Genetically Engineered Mouse Models of Liver Tumorigenesis Reveal a Wide Histological Spectrum of Neoplastic and Non-Neoplastic Liver Lesions

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
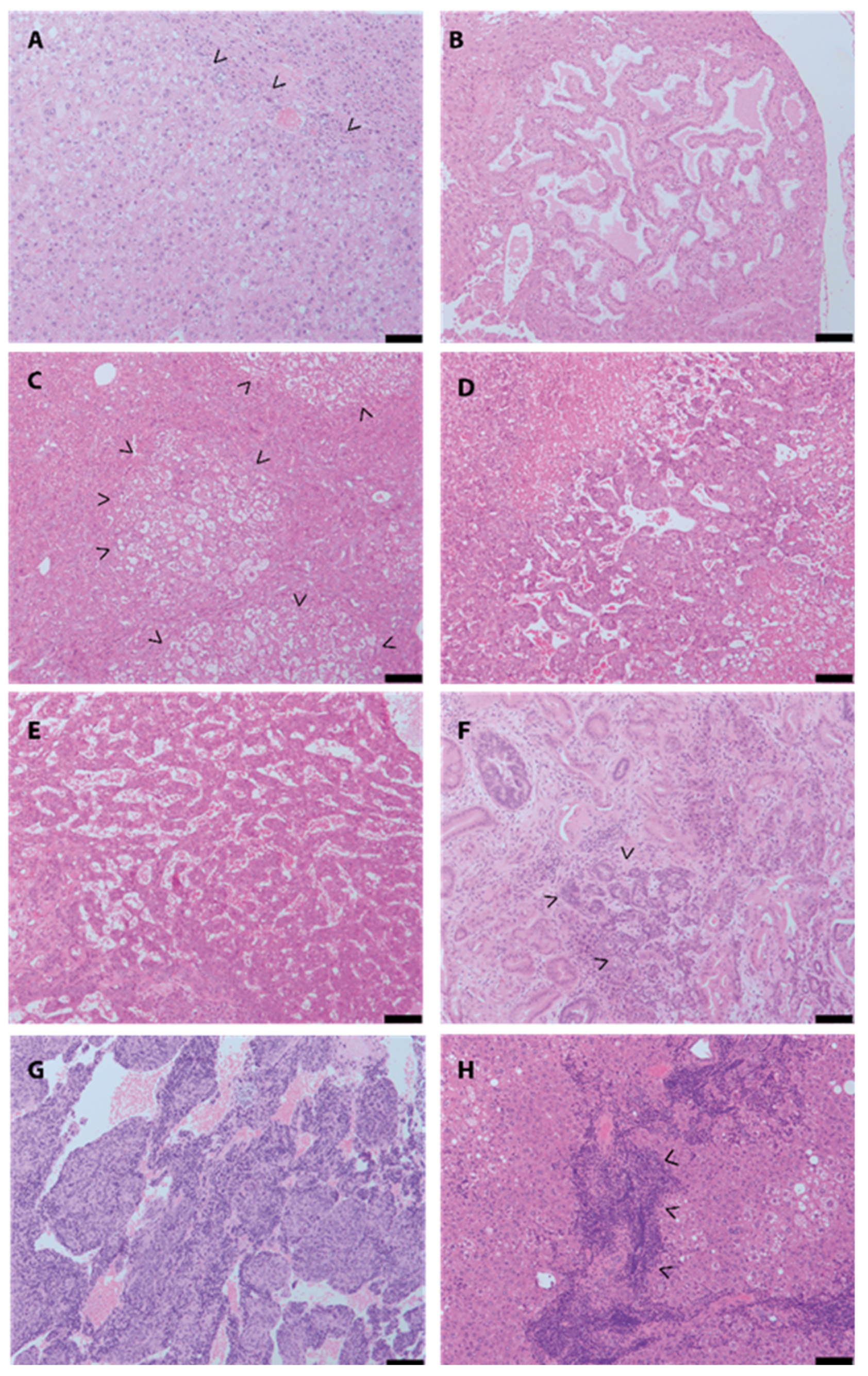
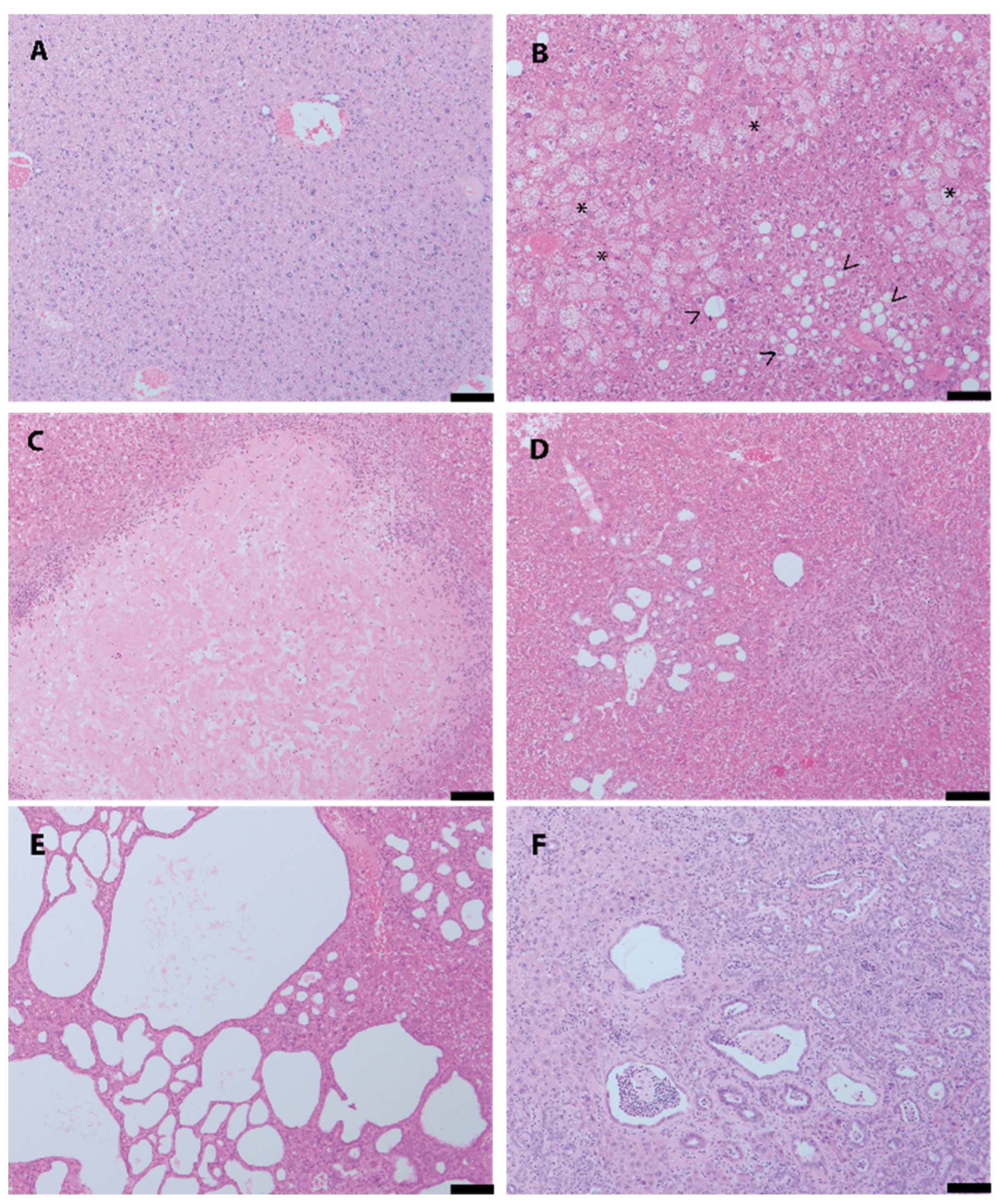
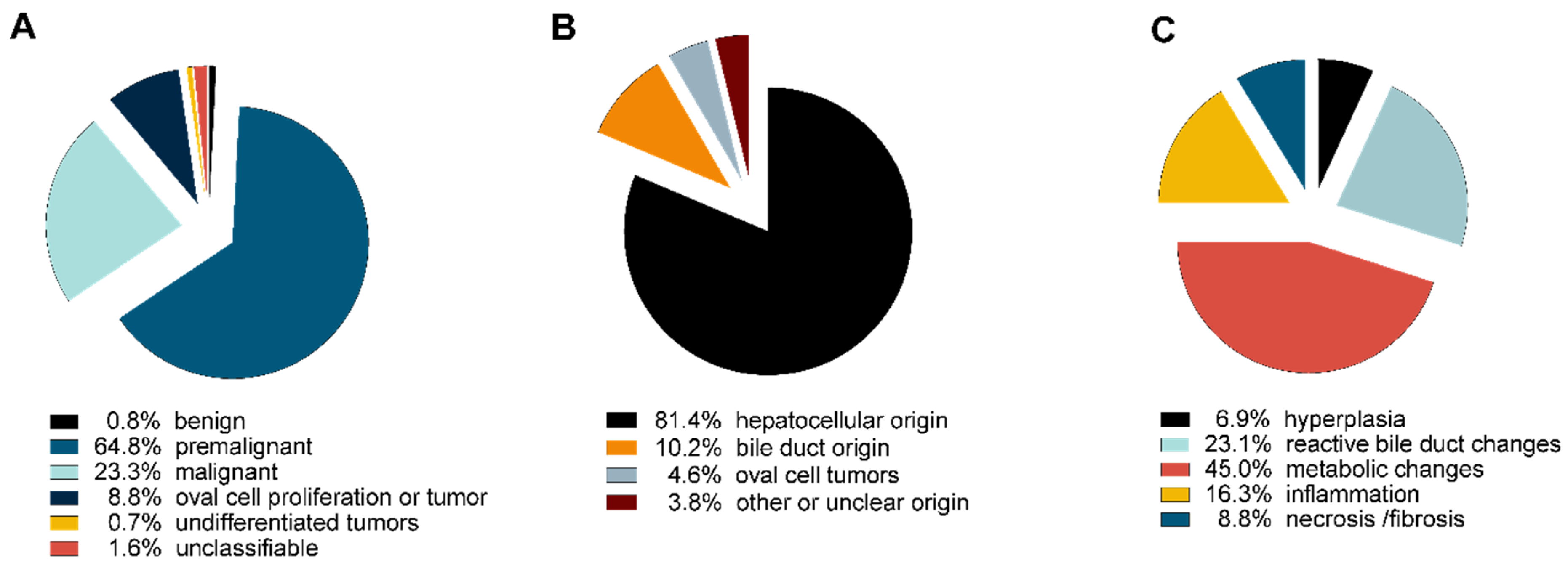
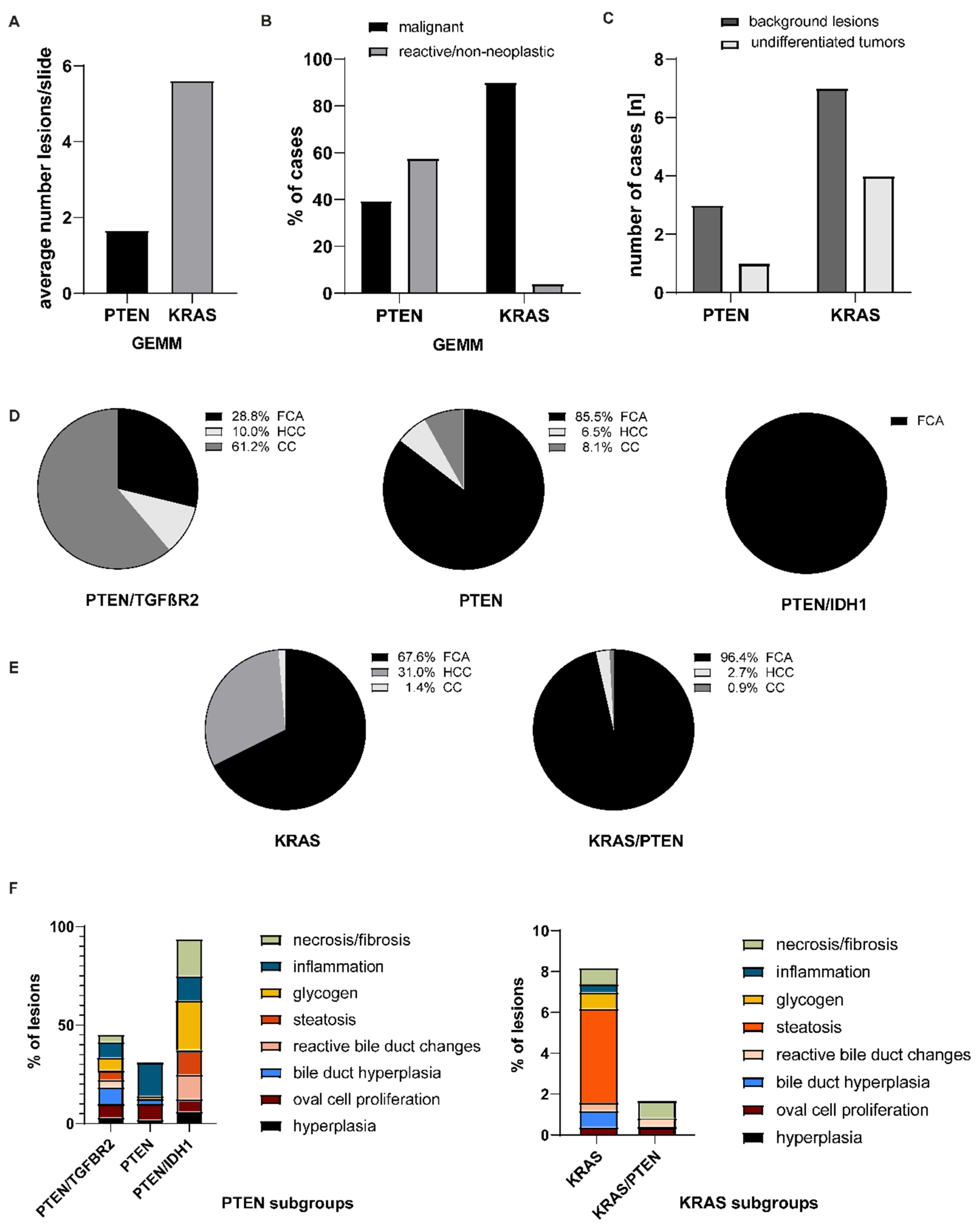
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gengenbacher, N.; Singhal, M.; Augustin, H.G. Preclinical mouse solid tumour models: Status quo, challenges and perspectives. Nat. Rev. Cancer 2017, 17, 751–765. [Google Scholar] [CrossRef]
- Bakiri, L.; Wagner, E.F. Mouse models for liver cancer. Mol. Oncol. 2013, 7, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 536–554. [Google Scholar] [CrossRef] [PubMed]
- Carlessi, R.; Kohn-Gaone, J.; Olynyk, J.K.; Tirnitz-Parker, J.E.E. Mouse Models of Hepatocellular Carcinoma. In Hepatocellular Carcinoma; Tirnitz-Parker, J.E.E., Ed.; Codon Publications: Brisbane, Australia, 2019. [Google Scholar] [CrossRef]
- Chen, K.; Ma, J.; Jia, X.; Ai, W.; Ma, Z.; Pan, Q. Advancing the understanding of NAFLD to hepatocellular carcinoma development: From experimental models to humans. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Ro, S.W.; Seo, S.H.; Jeon, Y.; Moon, H.; Kim, D.Y.; Kim, S.U. Genetically Engineered Mouse Models for Liver Cancer. Cancers 2019, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N.; Campbell, J.S. Mouse models of hepatocellular carcinoma. Semin. Liver Dis. 2010, 30, 87–98. [Google Scholar] [CrossRef]
- He, L.; Tian, D.A.; Li, P.Y.; He, X.X. Mouse models of liver cancer: Progress and recommendations. Oncotarget 2015, 6, 23306–23322. [Google Scholar] [CrossRef]
- Knoblaugh, S.E.; Hohl, T.M.; La Perle, K.M.D. Pathology Principles and Practices for Analysis of Animal Models. ILAR J. 2018, 59, 40–50. [Google Scholar] [CrossRef]
