Immune Checkpoint Inhibitors in pMMR Metastatic Colorectal Cancer: A Tough Challenge


 ,
, 
Abstract
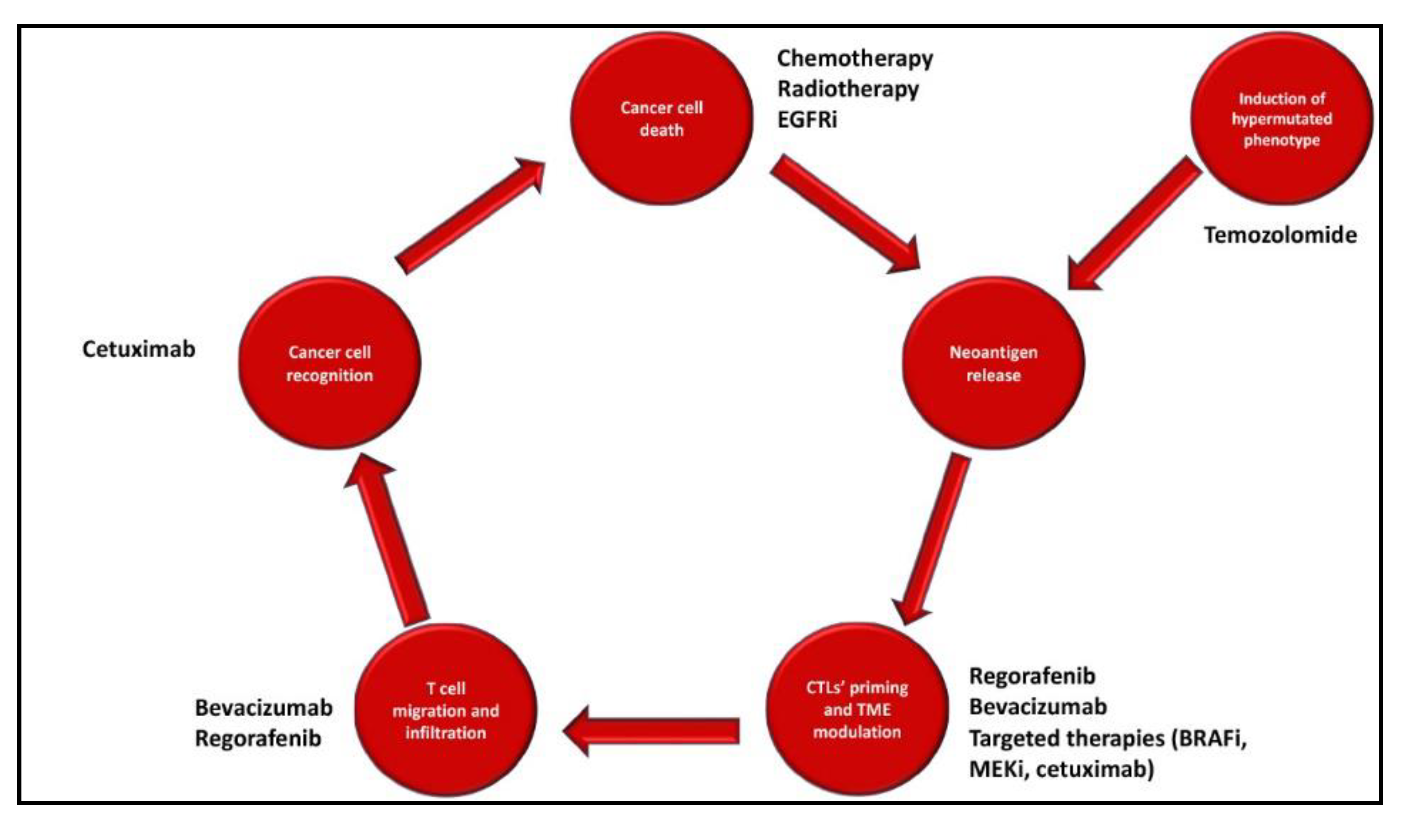
1. Introduction
2. Anti-VEGF and Chemotherapy + Immune Checkpoint Inhibitors
3. Anti-EGFR ± Chemotherapy + Immune Checkpoint Inhibitors
4. Temozolomide + Immune Checkpoint Inhibitors
5. Anti-TAMs + Immune Checkpoint Inhibitors
6. Radiotherapy + Immune Checkpoint Inhibitors
6.1. Locally Advanced Rectal Cancer
6.2. Metastatic Disease
6.3. MAPK Signaling
6.3.1. KRAS Targeting
6.3.2. BRAF Inhibition
6.4. PIK3CA/AKT/mTOR Targeting
7. Conclusions
Funding
Conflicts of Interest
References
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Kalyan, A.; Kircher, S.; Shah, H.; Mulcahy, M.; Benson, A. Updates on immunotherapy for colorectal cancer. J. Gastrointest. Oncol. 2018, 9, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair—Deficient/microsatellite instability—High metastatic colorectal cancer. JCO 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.; Van Cutsem, E.; McDermott, R.S.; Hill, A.G.; et al. Nivolumab (NIVO) + low-dose ipilimumab (IPI) in previously treated patients (pts) with microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC): Long-term follow-up. JCO 2019, 37, 635. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.M.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus chemotherapy for microsatellite instability-high/mismatch repair deficient metastatic colorectal cancer: The phase 3 KEYNOTE-177 Study. In Proceedings of the ASCO Annual Meeting 2020, Virtual Scientific Program, Chicago, IL, USA, 29–31 May 2020. [Google Scholar]
- FDA Approves Pembrolizumab for First-Line Treatment of MSI-H/dMMR Colorectal Cancer. Available online: https://www.ascopost.com/issues/july-10-2020/fda-approves-pembrolizumab-for-the-first-line-treatment-of-msi-hdmmr-colorectalcancer/#:~:text=On%20June%2029%2C%20the%20U.S.,deficient%20(dMMR)%20colorectal%20cancer (accessed on 17 July 2020).
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef]
- Becht, E.; de Reyniès, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautès-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef]
- Salem, M.E.; Bodor, J.N.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Korn, W.M.; Shields, A.F.; Worrilow, W.M.; Kim, E.S.; et al. Relationship between MLH1, PMS2, MSH2 and MSH6 gene-specific alterations and tumor mutational burden in 1057 microsatellite instability-high solid tumors. Int. J. Cancer 2020. [Google Scholar] [CrossRef]
- FDA Approves Pembrolizumab for Adults and Children with TMB-H Solid Tumors. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors (accessed on 17 July 2020).
