Proteotoxic Stress and Cell Death in Cancer Cells



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Proteotoxic Stress: An Introduction
2. The Protein Quality System
3. Cellular Responses to the Unfolded Proteins
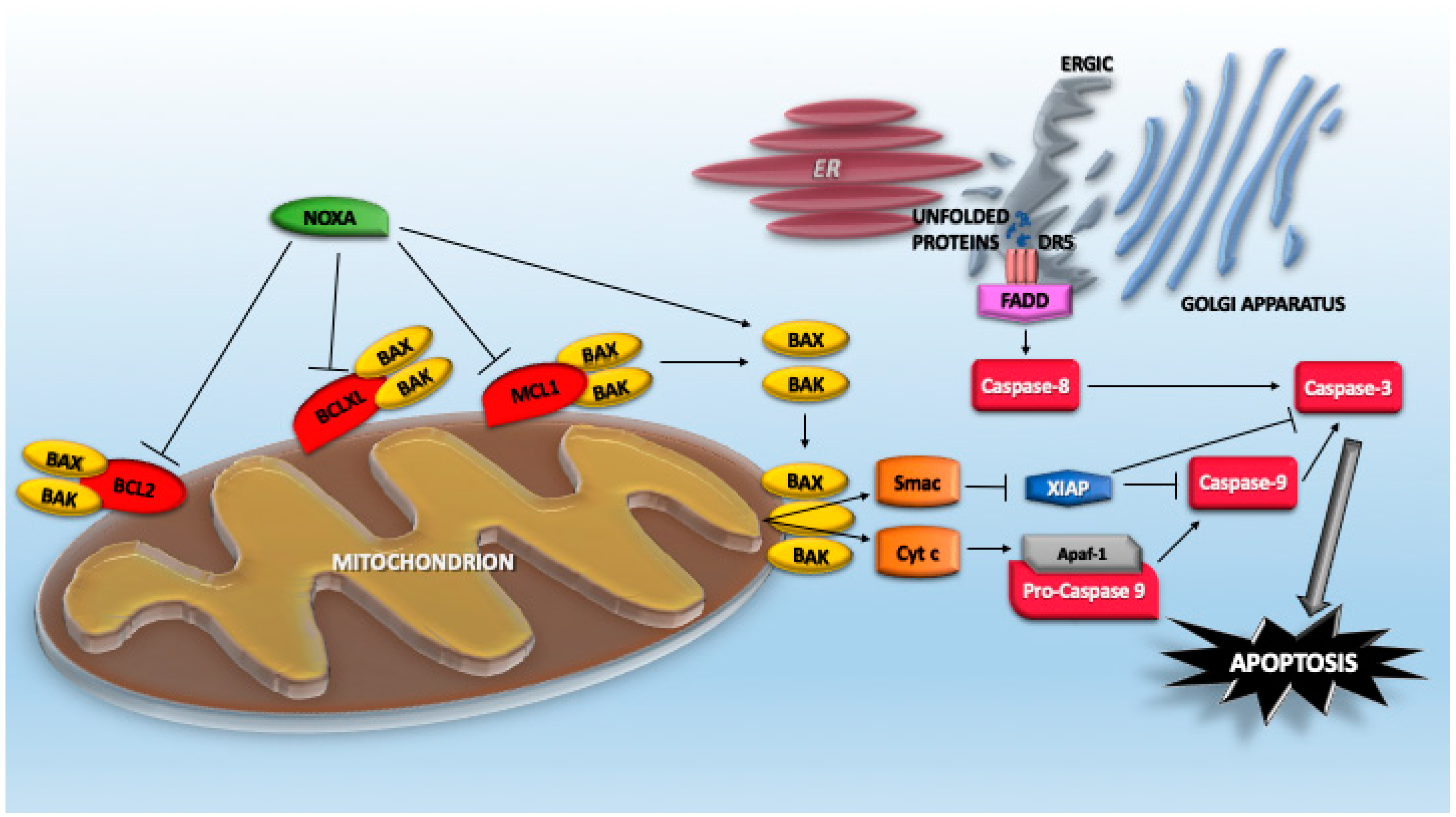
4. Cell Death Pathways Activated by Proteotoxic Stress
4.1. The Extrinsic Pathway of Caspase Activation
4.2. The ATF Network
4.3. The BCL2 Family Members
4.4. Additional Cell Death Responses
5. Proteotoxic Stress in Cancer Cells
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Ellis, R.J.; Minton, A.P. Protein aggregation in crowded environments. Biol. Chem. 2006, 387, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, S.; Nyayanit, D.; Gadgil, C. A systems view of the protein expression process. Syst. Synth. Biol. 2011, 5, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Taylor, R.C.; Dillin, A. Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 2011, 3, a004440. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Higuchi-Sanabria, R.; Frankino, P.A.; Paul, J.W., III; Tronnes, S.U.; Dillin, A. A futile battle? Protein quality control and the stress of aging. Dev. Cell 2018, 44, 139–163. [Google Scholar] [CrossRef]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and its co-chaperones in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Balchin, D.; Miličić, G.; Strauss, M.; Hayer-Hartl, M.; Hartl, F.U. Pathway of actin folding directed by the eukaryotic chaperonin TRiC. Cell 2018, 174, 1507–1521. [Google Scholar] [CrossRef] [PubMed]
- Preissler, S.; Deuerling, E. Ribosome-associated chaperones as key players in proteostasis. Trends Biochem. Sci. 2012, 37, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Kramer, G.; Shiber, A.; Bukau, B. Mechanisms of cotranslational maturation of newly synthesized proteins. Annu. Rev. Biochem. 2019, 88, 337–364. [Google Scholar] [CrossRef]
- Hayer-Hartl, M.; Bracher, A.; Hartl, F.U. The GroELGroES chaperonin machine: A nano-cage for protein folding. Trends Biochem. Sci. 2016, 41, 62–76. [Google Scholar] [CrossRef]
- Carra, S.; Alberti, S.; Arrigo, P.A.; Benesch, J.L.; Benjamin, I.J.; Boelens, W.; Bartelt-Kirbach, B.; Brundel, B.; Buchner, J.; Bukau, B.; et al. The growing world of small heat shock proteins: From structure to functions. Cell Stress Chaperones 2017, 22, 601–611. [Google Scholar] [CrossRef]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef]
- Powers, E.T.; Balch, W.E. Diversity in the origins of proteostasis networks—A driver for protein function in evolution. Nat. Rev. Mol. Cell Biol. 2013, 14, 237–248. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Tucker, G.; Peng, J.; Krykbaeva, I.; Lin, Z.Y.; Larsen, B.; Choi, H.; Berger, B.; Gingras, A.C.; Lindquist, S. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 2014, 158, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.; Pillarsetty, N.; Koren, J.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397–401. [Google Scholar] [CrossRef] [PubMed]
- De Thonel, A.; Mezger, V.; Garrido, C. Implication of Heat Shock Factors in tumorigenesis: Therapeutical potential. Cancers 2011, 3, 1158–1181. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Alberti, S.; Höhfeld, J. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim. Biophys. Acta 2004, 1695, 171–188. [Google Scholar] [CrossRef] [PubMed]
- Kundrat, L.; Regan, L. Balance between folding and degradation for Hsp90-dependent client proteins: A key role for CHIP. Biochemistry 2010, 49, 7428–7438. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Ciechanover, A. The ubiquitin proteolytic system: From a vague idea, through basic mechanisms, and onto human diseases and drug targeting. Neurology 2006, 66, S7–S19. [Google Scholar] [CrossRef]
- Demarchi, F.; Brancolini, C. Altering protein turnover in tumor cells: New opportunities for anti-cancer therapies. Drug Resist. Updates 2005, 8, 359–368. [Google Scholar] [CrossRef]
- Chowdhury, M.; Enenkel, C. Intracellular dynamics of the Ubiquitin-Proteasome-System. F1000Research 2015, 4, 367. [Google Scholar] [CrossRef]
- Morreale, F.E.; Walden, H. Types of ubiquitin ligases. Cell 2016, 165, 248.e1. [Google Scholar] [CrossRef] [PubMed]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The ubiquitin-proteasome system. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef]
- D’Arcy, P.; Wang, X.; Linder, S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol. Ther. 2015, 147, 32–54. [Google Scholar] [CrossRef]
- Sgorbissa, A.; Potu, H.; Brancolini, C. Isopeptidases in anticancer therapy: Looking for inhibitors. Am. J. Transl. Res. 2010, 2, 235–247. [Google Scholar]
- Rehman, S.A.A.; Kristariyanto, Y.A.; Choi, S.Y.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 is a member of an evolutionarily conserved and structurally distinct new family of deubiquitinating enzymes. Molecular. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef]
- Kuo, C.L.; Goldberg, A.L. Ubiquitinated proteins promote the association of proteasomes with the deubiquitinating enzyme Usp14 and the ubiquitin ligase Ube3c. Proc. Natl. Acad. Sci. USA 2017, 114, E3404–E3413. [Google Scholar] [CrossRef]