- Steiger, K.; Ballke, S.; Yen, H.Y.; Seelbach, O.; Alkhamas, A.; Boxberg, M.; Schwamborn, K.; Knolle, P.A.; Weichert, W.; Mogler, C. Histopathological research laboratories in translational research: Conception and integration into the infrastructure of pathological institutes. Der Pathol. 2019, 40, 172–178. [Google Scholar] [CrossRef]
- Ward, J.M.; Schofield, P.N.; Sundberg, J.P. Reproducibility of histopathological findings in experimental pathology of the mouse: A sorry tail. Lab. Anim. 2017, 46, 146–151. [Google Scholar] [CrossRef]
- Kubota, T. Metastatic models of human cancer xenografted in the nude mouse: The importance of orthotopic transplantation. J. Cell. Biochem. 1994, 56, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Leenders, M.W.; Nijkamp, M.W.; Borel Rinkes, I.H. Mouse models in liver cancer research: A review of current literature. World J. Gastroenterol. 2008, 14, 6915–6923. [Google Scholar] [CrossRef] [PubMed]
- Molina-Sanchez, P.; Lujambio, A. Experimental Models for Preclinical Research in Hepatocellular Carcinoma. In Hepatocellular Carcinoma: Translational Precision Medicine Approaches; Hoshida, Y., Ed.; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; Peitta, G.; Seddaiu, M.A.; Feo, F.; Calvisi, D.F. Experimental Models to Define the Genetic Predisposition to Liver Cancer. Cancers 2019, 11, 1450. [Google Scholar] [CrossRef]
- Sanchez, A.; Fabregat, I. Genetically modified animal models recapitulating molecular events altered in human hepatocarcinogenesis. Clin. Transl. Oncol. 2009, 11, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Tennant, B.C. Animal models of hepadnavirus-associated hepatocellular carcinoma. Clin. Liver Dis. 2001, 5, 43–68. [Google Scholar] [CrossRef]