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: Results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kronbichler, A.; Eisenhut, M.; Hong, S.H.; van der Vliet, H.J.; Kang, J.; Shin, J.I.; Gamerith, G. Tumor mutational burden and efficacy of immune checkpoint inhibitors: A systematic review and meta-analysis. Cancers 2019, 11, 1798. [Google Scholar] [CrossRef]
- O’Neil, B.H.; Wallmark, J.; Lorente, D.; Elez, E.; Raimbourg, J.; Gomez-Roca, C.; Ejadi, S.; Piha-Paul, S.A.; Moss, R.A.; Siu, L.L.; et al. 502 Pembrolizumab (MK-3475) for patients (pts) with advanced colorectal carcinoma (CRC): Preliminary results from KEYNOTE-028. Eur. J. Cancer 2015, 51, S103. [Google Scholar] [CrossRef]
- Picard, E.; Verschoor, C.P.; Ma, G.W.; Pawelec, G. Relationships between immune landscapes, genetic subtypes and responses to immunotherapy in colorectal cancer. Front. Immunol. 2020, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Tabernero, J.; Arnold, D.; De Gramont, A.; Ducreux, M.P.; O#x2019;Dwyer, P.J.; van Cutsem, E.; Bosanac, I.; Srock, S.; Mancao, C.; et al. CCTG CO.26: Updated analysis and impact of plasma-detected microsatellite stability (MSS) and tumor mutation burden (TMB) in a phase II trial of durvalumab (D) plus tremelimumab (T) and best supportive care (BSC) versus BSC alone in patients (pts) with refractory metastatic colorectal carcinoma (rmCRC). In Proceedings of the ASCO Annual Meeting 2019, Chicago, IL, USA, 31 May–4 June 2019. [Google Scholar]
- Chen, E.X.; Jonker, D.J.; Loree, J.M.; Kennecke, H.F.; Berry, S.R.; Couture, F.; Ahmad, C.E.; Goffin, J.R.; Kavan, P.; Harb, M.; et al. Effect of combined immune checkpoint inhibition vs best supportive care alone in patients with advanced colorectal cancer: The canadian cancer trials group CO.26 study. JAMA Oncol. 2020, 6, 831–838. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Shahda, S.; Noonan, A.M.; Bekaii-Saab, T.S.; O’Neil, B.H.; Sehdev, A.; Shaib, W.L.; Helft, P.R.; Loehrer, P.J.; Tong, Y.; Liu, Z.; et al. A phase II study of pembrolizumab in combination with mFOLFOX6 for patients with advanced colorectal cancer. JCO 2017, 35, 3541. [Google Scholar] [CrossRef]
- Grothey, A.; Tabernero, J.; Arnold, D.; De Gramont, A.; Ducreux, M.P.; O’Dwyer, P.J.; van Cutsem, E.; Bosanac, I.; Srock, S.; Mancao, C.; et al. Fluoropyrimidine (FP) + bevacizumab (BEV) + atezolizumab vs FP/BEV in BRAFwt metastatic colorectal cancer (mCRC): Findings from Cohort 2 of MODUL—A multicenter randomized trial of biomarker driven maintenance treatment following first line induction therapy. In Proceedings of the ESMO Congress 2018, Munich, Germany, 19–23 October 2018. [Google Scholar]
- Mettu, N.B.; Twohy, E.; Ou, F.-S.; Halfdanarson, T.R.; Lenz, H.J.; Breakstone, R.; Boland, P.M.; Crysler, O.; Wu, C.; Grothey, A.; et al. 533PD—BACCI: A phase II randomized, double-blind, multicenter, placebo-controlled study of capecitabine (C) bevacizumab (B) plus atezolizumab (A) or placebo (P) in refractory metastatic colorectal cancer (mCRC): An ACCRU network study. Ann. Oncol. 2019, 30, v203. [Google Scholar] [CrossRef]
- Wallin, J.; Pishvaian, M.J.; Hernandez, G.; Yadav, M.; Jhunjhunwala, S.; Delamarre, L.; He, X.; Powderly, J.; Lieu, C.; Eckhardt, S.G.; et al. Abstract 2651: Clinical activity and immune correlates from a phase Ib study evaluating atezolizumab (anti-PDL1) in combination with FOLFOX and bevacizumab (anti-VEGF) in metastatic colorectal carcinoma. Cancer Res. 2016, 76, 2651. [Google Scholar] [CrossRef]
- FOLFOXIRI + Bev + Atezo vs FOLFOXIRI + Bev as First-Line Treatment of Unresectable Metastatic Colorectal Cancer Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT03721653 (accessed on 17 July 2020).
- Stein, A.; Binder, M.; Goekkurt, E.; Lorenzen, S.; Riera-Knorrenschild, J.; Depenbusch, R.; Ettrich, T.J.; Doerfel, S.; Al-Batran, S.-E.; Karthaus, M.; et al. Avelumab and cetuximab in combination with FOLFOX in patients with previously untreated metastatic colorectal cancer (MCRC): Final results of the phase II AVETUX trial (AIO-KRK-0216). In Proceedings of the 2020 Gastrointestinal Cancer Symposium, San Francisco, CA, USA, 15–17 January 2020. [Google Scholar]
- Troiani, T.; Martinelli, E.; Ciardiello, D.; Zanaletti, N.; Cardone, C.; Borrelli, C.; Avallone, A.; Falcone, A.; Maiello, E.; Bordonaro, R.; et al. Phase II study of avelumab in combination with cetuximab in pre-treated RAS wild-type metastatic colorectal cancer patients: CAVE (cetuximab-avelumab) Colon. In Proceedings of the 2019 Gastrointestinal Cancer Symposium, San Francisco, CA, USA, 17–19 January 2019. [Google Scholar]
- Fukuoka, S.; Hara, H.; Takahashi, N.; Kojima, T.; Kawazoe, A.; Asayama, M.; Yoshii, T.; Kotani, D.; Tamura, H.; Mikamoto, Y.; et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: An open-label, dose-escalation, and dose-expansion phase IB trial (REGONIVO, EPOC1603). J. Clin. Oncol. 2020, 38, 2053–2061. [Google Scholar] [CrossRef]
- Kim, R. Phase I/IB study of regorafenib and nivolumab in mismatch repair (MMR) proficient advanced refractory colorectal cancer. In Proceedings of the ESMO World GI 2020—Virtual, Barcelona, Spain, 1–4 July 2020. [Google Scholar]
- Cousin, S.; Bellera, C.A.; Guégan, J.P.; Gomez-Roca, C.A.; Metges, J.-P.; Adenis, A.; Pernot, S.; Cantarel, C.; Kind, M.; Toulmonde, M. REGOMUNE: A phase II study of regorafenib plus avelumab in solid tumors—Results of the non-MSI-H metastatic colorectal cancer (mCRC) cohort. In Proceedings of the ASCO Annual Meeting 2020, Virtual Scientific Program, Chicago, IL, USA, 29–31 May 2020. [Google Scholar]
- Parikh, A.R.; Clark, J.W.; Wo, J.Y.-L.; Yeap, B.Y.; Allen, J.N.; Blaszkowsky, L.S.; Ryan, D.P.; Giantonio, B.J.; Weekes, C.D.; Zhu, A.X.; et al. A phase II study of ipilimumab and nivolumab with radiation in microsatellite stable (MSS) metastatic colorectal adenocarcinoma (mCRC). In Proceedings of the ASCO Annual Meeting 2019, Chicago, IL, USA, 31 May–4 June 2019. [Google Scholar]
- Nivolumab Plus FOLFOXIRI/Bevacizumab in First Line Chemotherapy of Advanced Colorectal Cancer RASm/BRAFm Patients (NIVACOR). Available online: https://clinicaltrials.gov/ct2/show/NCT04072198 (accessed on 17 July 2020).
- Martinelli, E.; Ciardiello, D.; Martini, G.; Troiani, T.; Cardone, C.; Vitiello, P.P.; Normanno, N.; Rachiglio, A.M.; Maiello, E.; Latiano, T.; et al. Implementing anti-epidermal growth factor receptor (EGFR) therapy in metastatic colorectal cancer: Challenges and future perspectives. Ann. Oncol. 2020, 31, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Pembrolizumab in MMR-Proficient Metastatic Colorectal Cancer Pharmacologically Primed to Trigger Hypermutation Status (ARETHUSA). Available online: https://clinicaltrials.gov/ct2/show/NCT03519412 (accessed on 17 July 2020).
