal Adenocarcinoma. Cancers. 2020; 12(9):2477. https://doi.org/10.3390/cancers12092477
Chicago/Turabian StyleLee, Jae-Seon, Su-Jin Oh, Hyun-Jung Choi, Joon Hee Kang, Seon-Hyeong Lee, Ji Sun Ha, Sang Myung Woo, Hyonchol Jang, Ho Lee, and Soo-Youl Kim. 2020. "ATP Production Relies on Fatty Acid Oxidation Rather than Glycolysis in Pancreatic Ductal Adenocarcinoma" Cancers 12, no. 9: 2477. https://doi.org/10.3390/cancers12092477
APA StyleLee, J.-S., Oh, S.-J., Choi, H.-J., Kang, J. H., Lee, S.-H., Ha, J. S., Woo, S. M., Jang, H., Lee, H., & Kim, S.-Y. (2020). ATP Production Relies on Fatty Acid Oxidation Rather than Glycolysis in Pancreatic Ductal Adenocarcinoma. Cancers, 12(9), 2477. https://doi.org/10.3390/cancers12092477