- Whitlock, R.S.; Yang, T.; Vasudevan, S.A.; Woodfield, S.E. Animal Modeling of Pediatric Liver Cancer. Cancers 2020, 12, 273. [Google Scholar] [CrossRef] [PubMed]
- Thoolen, B.; Maronpot, R.R.; Harada, T.; Nyska, A.; Rousseaux, C.; Nolte, T.; Malarkey, D.E.; Kaufmann, W.; Kuttler, K.; Deschl, U.; et al. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol. Pathol. 2010, 38, 5S–81S. [Google Scholar] [CrossRef]
- Mazer, B.L.; Homer, R.J.; Rimm, D.L. False-positive pathology: Improving reproducibility with the next generation of pathologists. Lab. Investig. 2019, 99, 1260–1265. [Google Scholar] [CrossRef]
- Pisani, G.B.; Valenti, J.L.; Quintana, A.B. Hepatic preneoplasia induction in male Wistar rats: Histological studies up to five months post treatment. Rev. Esp. De Enferm. Dig.: Organo Of. De La Soc. Esp. De Patol. Dig. 2016, 108, 457–463. [Google Scholar] [CrossRef]
- Ruebner, B.H.; Gershwin, M.E.; Hsieh, L.; Dunn, P. Ultrastructure of spontaneous neoplasms induced by diethylnitrosamine and dieldrin in the C3H mouse. J. Environ. Pathol. Toxicol. 1980, 4, 237–254. [Google Scholar]
- Solt, D.B.; Medline, A.; Farber, E. Rapid emergence of carcinogen-induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am. J. Pathol. 1977, 88, 595–618. [Google Scholar] [PubMed]
- Popp, J.A.; Goldsworthy, T.L. Defining foci of cellular alteration in short-term and medium-term rat liver tumor models. Toxicol. Pathol. 1989, 17, 561–568. [Google Scholar] [CrossRef]
- Bannasch, P.; Enzmann, H.; Klimek, F.; Weber, E.; Zerban, H. Significance of sequential cellular changes inside and outside foci of altered hepatocytes during hepatocarcinogenesis. Toxicol. Pathol. 