- Aleo, E.; Henderson, C.J.; Fontanini, A.; Solazzo, B.; Brancolini, C. Identification of new compounds that trigger apoptosome-independent caspase activation and apoptosis. Cancer Res. 2006, 66, 9235–9244. [Google Scholar] [CrossRef]
- Tian, Z.; D’Arcy, P.; Wang, X.; Ray, A.; Tai, Y.T.; Hu, Y.; Carrasco, R.D.; Richardson, P.; Linder, S.; Chauhan, D.; et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014, 123, 706–716. [Google Scholar] [CrossRef]
- Zhang, X.; Pellegrini, P.; Saei, A.A.; Hillert, E.K.; Mazurkiewicz, M.; Olofsson, M.H.; Zubarev, R.A.; D’Arcy, P.; Linder, S. The deubiquitinase inhibitor b-AP15 induces strong proteotoxic stress and mitochondrial damage. Biochem. Pharmacol. 2018, 156, 291–301. [Google Scholar] [CrossRef]
- Baumeister, W.; Walz, J.; Zühl, F.; Seemüller, E. The proteasome: Paradigm of a self-compartmentalizing protease. Cell 1998, 92, 367–380. [Google Scholar] [CrossRef]
- Voges, D.; Zwickl, P.; Baumeister, W. The 26S proteasome: A molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999, 68, 1015–1068. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Akopian, T.N.; Castillo, V.; Goldberg, A.L. Proteasome active sites allosterically regulate each other, suggesting a cyclical bite-chew mechanism for protein breakdown. Mol. Cell 1999, 4, 395–402. [Google Scholar] [CrossRef]
- Beck, F.; Unverdorben, P.; Bohn, S.; Schweitzer, A.; Pfeifer, G.; Sakata, E.; Nickell, S.; Plitzko, J.M.; Villa, E.; Baumeister, W.; et al. Near-atomic resolution structural model of the yeast 26S proteasome. Proc. Natl. Acad. Sci. USA 2012, 109, 14870–14875. [Google Scholar] [CrossRef] [PubMed]
- de la Peña, A.H.; Goodall, E.A.; Gates, S.N.; Lander, G.C.; Martin, A. Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science 2018, 362, eaav0725. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019, 565, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; Matouschek, A. Substrate selection by the proteasome through initiation regions. Protein Sci. 2019, 28, 1222–1232. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of deubiquitinase specificityand regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Seguin, S.J.; Lambert, H.; Landry, J. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J. Biol. Chem. 2008, 283, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Fürst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; Münch, C.; Hanssum, A.; Bertolotti, A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol. Cell 2012, 48, 242–253. [Google Scholar] [CrossRef]
- Zhang, T.; Shen, S.; Qu, J.; Ghaemmaghami, S. Global analysis of cellular protein flux quantifies the selectivity of basal autophagy. Cell Rep. 2016, 14, 2426–2439. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Li, J.; Zhang, D.; Wiersma, M.; Brundel, B.J.J.M. Role of autophagy in proteostasis: Friend and foe in cardiac diseases. Cells 2018, 7, 279. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A.; Varshavsky, A. Basic medical research award. The ubiquitin system. Nat. Med. 2000, 6, 1073–1081. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.C.; Wilkinson, A.S.; Scott, F.L.; Csomos, R.A.; Salvesen, G.S.; Duckett, C.S. Neutralization of Smac/Diablo by Inhibitors of Apoptosis (IAPs): A caspase-independent mechanism for apoptotic inhibition. J. Biol. Chem. 2004, 279, 51082–51090. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The Unfolded Protein Response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Auner, H.W.; Moody, A.M.; Ward, T.H.; Kraus, M.; Milan, E.; May, P.; Chaidos, A.; Driessen, C.; Cenci, S.; Dazzi, F.; et al. Combined inhibition of p97 and the proteasome causes lethal disruption of the secretory apparatus in multiple myeloma cells. PLoS ONE 2013, 8, e74415. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, N.S.; Connell, P.; Christians, E.S.; Yan, L.J.; Taylor, R.P.; Orosz, A.; Zhang, X.Q.; Stevenson, T.J.; Peshock, R.M.; Leopold, J.A.; et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 2007, 130, 427–439. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef]
- Kannan, S.; Muthusamy, V.R.; Whitehead, K.J.; Wang, L.; Gomes, A.V.; Litwin, S.E.; Kensler, T.W.; Abel, E.D.; Hoidal, J.R.; Rajasekaran, N.S. Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy. Cardiovasc. Res. 2013, 100, 63–73. [Google Scholar] [CrossRef]
- Ri, M. Endoplasmic-reticulum stress pathway-associated mechanisms of action of proteasome inhibitors in multiple myeloma. Int. J. Hematol. 2016, 104, 273–280. [Google Scholar] [CrossRef]
- Lafont, E. Stress management: Death receptor signalling and cross-talks with the unfolded protein response in cancer. Cancers 2020, 12, 1113. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Calfon, M.; Urano, F.; Novoa, I.; Ron, D. Transcriptional and translational control in the mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol. 2002, 18, 575–599. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef]