- NIVOLUMAB Plus IPILIMUMAB and TEMOZOLOMIDE in Microsatellite Stable, MGMT Silenced Metastatic Colorectal Cancer (MAYA). Available online: https://clinicaltrials.gov/ct2/show/NCT03832621 (accessed on 17 July 2020).
- Study on the Effectiveness and Safety of the Combination of the Two Drugs Regorafenib and Nivolumab in Patients with Colorectal Cancer (Cancer of the Colon or Rectum Classified as Proficient Mismatch Repair and Microsatellite Stable). Available online: https://clinicaltrials.gov/ct2/show/NCT04126733 (accessed on 17 July 2020).
- Regorafenib and Pembrolizumab in Treating Participants with Advanced or Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03657641 (accessed on 17 July 2020).
- Efficacy and Safety of Pembrolizumab (MK-3475) Plus Lenvatinib (E7080/MK-7902) in Previously Treated Participants with Select Solid Tumors (MK-7902-005/E7080-G000-224/LEAP-005). Available online: https://clinicaltrials.gov/ct2/show/NCT03797326 (accessed on 17 July 2020).
- Immunotherapy in Locally Advanced Rectal Cancer (AVANA). Available online: https://clinicaltrials.gov/ct2/show/NCT03854799 (accessed on 17 July 2020).
- The Combination of Immunotherapy and Neoadjuvant Chemoradiotherapy in MSS Locally Advanced Rectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04411537 (accessed on 17 July 2020).
- Trial of Nivolumab with FOLFOX after Chemoradiation in Rectal Cancer Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT03921684 (accessed on 17 July 2020).
- Neo-adjuvant Pembrolizumab and Radiotherapy in Localised MSS Rectal Cancer (PEMREC). Available online: https://clinicaltrials.gov/ct2/show/NCT04109755 (accessed on 17 July 2020).
- Neoadjuvant Treatment in Rectal Cancer with Radiotherapy Followed by Atezolizumab and Bevacizumab (TARZAN). Available online: https://clinicaltrials.gov/ct2/show/NCT04017455 (accessed on 17 July 2020).
- Assess the Efficacy of Pembrolizumab Plus Radiotherapy or Ablation in Metastatic Colorectal Cancer Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT02437071 (accessed on 17 July 2020).
- A Clinical Trial of Durvalumab and Tremelimumab, Administered with Radiation Therapy or Ablation in Patients with Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03122509 (accessed on 17 July 2020).
- Atezolizumab with Stereotactic Ablative Radiotherapy in Patients with Metastatic Tumours (SABR-PDL1). Available online: https://clinicaltrials.gov/ct2/show/NCT02992912 (accessed on 17 July 2020).
- Durvalumab and Tremelimumab With or Without High or Low-Dose Radiation Therapy in Treating Patients with Metastatic Colorectal or Non-small Cell Lung Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02888743 (accessed on 17 July 2020).
- Regorafenib and Nivolumab in Combination with Radiotherapy for pMMR/MSS Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04030260 (accessed on 17 July 2020).
- Phase II Study of Toripalimab Plus Stereotactic Body Radiotherapy in Colorectal Cancer Patients with Oligometastasis. Available online: https://clinicaltrials.gov/ct2/show/NCT03927898 (accessed on 17 July 2020).
- PI Pembro in Combination with Stereotactic Body Radiotherapy for Liver Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02837263 (accessed on 17 July 2020).
- Camrelizumab Combined with Apatinib, XELOX, RFA in the Treatment of Liver Metastases of Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04202978 (accessed on 17 July 2020).
- Immunotherapy with Y90-RadioEmbolization for Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04108481 (accessed on 12 August 2020).
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C inhibitor MRTX849 Provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR blockade reverts resistance to KRAS G12C inhibition in colorectal cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Encorafenib, Binimetinib, and Nivolumab in Treating Patients with Microsatellite Stable BRAFV600E Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04044430 (accessed on 17 July 2020).
- Study of PI3Kinase Inhibition (Copanlisib) and Anti-PD-1 Antibody Nivolumab in Relapsed/Refractory Solid Tumors with Expansions in Mismatch-repair Proficient (MSS) Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03711058 (accessed on 17 July 2020).
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W.L. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016, 13, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef]
- Grimaldi, A.; Cammarata, I.; Martire, C.; Focaccetti, C.; Piconese, S.; Buccilli, M.; Mancone, C.; Buzzacchino, F.; Berrios, J.R.G.; D’Alessandris, N.; et al. Combination of chemotherapy and PD-1 blockade induces T cell responses to tumor non-mutated neoantigens. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Zitvogel, L.; Kroemer, G. The secret ally: Immunostimulation by anticancer drugs. Nat. Rev. Drug Discov. 2012, 11, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Kanterman, J.; Sade-Feldman, M.; Biton, M.; Ish-Shalom, E.; Lasry, A.; Goldshtein, A.; Hubert, A.; Baniyash, M. Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Res. 2014, 74, 6022–6035. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Aparicio, M.; Alzuguren, P.; Mauleon, I.; Medina-Echeverz, J.; Hervas-Stubbs, S.; Mancheno, U.; Berraondo, P.; Crettaz, J.; Gonzalez-Aseguinolaza, G.; Prieto, J.; et al. Oxaliplatin in combination with liver-specific expression of interleukin 12 reduces the immunosuppressive microenvironment of tumours and eradicates metastatic colorectal cancer in mice. Gut 2011, 60, 341–349. [Google Scholar] [CrossRef]
- Galaine, J.; Turco, C.; Vauchy, C.; Royer, B.; Mercier-Letondal, P.; Queiroz, L.; Loyon, R.; Mouget, V.; Boidot, R.; Laheurte, C.; et al. CD4 T cells target colorectal cancer antigens upregulated by oxaliplatin. Int. J. Cancer 2019, 145, 3112–3125. [Google Scholar] [CrossRef] [PubMed]
- Dosset, M.; Vargas, T.R.; Lagrange, A.; Boidot, R.; Végran, F.; Roussey, A.; Chalmin, F.; Dondaine, L.; Paul, C.; Lauret Marie-Joseph, E.; et al. PD-1/PD-L1 pathway: An adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology 2018, 7, e1433981. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Osada, T.; Chong, G.; Tansik, R.; Hong, T.; Spector, N.; Kumar, R.; Hurwitz, H.I.; Dev, I.; Nixon, A.B.; Lyerly, H.K.; et al. The Effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol. Immunother. 2008, 57, 1115–1124. [Google Scholar] [CrossRef]
- Gavalas, N.G.; Tsiatas, M.; Tsitsilonis, O.; Politi, E.; Ioannou, K.; Ziogas, A.C.; Rodolakis, A.; Vlahos, G.; Thomakos, N.; Haidopoulos, D.; et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. Br. J. Cancer 2012, 107, 1869–1875. [Google Scholar] [CrossRef]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef]
- Elamin, Y.Y.; Rafee, S.; Toomey, S.; Hennessy, B.T. Immune effects of bevacizumab: Killing two birds with one stone. Cancer Microenviron. 2015, 8, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Cremolini, C.; Masi, G.; Lonardi, S.; Zagonel, V.; Salvatore, L.; Cortesi, E.; Tomasello, G.; Ronzoni, M.; Spadi, R.; et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N. Engl. J. Med. 2014, 371, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Moretto, R.; Corallo, S.; Belfiore, A.; Rossini, D.; Boccaccino, A.; Lonardi, S.; Centonze, G.; Morano, F.; Germani, M.M.; Loupakis, F.; et al. Prognostic impact of immune-microenvironment in colorectal liver metastases resected after triplets plus a biologic agent: A pooled analysis of five prospective trials. Eur. J. Cancer 2020, 135, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Antoniotti, C.; Rossini, D.; Lonardi, S.; Loupakis, F.; Pietrantonio, F.; Bordonaro, R.; Latiano, T.P.; Tamburini, E.; Santini, D.; et al. Upfront FOLFOXIRI plus bevacizumab and reintroduction after progression versus mFOLFOX6 plus bevacizumab followed by FOLFIRI plus bevacizumab in the treatment of patients with metastatic colorectal cancer (TRIBE2): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2020, 21, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Colon Cancer (Version 4.2020). Available online: https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf (accessed on 17 July 2020).