1989, 17, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Freimuth, J.; Gassler, N.; Moro, N.; Gunther, R.W.; Trautwein, C.; Liedtke, C.; Krombach, G.A. Application of magnetic resonance imaging in transgenic and chemical mouse models of hepatocellular carcinoma. Mol. Cancer 2010, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Friemel, J.; Frick, L.; Unger, K.; Egger, M.; Parrotta, R.; Boge, Y.T.; Adili, A.; Karin, M.; Luedde, T.; Heikenwalder, M.; et al. Characterization of HCC Mouse Models: Towards an Etiology-Oriented Subtyping Approach. Mol. Cancer Res. 2019, 17, 1493–1502. [Google Scholar] [CrossRef]
- Lee, J.S.; Chu, I.S.; Mikaelyan, A.; Calvisi, D.F.; Heo, J.; Reddy, J.K.; Thorgeirsson, S.S. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 2004, 36, 1306–1311. [Google Scholar] [CrossRef]
- Ye, H.; Zhang, C.; Wang, B.J.; Tan, X.H.; Zhang, W.P.; Teng, Y.; Yang, X. Synergistic function of Kras mutation and HBx in initiation and progression of hepatocellular carcinoma in mice. Oncogene 2014, 33, 5133–5138. [Google Scholar] [CrossRef]
- Ikenoue, T.; Terakado, Y.; Nakagawa, H.; Hikiba, Y.; Fujii, T.; Matsubara, D.; Noguchi, R.; Zhu, C.; Yamamoto, K.; Kudo, Y.; et al. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci. Rep. 2016, 6, 23899. [Google Scholar] [CrossRef]
- Rad, R.; Rad, L.; Wang, W.; Strong, A.; Ponstingl, H.; Bronner, I.F.; Mayho, M.; Steiger, K.; Weber, J.; Hieber, M.; et al. A conditional piggyBac transposition system for genetic screening in mice identifies oncogenic networks in pancreatic cancer. Nat. Genet. 2015, 47, 47–56. [Google Scholar] [CrossRef]
- Weber, J.; de la Rosa, J.; Grove, C.S.; Schick, M.; Rad, L.; Baranov, O.; Strong, A.; Pfaus, A.; Friedrich, M.J.; Engleitner, T.; et al. PiggyBac transposon tools for recessive screening identify B-cell lymphoma drivers in mice. Nat. Commun. 2019, 10, 1415. [Google Scholar] [CrossRef]