- Xing, H.Y.; Cai, Y.Q.; Wang, X.F.; Wang, L.L.; Li, P.; Wang, G.Y.; Chen, J.H. The cytoprotective effect of hyperoside against oxidative stress is mediated by the Nrf2-ARE signaling pathway through GSK-3β inactivation. PLoS ONE 2015, 10, e0145183. [Google Scholar] [CrossRef]
- Zhang, W.; Hietakangas, V.; Wee, S.; Lim, S.C.; Gunaratne, J.; Cohen, S.M. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev. 2013, 27, 441–449. [Google Scholar] [CrossRef]
- You, S.; Li, H.; Hu, Z.; Zhang, W. eIF2α kinases PERK and GCN2 act on FOXO to potentiate FOXO activity. Genes Cells 2018, 23, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Bobrovnikova-Marjon, E.; Pytel, D.; Riese, M.J.; Vaites, L.P.; Singh, N.; Koretzky, G.A.; Witze, E.S.; Diehl, J.A. PERK utilizes intrinsic lipid kinase activity to generate phosphatidic acid, mediate Akt activation, and promote adipocyte differentiation. Mol. Cell. Biol. 2012, 32, 2268–2278. [Google Scholar] [CrossRef] [PubMed]
- Pytel, D.; Gao, Y.; Mackiewicz, K.; Katlinskaya, Y.V.; Staschke, K.A.; Paredes, M.C.G.; Yoshida, A.; Qie, S.; Zhang, G.; Chajewski, O.S.; et al. PERK is a haploinsufficient tumor suppressor: Gene dose determines tumor-suppressive versus tumor promoting properties of PERK in melanoma. PLoS Genet. 2016, 12, e1006518. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D.J. The integrated stress response and proteotoxicity in cancer therapy. Biochem. Biophys. Res. Commun. 2017, 482, 450–453. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Schroeder, M.D.; Zhu, K.; Xiong, K.; McConkey, D.J. HRI-mediated translational repression reduces proteotoxicity and sensitivity to Bortezomib in human pancreatic cancer cells. Oncogene 2018, 37, 4413–4427. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP indevelopment, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Couillault, C.; Fourquet, P.; Pophillat, M.; Ewbank, J.J. A UPR-independent infection-specific role for a BiP/GRP78 protein in the control of antimicrobial peptide expression in C. elegans epidermis. Virulence 2012, 3, 299–308. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Hillary, R.F.; FitzGerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef] [PubMed]
- Ron, D. Translational control in the endoplasmic reticulum stress response. J. Clin. Investig. 2002, 110, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Guang, M.H.Z.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting proteotoxic stress in cancer: A review of the role that protein quality control pathways play in oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Cano-González, A.; Mauro-Lizcano, M.; Iglesias-Serret, D.; Gil, J.; López-Rivas, A. Involvement of both caspase-8 and Noxa-activated pathways in Endoplasmic Reticulum stress-induced apoptosis in triple-negative breast tumor cells. Cell Death Dis. 2018, 9, 134. [Google Scholar] [CrossRef]
- Zhu, G.Y.; Li, Y.W.; Tse, A.K.W.; Hau, D.K.P.; Leung, C.H.; Yu, Z.L.; Fong, W.L. 20(s)-protopanaxadiol, a metabolite of ginsenosides, induced cell apoptosis through Endoplasmic Reticulum stress in human hepatocarcinoma HepG2 cells. Eur. J. Pharmacol. 2011, 668, 88–98. [Google Scholar] [CrossRef]
- Manini, I.; Sgorbissa, A.; Potu, H.; Tomasella, A.; Brancolini, C. The DeISGylase USP18 limits TRAIL-induced apoptosis through the regulation of TRAIL levels: Cellular levels of TRAIL influences responsiveness to TRAIL-induced apoptosis. Cancer Biol. Ther. 2013, 14, 1158–1166. [Google Scholar] [CrossRef]
- Martín-Pérez, R.; Palacios, C.; Yerbes, R.; Cano-González, A.; Iglesias-Serret, D.; Gil, J.; Reginato, M.J.; López-Rivas, A. Activated ERBB2/HER2 licenses sensitivity to apoptosis upon Endoplasmic Reticulum stress through a PERK-dependent pathway. Cancer Res. 2014, 74, 1766–1777. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef]
- Chang, T.K.; Lawrence, D.A.; Lu, M.; Tan, J.; Harnoss, J.M.; Marsters, S.A.; Liu, P.; Sandoval, W.; Martin, S.E.; Ashkenazi, A. Coordination between two branches of the unfolded protein response determines apoptotic cell fate. Mol. Cell 2018, 71, 629–636.e5. [Google Scholar] [CrossRef]
- Zhu, Z.C.; Liu, J.W.; Yang, C.; Li, M.J.; Wu, R.J.; Xiong, Z.Q. Targeting KPNB1 overcomes TRAIL resistance by regulating DR5, Mcl-1 and FLIP in glioblastoma cells. Cell Death Dis. 2019, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.; Marsters, S.A.; Ashkenazi, A.; Walter, P. Misfolded proteins bind and activate Death Receptor 5 to trigger apoptosis during unresolved endoplasmic reticulum stress. Elife 2020, 9, e52291. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, G.P.; O’Connor, H.; Henry, C.M.; Davidovich, P.; Clancy, D.M.; Albert, M.L.; Cullen, S.P.; Martin, S.J. TRAIL receptors serve as stress-associated molecular patterns to promote ER-stress-induced inflammation. Dev. Cell 2020, 52, 714–730.e5. [Google Scholar] [CrossRef] [PubMed]
- Estornes, Y.; Aguileta, M.A.; Dubuisson, C.; De Keyser, J.; Goossens, V.; Kersse, K.; Samali, A.; Vandenabeele, P.; Bertrand, M.J.M. RIPK1 promotes death receptor-independent caspase-8-mediated apoptosis under unresolved ER stress conditions. Cell Death Dis. 2015, 6, e1798. [Google Scholar] [CrossRef]