- Wang, L.; Wei, Y.; Fang, W.; Lu, C.; Chen, J.; Cui, G.; Diao, H. Cetuximab enhanced the cytotoxic activity of immune cells during treatment of colorectal cancer. CPB 2017, 44, 1038–1050. [Google Scholar] [CrossRef]
- Kimura, H.; Sakai, K.; Arao, T.; Shimoyama, T.; Tamura, T.; Nishio, K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci. 2007, 98, 1275–1280. [Google Scholar] [CrossRef]
- Trotta, A.M.; Ottaiano, A.; Romano, C.; Nasti, G.; Nappi, A.; De Divitiis, C.; Napolitano, M.; Zanotta, S.; Casaretti, R.; D’Alterio, C.; et al. Prospective evaluation of cetuximab-mediated antibody-dependent cell cytotoxicity in metastatic colorectal cancer patients predicts treatment efficacy. Cancer Immunol. Res. 2016, 4, 366–374. [Google Scholar] [CrossRef]
- Trivedi, S.; Srivastava, R.M.; Concha-Benavente, F.; Ferrone, S.; Garcia-Bates, T.M.; Li, J.; Ferris, R.L. Anti-EGFR targeted monoclonal antibody isotype influences anti-tumor cellular immunity in head and neck cancer patients. Clin. Cancer Res. 2016, 22, 5229–5237. [Google Scholar] [CrossRef]
- Laurent-Puig, P.; Pekin, D.; Normand, C.; Kotsopoulos, S.K.; Nizard, P.; Perez-Toralla, K.; Rowell, R.; Olson, J.; Srinivasan, P.; Le Corre, D.; et al. Clinical relevance of KRAS-mutated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin. Cancer Res. 2015, 21, 1087–1097. [Google Scholar] [CrossRef]
- Tougeron, D.; Lecomte, T.; Pagès, J.C.; Villalva, C.; Collin, C.; Ferru, A.; Tourani, J.M.; Silvain, C.; Levillain, P.; Karayan-Tapon, L. Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2013, 24, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Modest, D.P.; Martens, U.M.; Riera-Knorrenschild, J.; Greeve, J.; Florschütz, A.; Wessendorf, S.; Ettrich, T.; Kanzler, S.; Nörenberg, D.; Ricke, J.; et al. FOLFOXIRI plus panitumumab as first-line treatment of RAS wild-type metastatic colorectal cancer: The randomized, open-label, phase II VOLFI study (AIO KRK0109). J. Clin. Oncol. 2019, 37, 3401–3411. [Google Scholar] [CrossRef] [PubMed]
- PhII Trial Panitumumab, Nivolumab, Ipilimumab in Kras/Nras/BRAF Wild-Type MSS Refractory mCRC. Available online: https://clinicaltrials.gov/ct2/show/NCT03442569 (accessed on 17 July 2020).
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for patients with RAS and BRAF Wild-type metastatic colorectal cancer with acquired resistance to first-line cetuximab and irinotecan: A phase 2 single-arm clinical trial. JAMA Oncol. 2019, 5, 343–350. [Google Scholar] [CrossRef]
- Osawa, H.; Shinozaki, E.; Nakamura, M.; Ohhara, Y.; Shindo, Y.; Shiozawa, M.; Uetake, H.; Matsumoto, H.; Ureshino, N.; Satake, H.; et al. 481PPhase II study of cetuximab rechallenge in patients with ras wild-type metastatic colorectal cancer: E-rechallenge trial. In Proceedings of the ESMO Congress 2018, Munich, Germany, 19–23 October 2018. [Google Scholar]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Newlands, E.S.; Stevens, M.F.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Esteller, M.; Herman, J.G. Generating mutations but providing chemosensitivity: The role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene 2004, 23, 1–8. [Google Scholar] [CrossRef]
- Esteller, M.; Sanchez-Cespedes, M.; Rosell, R.; Sidransky, D.; Baylin, S.B.; Herman, J.G. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999, 59, 67–70. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Dunn, J.; Baborie, A.; Alam, F.; Joyce, K.; Moxham, M.; Sibson, R.; Crooks, D.; Husband, D.; Shenoy, A.; Brodbelt, A.; et al. Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br. J. Cancer 2009, 101, 124–131. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Lobefaro, R.; Antista, M.; Lonardi, S.; Raimondi, A.; Morano, F.; Mosconi, S.; Rimassa, L.; Murgioni, S.; Sartore-Bianchi, A.; et al. Capecitabine and temozolomide versus FOLFIRI in RAS-mutated, MGMT-methylated metastatic colorectal cancer. Clin. Cancer Res. 2020, 26, 1017–1024. [Google Scholar] [CrossRef]
- Calegari, M.A.; Inno, A.; Monterisi, S.; Orlandi, A.; Santini, D.; Basso, M.; Cassano, A.; Martini, M.; Cenci, T.; de Pascalis, I.; et al. A phase 2 study of temozolomide in pretreated metastatic colorectal cancer with MGMT promoter methylation. Br. J. Cancer 2017, 116, 1279–1286. [Google Scholar] [CrossRef]
- Hochhauser, D.; Glynne-Jones, R.; Potter, V.; Grávalos, C.; Doyle, T.J.; Pathiraja, K.; Zhang, Q.; Zhang, L.; Sausville, E.A. A phase II study of temozolomide in patients with advanced aerodigestive tract and colorectal cancers and methylation of the O6-methylguanine-DNA methyltransferase promoter. Mol. Cancer Ther. 2013, 12, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Barault, L.; Moutinho, C.; Cassingena, A.; Bencardino, K.; Ghezzi, S.; Palmeri, L.; Bonazzina, E.; Tosi, F.; Ricotta, R.; et al. Tumor MGMT promoter hypermethylation changes over time limit temozolomide efficacy in a phase II trial for metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1062–1067. [Google Scholar] [CrossRef]
- Barault, L.; Amatu, A.; Bleeker, F.E.; Moutinho, C.; Falcomatà, C.; Fiano, V.; Cassingena, A.; Siravegna, G.; Milione, M.; Cassoni, P.; et al. Digital PCR quantification of MGMT methylation refines prediction of clinical benefit from alkylating agents in glioblastoma and metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1994–1999. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Pietrantonio, F.; Amatu, A.; Milione, M.; Cassingena, A.; Ghezzi, S.; Caporale, M.; Berenato, R.; Falcomatà, C.; Pellegrinelli, A.; et al. Digital PCR assessment of MGMT promoter methylation coupled with reduced protein expression optimises prediction of response to alkylating agents in metastatic colorectal cancer patients. Eur. J. Cancer 2017, 71, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [PubMed]
- Hervieu, A.; Rébé, C.; Végran, F.; Chalmin, F.; Bruchard, M.; Vabres, P.; Apetoh, L.; Ghiringhelli, F.; Mignot, G. Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth. J. Investig. Dermatol. 2013, 133, 499–508. [Google Scholar] [CrossRef]
- Vikas, P.; Borcherding, N.; Chennamadhavuni, A.; Garje, R. Therapeutic potential of combining PARP inhibitor and immunotherapy in solid tumors. Front. Oncol. 2020, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Arena, S.; Siena, S.; Bardelli, A.; Sartore-Bianchi, A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann. Oncol. 2020. [Google Scholar] [CrossRef]
- Basket Combination Study of Inhibitors of DNA Damage Response, Angiogenesis and Programmed Death Ligand 1 in Patients with Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03851614 (accessed on 12 August 2020).