- Das Thakur, M.; Pryer, N.K.; Singh, M. Mouse tumour models to guide drug development and identify resistance mechanisms. J. Pathol. 2014, 232, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, I.J. Generating Genetically Modified Mice: A Decision Guide. Methods Mol. Biol. 2017, 1642, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.D.; Santagostino, S.F.; Foreman, O. Applications and considerations for the use of genetically engineered mouse models in drug development. Cell Tissue Res. 2019, 380, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Xu, Z.; Wang, J.; Cigliano, A.; Pilo, M.G.; Ribback, S.; Zhang, S.; Qiao, Y.; Che, L.; Pascale, R.M.; et al. Functional role of SGK3 in PI3K/Pten driven liver tumor development. BMC Cancer 2019, 19, 343. [Google Scholar] [CrossRef]
- Hill, M.A.; Alexander, W.B.; Guo, B.; Kato, Y.; Patra, K.; O’Dell, M.R.; McCall, M.N.; Whitney-Miller, C.L.; Bardeesy, N.; Hezel, A.F. Kras and Tp53 Mutations Cause Cholangiocyte And Hepatocyte-Derived Cholangiocarcinoma. Cancer Res. 2018, 78, 4445–4451. [Google Scholar] [CrossRef]
- Kachaylo, E.; Tschuor, C.; Calo, N.; Borgeaud, N.; Ungethum, U.; Limani, P.; Piguet, A.C.; Dufour, J.F.; Foti, M.; Graf, R.; et al. PTEN Down-Regulation Promotes beta-Oxidation to Fuel Hypertrophic Liver Growth After Hepatectomy in Mice. Hepatology 2017, 66, 908–921. [Google Scholar] [CrossRef]
- Lin, Y.K.; Fang, Z.; Jiang, T.Y.; Wan, Z.H.; Pan, Y.F.; Ma, Y.H.; Shi, Y.Y.; Tan, Y.X.; Dong, L.W.; Zhang, Y.J.; et al. Combination of Kras activation and PTEN deletion contributes to murine hepatopancreatic ductal malignancy. Cancer Lett. 2018, 421, 161–169. [Google Scholar] [CrossRef]
- Calderaro, J.; Ziol, M.; Paradis, V.; Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 2019, 71, 616–630. [Google Scholar] [CrossRef]