- Von Karstedt, S.; Walczak, H. An unexpected turn of fortune: Targeting TRAIL-Rs in KRAS-driven cancer. Cell Death Discov. 2020, 6, 14. [Google Scholar] [CrossRef]
- Pitale, P.M.; Gorbatyuk, O.; Gorbatyuk, M. Neurodegeneration: Keeping ATF4 on a tight leash. Front. Cell. Neurosci. 2017, 11, 410. [Google Scholar] [CrossRef]
- Rzymski, T.; Milani, M.; Singleton, D.C.; Harris, A.L. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 2009, 8, 3838–3847. [Google Scholar] [CrossRef]
- Lassot, I.; Ségéral, E.; Berlioz-Torrent, C.; Durand, H.; Groussin, L.; Hai, T.; Benarous, R.; Margottin-Goguet, F. ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(betaTrCP) ubiquitin ligase. Mol. Cell. Biol. 2001, 21, 2192–2202. [Google Scholar] [CrossRef]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A.; et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205. [Google Scholar] [CrossRef]
- Cnop, M.; Toivonen, S.; Igoillo-Esteve, M.; Salpea, P. Endoplasmic reticulum stress and eIF2α phosphorylation: The Achilles heel of pancreatic β cells. Mol. Metab. 2017, 6, 1024–1039. [Google Scholar] [CrossRef]
- Palam, L.R.; Baird, T.D.; Wek, R.C. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J. Biol. Chem. 2011, 286, 10939–10949. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.S.; Liu, D.; Li, T.; de Zavalia, N.; Zhu, L.; Li, J.; Karthikeyan, R.; Alain, T.; Liu, A.C.; Storch, K.F.; et al. The eIF2α kinase GCN2 modulates period and rhythmicity of the circadian clock by translational control of Atf4. Neuron 2019, 104, 724–735.e6. [Google Scholar] [CrossRef] [PubMed]
- Ratan, R.R. The chemical biology of ferroptosis in the central nervous system. Cell Chem. Biol. 2020, 27, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, N.; Messah, C.; Han, J.; LaVail, M.M.; Kaufman, R.J.; Lin, J.H. Translational and posttranslational regulation of XIAP by eIF2alpha and ATF4 promotes ER stress-induced cell death during the unfolded protein response. Mol. Biol. Cell 2014, 25, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Imbriano, C.; Mantovani, R. Dynamic recruitment of transcription factors and epigenetic changes on the ER stress response gene promoters. Nucleic Acids Res. 2006, 34, 3116–3127. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef]
- Shiraishi, T.; Yoshida, T.; Nakata, S.; Horinaka, M.; Wakada, M.; Mizutani, Y.; Miki, T.; Sakai, T. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer Res. 2005, 65, 6364–6370. [Google Scholar] [CrossRef]
- Hai, T.; Wolford, C.C.; Chang, Y.S. ATF3, a Hub of the cellular adaptive-response network, in the pathogenesis of diseases: Is modulation of inflammation a unifying component? Gene Expr. 2010, 15, 1–11. [Google Scholar] [CrossRef]
- Edagawa, M.; Kawauchi, J.; Hirata, M.; Goshima, H.; Inoue, M.; Okamoto, T.; Murakami, A.; Maehara, Y.; Kitajima, S. Role of activating transcription factor 3 (ATF3) in endoplasmic reticulum (ER) Stress-induced sensitization of p53-deficient human colon cancer cells to tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis through Up-regulation of death receptor 5 (DR5) by zerumbone and celecoxib. J. Biol. Chem. 2014, 289, 21544–21561. [Google Scholar] [CrossRef]
- Li, T.; Su, L.; Lei, Y.; Liu, X.; Zhang, Y.; Liu, X. DDIT3 and KAT2A regulate TNFRSF10A and TNFRSF10B expression in endoplasmic reticulum stress-mediated apoptosis in human lung cancer cells. J. Biol. Chem. 2015, 290, 11108–11118. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Püschel, F.; León-Annicchiarico, C.L.; O’Connor, H.; Martin, S.J.; Palou-Gramón, D.; Lucendo, E.; Muñoz-Pinedo, C. Glucose deprivation induces ATF4-mediated apoptosis through TRAIL death receptors. Mol. Cell. Biol. 2017, 37, e00479-16. [Google Scholar] [CrossRef] [PubMed]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Vu, T.T.; Cook, W.; Naseri, M.; Zhan, K.; Nakajima, W.; Harada, H. p53-independent Noxa induction by cisplatin is regulated by ATF3/ATF4 in head and neck squamous cell carcinoma cells. Mol. Oncol. 2018, 12, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Morsi, R.Z.; Hage-Sleiman, R.; Kobeissy, H.; Dbaibo, G. Noxa: Role in cancer pathogenesis and treatment. Curr. Cancer Drug Targets 2018, 18, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Ahn, H.J.; Ryu, J.H.; Suk, K.; Park, J.H. BH3-only protein Noxa is a mediator of hypoxic cell death induced by Hypoxia-Inducible Factor 1alpha. J. Exp. Med. 2004, 199, 113–124. [Google Scholar] [CrossRef]
- Hershko, T.; Ginsberg, D. Up-regulation of Bcl-2 Homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J. Biol. Chem. 2004, 279, 8627–8634. [Google Scholar] [CrossRef]
- Qin, J.Z.; Ziffra, J.; Stennett, L.; Bodner, B.; Bonish, B.K.; Chaturvedi, V.; Bennett, F.; Pollock, P.M.; Trent, J.M.; Hendrix, M.J.C.; et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005, 65, 6282–6293. [Google Scholar] [CrossRef]
- Fernandez, Y.; Verhaegen, M.; Miller, T.P.; Rush, J.L.; Steiner, P.; Opipari, A.W.J.; Lowe, S.W.; Soengas, M.S. Differential regulation of Noxa in normal melanocytes and melanoma cells by proteasome inhibition: Therapeutic implications. Cancer Res. 2005, 65, 6294–6304. [Google Scholar] [CrossRef]