- Lo Russo, G.; Moro, M.; Sommariva, M.; Cancila, V.; Boeri, M.; Centonze, G.; Ferro, S.; Ganzinelli, M.; Gasparini, P.; Huber, V.; et al. Antibody-Fc/FcR interaction on macrophages as a Mechanism for hyperprogressive disease in non-small cell lung cancer subsequent to PD-1/PD-L1 blockade. Clin. Cancer Res. 2019, 25, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Arwert, E.N.; Harney, A.S.; Entenberg, D.; Wang, Y.; Sahai, E.; Pollard, J.W.; Condeelis, J.S. A unidirectional transition from migratory to perivascular macrophage is required for tumor cell intravasation. Cell Rep. 2018, 23, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Ann. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Abou-Elkacem, L.; Arns, S.; Brix, G.; Gremse, F.; Zopf, D.; Kiessling, F.; Lederle, W. Regorafenib inhibits growth, angiogenesis, and metastasis in a highly aggressive, orthotopic colon cancer model. Mol. Cancer Ther. 2013, 12, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-W.; Ou, D.-L.; Hsu, C.-L.; Lin, L.; Cheng, A.-L.; Hsu, C. FRI-471-Regorafenib may enhance efficacy of anti-program cell death-1 therapy in hepatocellular carcinoma through modulation of macrophage polarization. J. Hepatol. 2019, 70, e605–e606. [Google Scholar] [CrossRef]
- Wu, R.-Y.; Kong, P.-F.; Xia, L.-P.; Huang, Y.; Li, Z.-L.; Tang, Y.-Y.; Chen, Y.-H.; Li, X.; Senthilkumar, R.; Zhang, H.-L.; et al. Regorafenib promotes antitumor immunity via inhibiting PD-L1 and IDO1 expression in melanoma. Clin. Cancer Res. 2019, 25, 4530–4541. [Google Scholar] [CrossRef] [PubMed]
- Hoff, S.; Grünewald, S.; Röse, L.; Zopf, D. Immunomodulation by regorafenib alone and in combination with anti PD1 antibody on murine models of colorectal cancer. Ann. Oncol. 2017, 28, v423. [Google Scholar] [CrossRef]
- Wang, C.; Chevalier, D.; Saluja, J.; Sandhu, J.; Lau, C.; Fakih, M. Regorafenib and nivolumab or pembrolizumab combination and circulating tumor dna response assessment in refractory microsatellite stable colorectal cancer. Oncologist 2020, 25, e1188–e1194. [Google Scholar] [CrossRef]
- Lin, S.J.; Gagnon-Bartsch, J.A.; Tan, I.B.; Earle, S.; Ruff, L.; Pettinger, K.; Ylstra, B.; van Grieken, N.; Rha, S.Y.; Chung, H.C.; et al. Signatures of tumour immunity distinguish Asian and non-Asian gastric adenocarcinomas. Gut 2015, 64, 1721–1731. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S.; Ou, F.-S.; Ahn, D.H.; Boland, P.M.; Ciombor, K.K.; Heying, E.N.; Dockter, T.J.; Jacobs, N.L.; Pasche, B.C.; Cleary, J.M.; et al. Regorafenib dose-optimisation in patients with refractory metastatic colorectal cancer (ReDOS): A randomised, multicentre, open-label, phase 2 study. Lancet Oncol. 2019, 20, 1070–1082. [Google Scholar] [CrossRef]
- Argiles, G.; Margalef, N.M.; Valladares-Ayerbes, M.; de Prado, J.V.; Grávalos, C.; Alfonso, P.G.; Santos, C.; Tobeña, M.; Sastre, J.; Benavides, M.; et al. Results of REARRANGE trial: A randomized phase 2 study comparing different dosing approaches for regorafenib (REG) during the first cycle of treatment in patients (pts) with metastatic colorectal cancer (mCRC). Ann. Oncol. 2019, 30, iv135. [Google Scholar] [CrossRef]
- Shoji, H.; Iwasa, S.; Kuchiba, A.; Ogawa, G.; Kawasaki, M.; Nakamura, K.; Mori, M.; Honma, Y.; Takashima, A.; Kato, K. A phase II study of lenvatinib in patients with metastatic colorectal cancer refractory to standard chemotherapy: LEMON study (NCCH1503). In Proceedings of the ASCO Annual Meeting 2019, Chicago, IL, USA, 31 May–4 June 2019. [Google Scholar]
- Weichselbaum, R.R.; Liang, H.; Deng, L.; Fu, Y.-X. Radiotherapy and immunotherapy: A beneficial liaison? Nat. Rev. Clin. Oncol. 2017, 14, 365–379. [Google Scholar] [CrossRef]
- Pitroda, S.P.; Chmura, S.J.; Weichselbaum, R.R. Integration of radiotherapy and immunotherapy for treatment of oligometastases. Lancet Oncol. 2019, 20, e434–e442. [Google Scholar] [CrossRef]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Deng, L.; Chmura, S.; Burnette, B.; Liadis, N.; Darga, T.; Beckett, M.A.; Lingen, M.W.; Witt, M.; Weichselbaum, R.R.; et al. Radiation-induced equilibrium is a balance between tumor cell proliferation and T cell-mediated killing. J. Immunol. 2013, 190, 5874–5881. [Google Scholar] [CrossRef] [PubMed]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef]
- Rodríguez-Ruiz, M.E.; Rodríguez, I.; Mayorga, L.; Labiano, T.; Barbes, B.; Etxeberria, I.; Ponz-Sarvise, M.; Azpilikueta, A.; Bolaños, E.; Sanmamed, M.F.; et al. TGFβ blockade enhances radiotherapy abscopal efficacy effects in combination with anti-PD1 and anti-CD137 immunostimulatory monoclonal antibodies. Mol. Cancer Ther. 2019, 18, 621–631. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef]
- Chakraborty, M.; Abrams, S.I.; Coleman, C.N.; Camphausen, K.; Schlom, J.; Hodge, J.W. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004, 64, 4328–4337. [Google Scholar] [CrossRef]
- Lim, Y.J.; Koh, J.; Kim, S.; Jeon, S.-R.; Chie, E.K.; Kim, K.; Kang, G.H.; Han, S.-W.; Kim, T.-Y.; Jeong, S.-Y.; et al. Chemoradiation-induced alteration of programmed death-ligand 1 and CD8+ tumor-infiltrating lymphocytes identified patients with poor prognosis in rectal cancer: A matched comparison analysis. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 1216–1224. [Google Scholar] [CrossRef]
- Meillan, N.; Vernerey, D.; Lefèvre, J.H.; Manceau, G.; Svrcek, M.; Augustin, J.; Fléjou, J.-F.; Lascols, O.; Simon, J.-M.; Cohen, R.; et al. Mismatch repair system deficiency is associated with response to neoadjuvant chemoradiation in locally advanced rectal cancer. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 824–833. [Google Scholar] [CrossRef]
- Grapin, M.; Richard, C.; Limagne, E.; Boidot, R.; Morgand, V.; Bertaut, A.; Derangere, V.; Laurent, P.-A.; Thibaudin, M.; Fumet, J.D.; et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: A promising new combination. J. Immunother. Cancer 2019, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Rückert, M.; Weber, J.; Mayr, X.; Derer, A.; Lotter, M.; Bert, C.; Rödel, F.; Fietkau, R.; Gaipl, U.S. Hypofractionated irradiation has immune stimulatory potential and induces a timely restricted infiltration of immune cells in colon cancer tumors. Front. Immunol. 2017, 8, 231. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.-P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Glynne-Jones, R.; Wyrwicz, L.; Tiret, E.; Brown, G.; Rödel, C.; Cervantes, A.; Arnold, D. ESMO guidelines committee rectal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv22–iv40. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Rectal Cancer (Version 6.2020). Available online: https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf (accessed on 17 July 2020).