- Craig, A.J.; von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour evolution in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef]
- Huang, W.; Skanderup, A.J.; Lee, C.G. Advances in genomic hepatocellular carcinoma research. Gigascience 2018, 7, 7. [Google Scholar] [CrossRef]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis-(NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Lombardi, A.; Grimaldi, A.; Zappavigna, S.; Misso, G.; Caraglia, M. Hepatocarcinoma: Genetic and epigenetic features. Minerva Gastroenterol. Dietol. 2018, 64, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kumar, R.; Pandey, A.K. Hepatocellular Carcinoma: Causes, Mechanism of Progression and Biomarkers. Curr. Chem. Genom. Transl. Med. 2018, 12, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Umeda, S.; Kanda, M.; Kodera, Y. Emerging evidence of molecular biomarkers in hepatocellular carcinoma. Histol. Histopathol. 2018, 33, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.G.; Ioannidis, J.P. Reproducibility in science: Improving the standard for basic and preclinical research. Circ. Res. 2015, 116, 116–126. [Google Scholar] [CrossRef]
- Pulverer, B. Reproducibility blues. EMBO J. 2015, 34, 2721–2724. [Google Scholar] [CrossRef]
- Pusztai, L.; Hatzis, C.; Andre, F. Reproducibility of research and preclinical validation: Problems and solutions. Nat. Rev. Clin. Oncol. 2013, 10, 720–724. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lesions | FCA | HCC | CC | Undifferentiated | Oval Cell Tumor | Background Lesion | Unclassifiable | Bile Duct/Hepatocellular Adenoma | |
| GEMM | |||||||||
| PTEN/TGFβR2 | 13.8 | 4.8 | 29.3 | 0 | 0 | 1.6 | 3.7 | 0/0 | |
| PTEN | 51.7 | 3.9 | 4.9 | 0.5 | 2.0 | 0 | 2.4 | 0/0.49 | |
| PTEN/IDH1 | 6.3 | 0 | 0 | 0 | 0 | 0 | 0 | 0/0 | |
| KRAS | 53.4 | 24.5 | 1.1 | 1.5 | 7.6 | 1.9 | 0 | 1.1/0.8 | |