- Du, H.; Wolf, J.; Schafer, B.; Moldoveanu, T.; Chipuk, J.E.; Kuwana, T. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J. Biol. Chem. 2011, 286, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Vela, L.; Gonzalo, O.; Naval, J.; Marzo, I. Direct interaction of Bax and Bak proteins with Bcl-2 Homology Domain 3 (BH3)-only proteins in living cells revealed by fluorescence complementation. J. Biol. Chem. 2013, 288, 4935–4946. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Smith, A.; Meng, X.W.; Schneider, P.A.; Pang, Y.P.; Kaufmann, S.H. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J. Cell Biol. 2011, 194, 39–48. [Google Scholar] [CrossRef]
- Chen, H.C.; Kanai, M.; Inoue-Yamauchi, A.; Tu, H.C.; Huang, Y.; Ren, D.; Kim, H.; Takeda, S.; Reyna, D.E.; Chan, P.M.; et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat. Cell Biol. 2015, 17, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Reimertz, C.; Kogel, D.; Rami, A.; Chittenden, T.; Prehn, J.H. Gene expression during ER stress-induced apoptosis in neurons: Induction of the BH3-onlyprotein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J. Cell Biol. 2003, 162, 587–597. [Google Scholar] [CrossRef]
- Li, J.; Lee, B.; Lee, A.S. Endoplasmic reticulum stress-induced apoptosis: Multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and Noxa by p53. J. Biol. Chem. 2006, 281, 7260–7270. [Google Scholar] [CrossRef]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ichijo, H.; Korsmeyer, S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell. Biol. 1999, 19, 8469–8478. [Google Scholar] [CrossRef]
- Fan, M.; Goodwin, M.; Vu, T.; Brantley-Finley, C.; Gaarde, W.A.; Chambers, T.C. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J. Biol. Chem. 2001, 275, 29980–29985. [Google Scholar] [CrossRef]
- Llambi, F.; Wang, Y.M.; Victor, B.; Yang, M.; Schneider, D.M.; Gingras, S.; Parsons, M.J.; Zheng, J.H.; Brown, S.A.; Pelletier, S.; et al. BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 2016, 165, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Sopha, P.; Ren, H.Y.; Grove, D.E.; Cyr, D.M. Endoplasmic Reticulum stress-induced degradation of DNAJB12 stimulates BOK accumulation and primes cancer cells for apoptosis. J. Biol. Chem. 2017, 292, 11792–11803. [Google Scholar] [CrossRef] [PubMed]
- Lopes, U.G.; Erhardt, P.; Yao, R.; Cooper, G.M. p53-dependent induction of apoptosis by proteasome inhibitors. J. Biol. Chem. 1997, 272, 12893–12896. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, N.; Duggal, S.; Singh, S.K.; Porwal, K.; Srivastava, V.K.; Maurya, R.; Bhatt, M.L.; Mishra, D.P. Proteasome inhibition mediates p53 reactivation and anti-cancer activity of 6-gingerol in cervical cancer cells. Oncotarget 2015, 6, 43310–43325. [Google Scholar] [CrossRef] [PubMed]
- Brancolini, C. Inhibitors of the Ubiquitin-Proteasome System and the cell death machinery: How many pathways are activated? Curr. Mol. Pharmacol. 2008, 1, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Lamore, S.D.; Cabello, C.M.; Lesson, J.L.; Muñoz-Rodriguez, J.L.; Wondrak, G.T. Thiostrepton is an inducer of oxidative and proteotoxic stress that impairs viability of human melanoma cells but not primary melanocytes. Biochem. Pharmacol. 2012, 83, 1229–1240. [Google Scholar] [CrossRef]
- Arai, S.; Varkaris, A.; Nouri, M.; Chen, S.; Xie, L.; Balk, S.P. MARCH5 mediates NOXA-dependent MCL1 degradation driven by kinase inhibitors and integrated stress response activation. Elife 2020, 9, e54954. [Google Scholar] [CrossRef]
- Djajawi, T.M.; Liu, L.; Gong, J.N.; Huang, A.S.; Luo, M.J.; Xu, Z.; Okamoto, T.; Call, M.J.; Huang, D.C.S.; van Delft, M.F. MARCH5 requires MTCH2 to coordinate proteasomal turnover of the MCL1:NOXA complex. Cell Death Differ. 2020. [Google Scholar] [CrossRef]
- Vaux, D.L.; Silke, J. IAPs, RINGs and ubiquitylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 287–297. [Google Scholar] [CrossRef]
- Scomazzon, S.P.; Riccio, A.; Santopolo, S.; Lanzilli, G.; Coccia, M.; Rossi, A.; Santoro, M.G. The zinc-finger AN1-type domain 2a gene acts as a regulator of cell survival in human melanoma: Role of E3-ligase cIAP2. Mol. Cancer Res. 2019, 17, 2444–2456. [Google Scholar] [CrossRef]
- Pei, X.Y.; Dai, Y.; Grant, S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef] [PubMed]
- Ciotti, S.; Iuliano, L.; Cefalù, S.; Comelli, M.; Mavelli, I.; Di Giorgio, E.; Brancolini, C. GSK3β is a key regulator of the ROS-dependent necrotic death induced by the quinone DMNQ. Cell Death Dis. 2020, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Vanlangenakker, N.; Vanden Berghe, T.; Krysko, D.M.; Festjens, N.; Vandenabeele, P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr. Mol. Med. 2008, 8, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Fontanini, A.; Foti, C.; Potu, H.; Crivellato, E.; Maestro, R.; Bernardi, P.; Demarchi, F.; Brancolini, C. The isopeptidase inhibitor G5 triggers a caspase-independent necrotic death in cells resistant to apoptosis: A comparative study with the proteasome inhibitor bortezomib. J. Biol. Chem. 2009, 284, 8369–8381. [Google Scholar] [CrossRef]