- Azria, D.; Doyen, J.; Jarlier, M.; Martel-Lafay, I.; Hennequin, C.; Etienne, P.; Vendrely, V.; François, E.; de La Roche, G.; Bouché, O.; et al. Late toxicities and clinical outcome at 5 years of the ACCORD 12/0405-PRODIGE 02 trial comparing two neoadjuvant chemoradiotherapy regimens for intermediate-risk rectal cancer. Ann. Oncol. 2017, 28, 2436–2442. [Google Scholar] [CrossRef]
- Hong, Y.S.; Kim, S.Y.; Lee, J.S.; Nam, B.-H.; Kim, K.-P.; Kim, J.E.; Park, Y.S.; Park, J.O.; Baek, J.Y.; Kim, T.-Y.; et al. Oxaliplatin-based adjuvant chemotherapy for rectal cancer after preoperative chemoradiotherapy (ADORE): Long-term results of a randomized controlled trial. J. Clin. Oncol. 2019, 37, 3111–3123. [Google Scholar] [CrossRef]
- Hospers, G.; Bahadoer, R.R.; Dijkstra, E.A.; van Etten, B.; Marijnen, C.; Putter, H.; Meershoek-Klein Kranenbarg, E.; Roodvoets, A.G.; Nagtegaal, I.D.; Beets-Tan, R.G.; et al. Short-course radiotherapy followed by chemotherapy before TME in locally advanced rectal cancer: The randomized RAPIDO trial. In Proceedings of the ASCO Annual Meeting 2020, Virtual Scientific Program, Chicago, IL, USA, 29–31 May 2020. [Google Scholar]
- Conroy, T.; Lamfichekh, N.; Etienne, P.-L.; Rio, E.; Francois, E.; Mesgouez-Nebout, N.; Vendrely, V.; Artignan, X.; Bouché, O.; Gargot, D.; et al. Total neoadjuvant therapy with mFOLFIRINOX versus preoperative chemoradiation in patients with locally advanced rectal cancer: Final results of PRODIGE 23 phase III trial, a UNICANCER GI trial. In Proceedings of the ASCO Annual Meeting 2020, Virtual Scientific Program, Chicago, IL, USA, 29–31 May 2020. [Google Scholar]
- Abuodeh, Y.; Venkat, P.; Kim, S. Systematic review of case reports on the abscopal effect. Curr. Probl. Cancer 2016, 40, 25–37. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.-X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Rodriguez-Ruiz, M.E.; Rodriguez, I.; Barbes, B.; Mayorga, L.; Sanchez-Paulete, A.R.; Ponz-Sarvise, M.; Pérez-Gracia, J.L.; Melero, I. Brachytherapy attains abscopal effects when combined with immunostimulatory monoclonal antibodies. Brachytherapy 2017, 16, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Young, K.H.; Baird, J.R.; Savage, T.; Cottam, B.; Friedman, D.; Bambina, S.; Messenheimer, D.J.; Fox, B.; Newell, P.; Bahjat, K.S.; et al. Optimizing timing of immunotherapy improves control of tumors by hypofractionated radiation therapy. PLoS ONE 2016, 11, e0157164. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
- Maughan, T.S.; Adams, R.A.; Smith, C.G.; Meade, A.M.; Seymour, M.T.; Wilson, R.H.; Idziaszczyk, S.; Harris, R.; Fisher, D.; Kenny, S.L.; et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: Results of the randomised phase 3 MRC COIN trial. Lancet 2011, 377, 2103–2114. [Google Scholar] [CrossRef]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.-E.; Heintges, T.; Lerchenmüller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Blaj, C.; Schmidt, E.M.; Lamprecht, S.; Hermeking, H.; Jung, A.; Kirchner, T.; Horst, D. Oncogenic effects of high MAPK activity in colorectal cancer mark progenitor cells and persist irrespective of RAS mutations. Cancer Res. 2017, 77, 1763–1774. [Google Scholar] [CrossRef]
- Lal, N.; White, B.S.; Goussous, G.; Pickles, O.; Mason, M.J.; Beggs, A.D.; Taniere, P.; Willcox, B.E.; Guinney, J.; Middleton, G.W. KRAS mutation and consensus molecular subtypes 2 and 3 are independently associated with reduced immune infiltration and reactivity in colorectal cancer. Clin. Cancer Res. 2018, 24, 224–233. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, W.N.; Chang, C.-F.; Fischer, A.M.; Li, M.; Hedrick, S.M. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J. Immunol. 2008, 181, 7617–7629. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Pasam, A.; Dimopoulos, N.; Andrews, M.; Knights, A.; Puaux, A.-L.; Louahed, J.; Chen, W.; Woods, K.; Cebon, J.S. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol. Res. 2014, 2, 351–360. [Google Scholar] [CrossRef]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Knelson, E.H.; Sequist, L.V. A bright future for KRAS inhibitors. Nat. Cancer 2020, 1, 25–27. [Google Scholar] [CrossRef]
- CodeBreak 100: Activity of AMG 510, a novel small molecule inhibitor of KRASG12C, in patients with advanced colorectal cancer. In Proceedings of the ASCO Annual Meeting 2020, Virtual Scientific Program, Chicago, IL, USA, 29–31 May 2020.
- AMG 510 (pINN) Sotorasib Activity in Subjects with Advanced Solid Tumors with KRAS p.G12C Mutation (CodeBreak 101). Available online: https://clinicaltrials.gov/ct2/show/NCT04185883 (accessed on 17 July 2020).