| KRAS/PTEN | 89.4 | 2.5 | 0.8 | 0 | 5.5 | 0.9 | 0 | 0/0 | |
| Lesions | Hyperplasia | Oval Cell Proliferation | Bile Duct Hyperplasia | Reactive Bile Duct Changes | Steatosis | Glycogen Accumulation | Inflammation | Necrosis/Fibrosis | |
| GEMM | |||||||||
| PTEN/TGFβR2 | 3.2 | 6.9 | 8.5 | 3.7 | 4.8 | 6.4 | 8.0 | 3.7 | |
| PTEN | 2.0 | 8.3 | 2.4 | 1.5 | 17.0 | 0 | 0 | 0 | |
| PTEN/IDH1 | 6.3 | 6.3 | 0 | 12.5 | 12.5 | 25.0 | 12.5 | 18.8 | |
| KRAS | 0 | 0.4 | 0.8 | 0.4 | 4.6 | 0.8 | 0.4 | 0.8 | |
| KRAS/PTEN | 2.0 | 8.3 | 2.4 | 1.5 | 17.0 | 6.8 | 3.9 | 1.0 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steiger, K.; Gross, N.; Widholz, S.A.; Rad, R.; Weichert, W.; Mogler, C. Genetically Engineered Mouse Models of Liver Tumorigenesis Reveal a Wide Histological Spectrum of Neoplastic and Non-Neoplastic Liver Lesions. Cancers 2020, 12, 2265. https://doi.org/10.3390/cancers12082265
Steiger K, Gross N, Widholz SA, Rad R, Weichert W, Mogler C. Genetically Engineered Mouse Models of Liver Tumorigenesis Reveal a Wide Histological Spectrum of Neoplastic and Non-Neoplastic Liver Lesions. Cancers. 2020; 12(8):2265. https://doi.org/10.3390/cancers12082265
Chicago/Turabian StyleSteiger, Katja, Nina Gross, Sebastian A. Widholz, Roland Rad, Wilko Weichert, and Carolin Mogler. 2020. "Genetically Engineered Mouse Models of Liver Tumorigenesis Reveal a Wide Histological Spectrum of Neoplastic and Non-Neoplastic Liver Lesions" Cancers 12, no. 8: 2265. https://doi.org/10.3390/cancers12082265
APA StyleSteiger, K., Gross, N., Widholz, S. A., Rad, R., Weichert, W., & Mogler, C. (2020). Genetically Engineered Mouse Models of Liver Tumorigenesis Reveal a Wide Histological Spectrum of Neoplastic and Non-Neoplastic Liver Lesions. Cancers, 12(8), 2265. https://doi.org/10.3390/cancers12082265