- Tomasella, A.; Blangy, A.; Brancolini, C. A receptor-interacting protein 1 (RIP1)-independent necrotic death under the control of protein phosphatase PP2A that involves the reorganization of Actin cytoskeleton and the action of Cofilin-1. J. Biol. Chem. 2014, 289, 25699–25710. [Google Scholar] [CrossRef]
- Darling, N.J.; Balmanno, K.; Cook, S.J. ERK1/2 signalling protects against apoptosis following Endoplasmic Reticulum stress but cannot provide long-term protection against BAX/BAK-independent cell death. PLoS ONE 2017, 12, e0184907. [Google Scholar] [CrossRef]
- Ramdzan, Y.M.; Trubetskov, M.M.; Ormsby, A.R.; Newcombe, E.A.; Sui, X.; Tobin, M.J.; Bongiovanni, M.N.; Gras, S.L.; Dewson, G.; Miller, J.M.L.; et al. Huntingtin inclusions trigger cellular quiescence, deactivate apoptosis, and lead to delayed necrosis. Cell Rep. 2017, 19, 919–927. [Google Scholar] [CrossRef]
- Wang, W.; Li, S.; Wang, H.; Li, B.; Shao, L.; Lai, Y.; Horvath, G.; Wang, Q.; Yamamoto, M.; Janicki, J.S.; et al. Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins. J. Mol. Cell. Cardiol. 2014, 72, 305–315. [Google Scholar] [CrossRef]
- Han, J.H.; Park, J.; Myung, S.H.; Lee, S.H.; Kim, H.Y.; Kim, K.S.; Seo, Y.W.; Kim, T.H. Noxa mitochondrial targeting domain induces necrosis via VDAC2 and mitochondrial catastrophe. Cell Death Dis. 2019, 10, 519. [Google Scholar] [CrossRef]
- Schwarzer, R.; Laurien, L.; Pasparakis, M. New insights into the regulation of apoptosis, necroptosis, and pyroptosis by Receptor Interacting Protein Kinase 1 and caspase-8. Curr. Opin. Cell Biol. 2020, 63, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Coustry, F.; Posey, K.L.; Liu, P.; Alcorn, J.L.; Hecht, J.T. D469del-COMP retention in chondrocytes stimulates caspase-independent necroptosis. Am. J. Pathol. 2012, 180, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, F.; Naponelli, V.; Silva, A.; Modernelli, A.; Ramazzina, I.; Bonacini, M.; Tardito, S.; Gatti, R.; Uggeri, J.; Bettuzzi, S. Polyphenon E(R), a standardized green tea extract, induces endoplasmic reticulum stress, leading to death of immortalized PNT1a cells by anoikis and tumorigenic PC3 by necroptosis. Carcinogenesis 2014, 35, 828–839. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Rivera, D.; Delvaeye, T.; Roelandt, R.; Nerinckx, W.; Augustyns, K.; Vandenabeele, P.; Bertrand, M.J.M. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 2017, 24, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Luan, Q.; Jin, L.; Jiang, C.C.; Tay, K.H.; Lai, F.; Liu, X.Y.; Liu, Y.L.; Guo, S.T.; Li, C.Y.; Yan, X.G.; et al. RIPK1 regulates survival of human melanoma cells upon endoplasmic reticulum stress through autophagy. Autophagy 2015, 11, 975–994. [Google Scholar] [CrossRef]
- Cheng, S.B.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019, 10, 927. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Bebber, C.M.; Müller, F.; Clemente, L.P.; Weber, J.; von Karstedt, S. Ferroptosis in cancer cell biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, D.H.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-induced endoplasmic reticulum stress: Cross-talk between ferroptosis and apoptosis. Mol. Cancer Res. 2018, 16, 1073–1076. [Google Scholar] [CrossRef]
- Rahmani, M.; Davis, E.M.; Crabtree, T.R.; Habibi, J.R.; Nguyen, T.K.; Dent, P.; Grant, S. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol. Cell. Biol. 2007, 27, 5499–5513. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mi, Y.; Zhang, X.; Ma, Q.; Song, Y.; Zhang, L.; Wang, D.; Xing, J.; Hou, B.; Li, H.; et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J. Exp. Clin. Cancer Res. 2019, 38, 402. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Flohé, L. Regulatory phenomena in the glutathione peroxidase superfamily. Antioxid. Redox Signal. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Bakunts, A.; Orsi, A.; Lari, F.; Tadè, L.; Danieli, A.; Rato, C.; Valetti, C.; Sitia, R.; Raimondi, A.; et al. Inadequate BiP availability defines endoplasmic reticulum stress. Elife 2019, 8, e41168. [Google Scholar] [CrossRef]
- Li, J.; Labbadia, J.; Morimoto, R.I. Rethinking HSF1 in stress, development, and organismal health. Trends Cell Biol. 2017, 27, 895–905. [Google Scholar] [CrossRef]
- Gaglia, G.; Rashid, R.; Yapp, C.; Joshi, G.N.; Li, C.G.; Lindquist, S.L.; Sarosiek, K.A.; Whitesell, L.; Sorger, P.K.; Santagata, S. HSF1 phase transition mediates stress adaptation and cell fate decisions. Nat. Cell Biol. 2020, 22, 151–158. [Google Scholar] [CrossRef]
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef]
- Cenci, S.; Sitia, R. Managing and exploiting stress in the antibody factory. FEBS Lett. 2007, 581, 3652–3657. [Google Scholar] [CrossRef]
- Ruggero, D. Translational control in cancer etiology. Cold Spring Harb. Perspect. Biol. 2013, 5, a012336. [Google Scholar] [CrossRef]