- Loupakis, F.; Ruzzo, A.; Cremolini, C.; Vincenzi, B.; Salvatore, L.; Santini, D.; Masi, G.; Stasi, I.; Canestrari, E.; Rulli, E.; et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br. J. Cancer 2009, 101, 715–721. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Sweeney, C.; Herrick, J.; Albertsen, H.; Levin, T.R.; Murtaugh, M.A.; Wolff, R.K.; Slattery, M.L. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005, 65, 6063–6069. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef]
- Venderbosch, S.; Nagtegaal, I.D.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.; Quirke, P.; Seymour, M.T.; Richman, S.D.; et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin. Cancer Res. 2014, 20, 5322–5330. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.M.; Christensen, E.R.; Tester, D.J.; Kim, C.Y.; Roche, P.C.; Burgart, L.J.; Thibodeau, S.N. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998, 58, 3455–3460. [Google Scholar] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Hutchinson, L.; Deng, A.; Green, M.R. Common BRAF(V600E)-directed pathway mediates widespread epigenetic silencing in colorectal cancer and melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, 1250–1255. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E–mutated colorectal cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- FDA Approves Encorafenib in Combination with Cetuximab for Metastatic Colorectal Cancer with a BRAF V600E Mutation. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-encorafenib-combination-cetuximab-metastatic-colorectal-cancer-braf-v600e-mutation (accessed on 17 July 2020).
- Koustas, E.; Papavassiliou, A.G.; Karamouzis, M.V. The role of autophagy in the treatment of BRAF mutant colorectal carcinomas differs based on microsatellite instability status. PLoS ONE 2018, 13, e0207227. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Corcoran, R. Clinical efficacy of combined BRAF, MEK, and PD-1 inhibition in BRAFV600E colorectal cancer patients. In Proceedings of the ESMO World GI 2020—Virtual, Barcelona, Spain, 1–4 July 2020. [Google Scholar]
- Dabrafenib + Trametinib + PDR001 in Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03668431 (accessed on 17 July 2020).
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shi, Y.; Zhou, K.; Wang, L.; Yan, Z.; Liu, Y.; Xu, L.; Zhao, S.; Chu, H.; Shi, T.; et al. PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-M.; Wang, Y.; Wang, Y.-L.; Wang, Y.; Liu, T.; Ni, M.; Li, M.-S.; Lin, L.; Ge, F.-J.; Gong, C.; et al. PIK3CA mutations contribute to acquired cetuximab resistance in patients with metastatic colorectal cancer. Clin. Cancer Res. 2017, 23, 4602–4616. [Google Scholar] [CrossRef] [PubMed]
- Price, T.J.; Bruhn, M.A.; Lee, C.K.; Hardingham, J.E.; Townsend, A.R.; Mann, K.P.; Simes, J.; Weickhardt, A.; Wrin, J.W.; Wilson, K.; et al. Correlation of extended RAS and PIK3CA gene mutation status with outcomes from the phase III AGITG MAX STUDY involving capecitabine alone or in combination with bevacizumab plus or minus mitomycin C in advanced colorectal cancer. Br. J. Cancer 2015, 112, 963–970. [Google Scholar] [CrossRef]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the clinical development of PI3K inhibitors: Strategies to improve their impact in solid tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef]
- Putz, E.M.; Prchal-Murphy, M.; Simma, O.A.; Forster, F.; Koenig, X.; Stockinger, H.; Piekorz, R.P.; Freissmuth, M.; Müller, M.; Sexl, V.; et al. PI3Kδ is essential for tumor clearance mediated by cytotoxic T lymphocytes. PLoS ONE 2012, 7, e40852. [Google Scholar] [CrossRef]
- Ali, K.; Soond, D.R.; Pineiro, R.; Hagemann, T.; Pearce, W.; Lim, E.L.; Bouabe, H.; Scudamore, C.L.; Hancox, T.; Maecker, H.; et al. Inactivation of the PI3K p110δ breaks regulatory T cell-mediated immune tolerance to cancer. Nature 2014, 510, 407–411. [Google Scholar] [CrossRef]
- Ahmad, S.; Abu-Eid, R.; Shrimali, R.; Webb, M.; Verma, V.; Doroodchi, A.; Berrong, Z.; Samara, R.; Rodriguez, P.C.; Mkrtichyan, M.; et al. Differential PI3Kδ signaling in CD4+ T-cell subsets enables selective targeting of T regulatory cells to enhance cancer immunotherapy. Cancer Res. 2017, 77, 1892–1904. [Google Scholar] [CrossRef]
- Carnevalli, L.S.; Sinclair, C.; Taylor, M.A.; Gutierrez, P.M.; Langdon, S.; Coenen-Stass, A.M.L.; Mooney, L.; Hughes, A.; Jarvis, L.; Staniszewska, A.; et al. PI3Kα/δ inhibition promotes anti-tumor immunity through direct enhancement of effector CD8+ T-cell activity. J. Immunother. Cancer 2018, 6, 158. [Google Scholar] [CrossRef]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.E.; Song, X.; Wrasidlo, W.; et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study Nameand/or NTC | Phase | Study Population | Agent(s) | End-Points * | Results |
|---|---|---|---|---|---|
| Anti-VEGF and Chemotherapy + Immune Checkpoint Inhibitors | |||||
| NCT0237335672 [21] | II | 1st line mCRC MSI n.s. | FOLFOX + Pembrolizumab | 1: PFS 2: ORR, DCR | mPFS not reached ORR: 53%, DCR: 100% |
| MODUL cohort 2 NCT02291289 [22] | II | 1st line mCRC BRAF wt, MSI n.s. | FOLFOX + BV followed by FP + BV vs. FP + BV + Atezolizumab | 1: PFS 2: OS | PFS: 7.4 mos vs. 7.2 mos, HR 0.96, p = 0.727 OS: 51%, HR = 0.86, p = 0.28 |
| BACCI NCT02873195 [23] | II | Pretreated mCRC MSI n.s. | Capecitabine + BV + Atezolizumab vs. Capecitabine + BV | 1: PFS 2: 12 mo OS | mPFS: 4.4 mos vs. 3.3 mos. HR 0.72, p = 0.051 12 mo OS: 43% vs. 52%, HR 0.94, p = 0.4. |
| NCT01633970 [24] | Ib | 1st line mCRC MSI n.s. | FOLFOX + BV + Atezolizumab | 1: Safety 2: PFS, OS | mPFS: 14.1 mos OR: 52% |
| ATEZOTRIBE NCT03721653 [25] | II | 1st line mCRC MSI n.s. | FOLFOXIRI + BV + Atezolizumab vs. FOLFOXIRI + BV | 1: PFS 2: ORR, irORR, R0 resection rate | No results posted |
| Anti-EGFR ± Chemotherapy + Immune Checkpoint Inhibitors | |||||
| AVETUX NCT03174405 [26] | II | 1st line mCRC RAS/BRAF wt MSI n.s. | FOLFOX + Cetuximab + Avelumab | 1: 12 mos PFS 2: PFS, ORR | 12 mo PFS: 40% mPFS: 11.1 mos ORR: 79.5% |
| CAVE Eudract 2017-004392-32 [27] | II | Pretreated mCRC RAS wt, MSI n.s. | Cetuximab + Avelumab | 1: OS 2: ORR, PFS, Safety | No results posted |
| Anti-TAMs + Immune Checkpoint Inhibitors | |||||
| REGONIVO NCT03406871 [28] | Ib | Preatreatd mCRC MSI n.s. | Regorafenib + Nivolumab | 1: Safety 2: ORR, PFS, OS | ORR: 36% mPFS: 5.6 mos mOS: not reached |
| NCT03712943 [29] | I/Ib | Preatreatd mCRC pMMR | Regorafenib + Nivolumab | 1: Safety 2: ORR, PFS, OS | ORR: 5% mPFS: 4.3 mos mOS: 11 mos |
| REGOMUNE NCT03475953 [30] | II | Pretreated mCRC pMMR | Regorafenib + Avelumab | 1: ORR 2: PFS, OS | ORR: 0% mPFS: 3.6 mos mOS: 10.8 mos |
| Radiotherapy + Immune Checkpoint Inhibitors | |||||
| NCT03104439 [31] | II | Preatreatd mCRC pMMR | SBRT 8 Gy + Nivolumab + Ipilimumab | 1: DCR 2: ORR | DCR: 17.5% ORR: 7.5% |
| Study Name and/or NTC | Phase | Study Population | Agent(s) | Primary End-Point(s) |
|---|---|---|---|---|
| Chemotherapy + Anti-Angiogenetics + Immune Checkpoint Inhibitors | ||||
| NIVACOR NCT04072198 [32] | II | 1st line mCRC RAS/BRAF mt, MSI n.s. | FOLFOXIRI + BV + Nivolumab | ORR |
| Anti-EGFR ± Chemotherapy + Immune Checkpoint Inhibitors | ||||
| AVETRIC Eudract 2019-001501-24 [33] | II | 1st line mCRC RAS wt, MSI n.s. | FOLFOXIRI + Cetuximab + Avelumab | PFS |
| Temozolomide + Immune Checkpoint Inhibitors | ||||
| ARETHUSA NCT03519412 [34] | II | Pretreated mCRC RAS mt, MGMT-neg pMMR (Cohort P) | TMZ until progression followed by Pembrolizumab if TMB > 20 Muts/Mb after TMZ administration | ORR |
| MAYA NCT03832621 [35] | II | Pretreated mCRC MGMT-neg, pMMR | TMZ for 2 cycles followed by TMZ + Nivolumab + Ipilimumab if SD/PR/CR to TMZ monotherapy | 8 mo PFS |
| Anti-TAMs +Immune Checkpoint Inhibitors | ||||
| NCT04126733 [36] | II | Pretreated mCRC BRAF wt pMMR | Regorafenib + Nivolumab | ORR |
| NCT03657641 [37] | I/II | Pretreated mCRC MSI n.s. | Regorafenib + Pembrolizumab | Safety, PFS, OS |
| NCT03797326 [38] | II | Pretreated mCRC pMMR | Lenvatinib + Pembrolizumab | ORR, Safety |
| Radiotherapy + Immune Checkpoint Inhibitors | ||||
| AVANA NCT03854799 [39] | II | LARC MSI n.s. | Pre-op capecitabine + RT + Avelumab → Surgery | pCR |
| NCT04411537 [40] | II | LARC pMMR | Pre-op Nivolumab → CAPIRI + RT → Nivolumab → Surgery → post-op XELOX | pCR |
| NCT03921684 [41] | II | LARC MSI n.s. | Pre-op capecitabine + RT → FOLFOX + Nivolumab → Surgery | Safety, pCR |
| PEMREC NCT04109755 [42] | II | LARC pMMR | Pre-op Short-Course RT + Pembrolizumab → Surgery | TRG |
| TARZAN NCT04017455 [43] | II | LARC MSI n.s. | Pre-op bevacizumab + atezolizumab → Surgery | cCRR and near-cCRR |
| NCT02437071 [44] | II | Pretreated mCRC MSI n.s. | Pembrolizumab + RT or RFA | ORR out of field of radiation |
| NCT03122509 [45] | II | Pretreated mCRC pMMR | Durvalumab + Tremelimumab + RT or RFA | ORR out of field of radiation |
| SABR-PDL1 NCT02992912 [46] | II | Pretreated mCRC MSI n.s. | Atezolizumab + SBRT (45 Gy) | PFS |
| NCT02888743 [47] | II | Pretreated mCRC pMMR | Durvalumab + Tremelimumab + High dose RT (Arm B) or Low dose RT (Arm C) vs. Durvalumab + Tremelimumab (Arm A) | ORR out of field of RT of Arm B and ARM C vs. ORR in Arm A |
| NCT04030260 [48] | II | Pretreated mCRC pMMR | Regorafenib + Nivolumab + RT ± irinotecan | mPFS |
| NCT03927898 [49] | II | Oligometastatic CRC with resected primary, achieving SD or PR to first-line therapy, and with all lesions amenable to SBRT MSI n.s. | SBRT + Toripalimab | 12 mo PFS |
| NCT02837263 [50] | Ib | Pretreated CRLM, candidate for SBRT to at least one intrahepatic lesion and for surgery with potential curative intent. pMMR | SBRT + Pembrolizumab | 12 mo recurrence rate |
| NCT04202978 [51] | I/II | Neoadjuvant CRLM. MSI n.s. | CAPOX + Apatinib + Camrelizumab → RFA → Surgery | R0 resection rate |
| iRE-C NCT04108481 [52] | I/II | Liver-predominant mCRC | Yttrium-90 RadioEmbolization + Durvalumab | Safety |
| Target Therapy + Immune Checkpoint Inhibitors | ||||
| CodeBreak 100 NCT03600883 [53] | I/II | Pretreated mCRC KRAS G12C mt MSI n.s. | AMG510 (Sotorasib) + Pembrolizumab | Safety |
| CodeBreak 101 NCT04185883 [54] | Ib | Pretreated mCRC KRAS G12C mtMSI n.s. | AMG510 (Sotorasib) + anti-PD1 | Safety |
| NCT04044430 [55] | I/II | Pretreated or PD within 6 mos of post-op CT mCRC BRAF V600E mt pMMR | Encorafenib + Binimetinib + Nivolumab | Safety, ORR |
| NCT03668431 [56] | II | Pretreated mCRC BRAF V600E mt MSI n.s. | Dabrafenib + Trametinib + Spartalizumab | Safety, ORR |
| NCT03711058 [57] | I/II | Pretreated mCRC pMMR | Copanlisib + Nivolumab | Safety, ORR |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marmorino, F.; Boccaccino, A.; Germani, M.M.; Falcone, A.; Cremolini, C. Immune Checkpoint Inhibitors in pMMR Metastatic Colorectal Cancer: A Tough Challenge. Cancers 2020, 12, 2317. https://doi.org/10.3390/cancers12082317
Marmorino F, Boccaccino A, Germani MM, Falcone A, Cremolini C. Immune Checkpoint Inhibitors in pMMR Metastatic Colorectal Cancer: A Tough Challenge. Cancers. 2020; 12(8):2317. https://doi.org/10.3390/cancers12082317
Chicago/Turabian StyleMarmorino, Federica, Alessandra Boccaccino, Marco Maria Germani, Alfredo Falcone, and Chiara Cremolini. 2020. "Immune Checkpoint Inhibitors in pMMR Metastatic Colorectal Cancer: A Tough Challenge" Cancers 12, no. 8: 2317. https://doi.org/10.3390/cancers12082317
APA StyleMarmorino, F., Boccaccino, A., Germani, M. M., Falcone, A., & Cremolini, C. (2020). Immune Checkpoint Inhibitors in pMMR Metastatic Colorectal Cancer: A Tough Challenge. Cancers, 12(8), 2317. https://doi.org/10.3390/cancers12082317