- Yun, Y.S.; Kim, K.H.; Tschida, B.; Sachs, Z.; Noble-Orcutt, K.E.; Moriarity, B.S.; Ai, T.; Ding, R.; Williams, J.; Chen, L.; et al. mTORC1 coordinates protein synthesis and immunoproteasome formation via PRAS40 to prevent accumulation of protein stress. Mol. Cell 2016, 61, 625–639. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Hyer, M.L.; Milhollen, M.A.; Ciavarri, J.; Fleming, P.; Traore, T.; Sappal, D.; Huck, J.; Shi, J.; Gavin, J.; Brownell, J.; et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 2018, 24, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Chui, M.H.; Doodnauth, S.A.; Erdmann, N.; Tiedemann, R.E.; Sircoulomb, F.; Drapkin, R.; Shaw, P.; Rottapel, R. Chromosomal instability and mTORC1 activation through PTEN loss contribute to proteotoxic stress in ovarian carcinoma. Cancer Res. 2019, 79, 5536–5549. [Google Scholar] [CrossRef]
- Seo, J.H.; Rivadeneira, D.B.; Caino, M.C.; Chae, Y.C.; Speicher, D.W.; Tang, H.Y.; Vaira, V.; Bosari, S.; Palleschi, A.; Rampini, P.; et al. The mitochondrial unfoldase-peptidase complex ClpXP controls bioenergetics stress and metastasis. PLoS Biol. 2016, 14, e1002507. [Google Scholar] [CrossRef] [PubMed]
- Llinàs-Arias, P.; Rosselló-Tortella, M.; López-Serra, P.; Pérez-Salvia, M.; Setién, F.; Marin, S.; Muñoz, J.P.; Junza, A.; Capellades, J.; Calleja-Cervantes, M.E.; et al. Epigenetic loss of the endoplasmic reticulum-associated degradation inhibitor SVIP induces cancer cell metabolic reprogramming. JCI Insight 2019, 5, e125888. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef]
- Torres, E.M.; Sokolsky, T.; Tucker, C.M.; Chan, L.Y.; Boselli, M.; Dunham, M.J.; Amon, A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 2007, 317, 916–924. [Google Scholar] [CrossRef]
- Ohashi, A.; Ohori, M.; Iwai, K.; Nakayama, Y.; Nambu, T.; Morishita, D.; Kawamoto, T.; Miyamoto, M.; Hirayama, T.; Okaniwa, M.; et al. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat. Commun. 2015, 6, 7668. [Google Scholar] [CrossRef]
- Vermulst, M.; Denney, A.S.; Lang, M.J.; Hung, C.W.; Moore, S.; Moseley, M.A.; Thompson, J.W.; Madden, V.; Gauer, J.; Wolfe, K.J.; et al. Transcription errors induce proteotoxic stress and shorten cellular lifespan. Nat. Commun. 2015, 6, 8065. [Google Scholar] [CrossRef]
- Bastola, P.; Oien, D.B.; Cooley, M.; Chien, J. Emerging cancer therapeutic targets in protein homeostasis. AAPS J. 2018, 20, 94. [Google Scholar] [CrossRef]
- Xie, J.L.; Jarosz, D.F. Mutations, protein homeostasis, and epigenetic control of genome integrity. DNA Repair 2018, 71, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Pfau, S.J.; Amon, A. Chromosomal instability and aneuploidy in cancer: From yeast to man. EMBO Rep. 2012, 13, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.M.; Vaites, L.P.; Wells, J.N.; Santaguida, S.; Paulo, J.A.; Storchova, Z.; Harper, J.W.; Marsh, J.A.; Amon, A. Protein aggregation mediates stoichiometry of protein complexes in aneuploid cells. Genes Dev. 2019, 33, 1031–1047. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Passerini, V.; Durrbaum, M.; Stingele, S.; Storchova, Z. HSF1 deficiency and impaired HSP90-dependent protein folding are hallmarks of aneuploid human cells. EMBO J. 2014, 33, 2374–2387. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Tsai, H.J.; Gordon, M.R.; Li, R. Cellular stress associated with aneuploidy. Dev. Cell 2018, 44, 420–431. [Google Scholar] [CrossRef]
- Mukhopadhyay, C.; Triplett, A.; Bargar, T.; Heckman, C.; Wagner, K.U.; Naramura, M. Casitas B-cell lymphoma (Cbl) proteins protect mammary epithelial cells from proteotoxicity of active c-Src accumulation. Proc. Natl. Acad. Sci. USA 2016, 113, E8228–E8237. [Google Scholar] [CrossRef]
- Dodgson, S.E.; Santaguida, S.; Kim, S.; Sheltzer, J.; Amon, A. The pleiotropic deubiquitinase Ubp3 confers aneuploidy tolerance. Genes Dev. 2016, 30, 2259–2271. [Google Scholar] [CrossRef]
- Sannino, S.; Guerriero, C.J.; Sabnis, A.J.; Stolz, D.B.; Wallace, C.T.; Wipf, P.; Watkins, S.C.; Bivona, T.G.; Brodsky, J.L. Compensatory increases of select proteostasis networks after Hsp70 inhibition in cancer cells. J. Cell Sci. 2018, 131, jcs217760. [Google Scholar] [CrossRef]
- Chen, L.; Yang, X. TRIM11 cooperates with HSF1 to suppress the anti-tumor effect of proteotoxic stress drugs. Cell Cycle 2019, 18, 60–68. [Google Scholar] [CrossRef]
- Jena, K.K.; Mehto, S.; Kolapalli, S.P.; Nath, P.; Sahu, R.; Chauhan, N.R.; Sahoo, P.K.; Dhar, K.; Das, S.K.; Chauhan, S.; et al. TRIM16 governs the biogenesis and disposal of stress-induced protein aggregates to evade cytotoxicity: Implication for neurodegeneration and cancer. Autophagy 2019, 15, 924–926. [Google Scholar] [CrossRef]
- Harris, I.S.; Endress, J.E.; Coloff, J.L.; Selfors, L.M.; McBrayer, S.K.; Rosenbluth, J.M.; Takahashi, N.; Dhakal, S.; Koduri, V.; Oser, M.G.; et al. Deubiquitinases maintain protein homeostasis and survival of cancer cells upon glutathione depletion. Cell Metab. 2019, 29, 1166–1181.e6. [Google Scholar] [CrossRef] [PubMed]
- Carugo, A.; Minelli, R.; Sapio, L.; Soeung, M.; Carbone, F.; Robinson, F.S.; Tepper, J.; Chen, Z.; Lovisa, S.; Svelto, M.; et al. p53 is a master regulator of proteostasis in SMARCB1-deficient malignant rhabdoid tumors. Cancer Cell 2019, 35, 204–220.e9. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Sicari, D.; Fantuz, M.; Bellazzo, A.; Valentino, E.; Apollonio, M.; Pontisso, I.; Di Cristino, F.; Dal Ferro, M.; Bicciato, S.; Del Sal, G.; et al. Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene 2019, 38, 6184–6195. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in cancer: A promising therapeutic approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef]
- Leu, J.I.J.; Barnoud, T.; Zhang, G.; Tian, T.; Wei, Z.; Herlyn, M.; Murphy, M.E.; George, D.L. Inhibition of stress-inducible HSP70 impairs mitochondrial proteostasis and function. Oncotarget 2017, 8, 45656–45669. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins promote cancer: It’s a protection racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef]
- Vangala, J.R.; Radhakrishnan, S.K. Nrf1-mediated transcriptional regulation of the proteasome requires a functional TIP60 complex. J. Biol. Chem. 2019, 294, 2036–2045. [Google Scholar] [CrossRef]
- Dias, M.H.; Fonseca, C.S.; Zeidler, J.D.; Albuquerque, L.L.; da Silva, M.S.; Cararo-Lopes, E.; Reis, M.S.; Noël, V.; Dos Santos, E.O.; Prior, I.A.; et al. Fibroblast growth factor 2 lethally sensitizes cancer cells to stress-targeted therapeutic inhibitors. Mol. Oncol. 2019, 13, 290–306. [Google Scholar] [CrossRef]
- Anderson, D.J.; Le Moigne, R.; Djakovic, S.; Kumar, B.; Rice, J.; Wong, S.; Wang, J.; Yao, B.; Valle, E.; von Soly, S.K.; et al. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 2015, 28, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Bick, F.; Peters, K.; Mohta, V.; Tirosh, B.; Patterson, J.B.; Kharabi-Masouleh, B.; Huber, M. Infliction of proteotoxic stresses by impairment of the unfolded protein response or proteasomal inhibition as a therapeutic strategy for mast cell leukemia. Oncotarget 2017, 9, 2984–3000. [Google Scholar] [CrossRef] [PubMed]
- Mazurkiewicz, M.; Hillert, E.K.; Wang, X.; Pellegrini, P.; Olofsson, M.H.; Selvaraju, K.; D’Arcy, P.; Linder, S. Acute lymphoblastic leukemia cells are sensitive to disturbances in protein homeostasis induced by proteasome deubiquitinase inhibition. Oncotarget 2017, 8, 21115–21127. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pastorek, M.; Muller, P.; Coates, P.J.; Vojtesek, B. Intrinsic proteotoxic stress levels vary and act as a predictive marker for sensitivity of cancer cells to Hsp90 inhibition. PLoS ONE 2018, 13, e0202758. [Google Scholar] [CrossRef]
- Xu, D.; Liang, S.Q.; Yang, H.; Lüthi, U.; Riether, C.; Berezowska, S.; Marti, T.M.; Hall, S.R.R.; Bruggmann, R.; Kocher, G.J.; et al. Increased sensitivity to apoptosis upon endoplasmic reticulum stress-induced activation of the unfolded protein response in chemotherapy-resistant malignant pleural mesothelioma. Br. J. Cancer 2018, 119, 65–75. [Google Scholar] [CrossRef]
- Kijima, T.; Prince, T.L.; Tigue, M.L.; Yim, K.H.; Schwartz, H.; Beebe, K.; Lee, S.; Budzynski, M.A.; Williams, H.; Trepel, J.B.; et al. HSP90 inhibitors disrupt a transient HSP90-HSF1 interaction and identify a noncanonical model of HSP90-mediated HSF1 regulation. Sci. Rep. 2018, 8, 6976. [Google Scholar] [CrossRef]
- Tomasella, A.; Picco, R.; Ciotti, S.; Sgorbissa, A.; Bianchi, E.; Manfredini, R.; Benedetti, F.; Trimarco, V.; Frezzato, F.; Trentin, L.; et al. The isopeptidase inhibitor 2cPE triggers proteotoxic stress and ATM activation in chronic lymphocytic leukemia cells. Oncotarget 2016, 19, 45429–45443. [Google Scholar] [CrossRef][Green Version]
- Majera, D.; Skrott, Z.; Bouchal, J.; Bartkova, J.; Simkova, D.; Gachechiladze, M.; Steigerova, J.; Kurfurstova, D.; Gursky, J.; Korinkova, G.; et al. Targeting genotoxic and proteotoxic stress-response pathways in human prostate cancer by clinically available PARP inhibitors, vorinostat and disulfiram. Prostate 2019, 79, 352–362. [Google Scholar] [CrossRef]
- Yallowitz, A.; Ghaleb, A.; Garcia, L.; Alexandrova, E.M.; Marchenko, N. Heat Shock Factor 1 confers resistance to lapatinib in ERBB2-positive breast cancer cells. Cell Death Dis. 2018, 9, 621. [Google Scholar] [CrossRef]
- Kültz, D. Evolution of cellular stress response mechanisms. J. Exp. Zool A Ecol. Integr. Physiol. 2020, 333, 359–378. [Google Scholar] [CrossRef]
- Boos, F.; Labbadia, J.; Herrmann, J.M. How the mitoprotein-induced stress response safeguards the cytosol: A unified view. Trends Cell Biol. 2020, 30, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Tepe, J.J. Proteasome activation to combat proteotoxicity. Molecules 2019, 24, 2841. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brancolini, C.; Iuliano, L. Proteotoxic Stress and Cell Death in Cancer Cells. Cancers 2020, 12, 2385. https://doi.org/10.3390/cancers12092385
Brancolini C, Iuliano L. Proteotoxic Stress and Cell Death in Cancer Cells. Cancers. 2020; 12(9):2385. https://doi.org/10.3390/cancers12092385
Chicago/Turabian StyleBrancolini, Claudio, and Luca Iuliano. 2020. "Proteotoxic Stress and Cell Death in Cancer Cells" Cancers 12, no. 9: 2385. https://doi.org/10.3390/cancers12092385
APA StyleBrancolini, C., & Iuliano, L. (2020). Proteotoxic Stress and Cell Death in Cancer Cells. Cancers, 12(9), 2385. https://doi.org/10.3390/cancers12092385