Interplay of tRNA-Derived Fragments and T Cell Activation in Breast Cancer Patient Survival

 , , and
, , and
Abstract
1. Introduction
2. Results
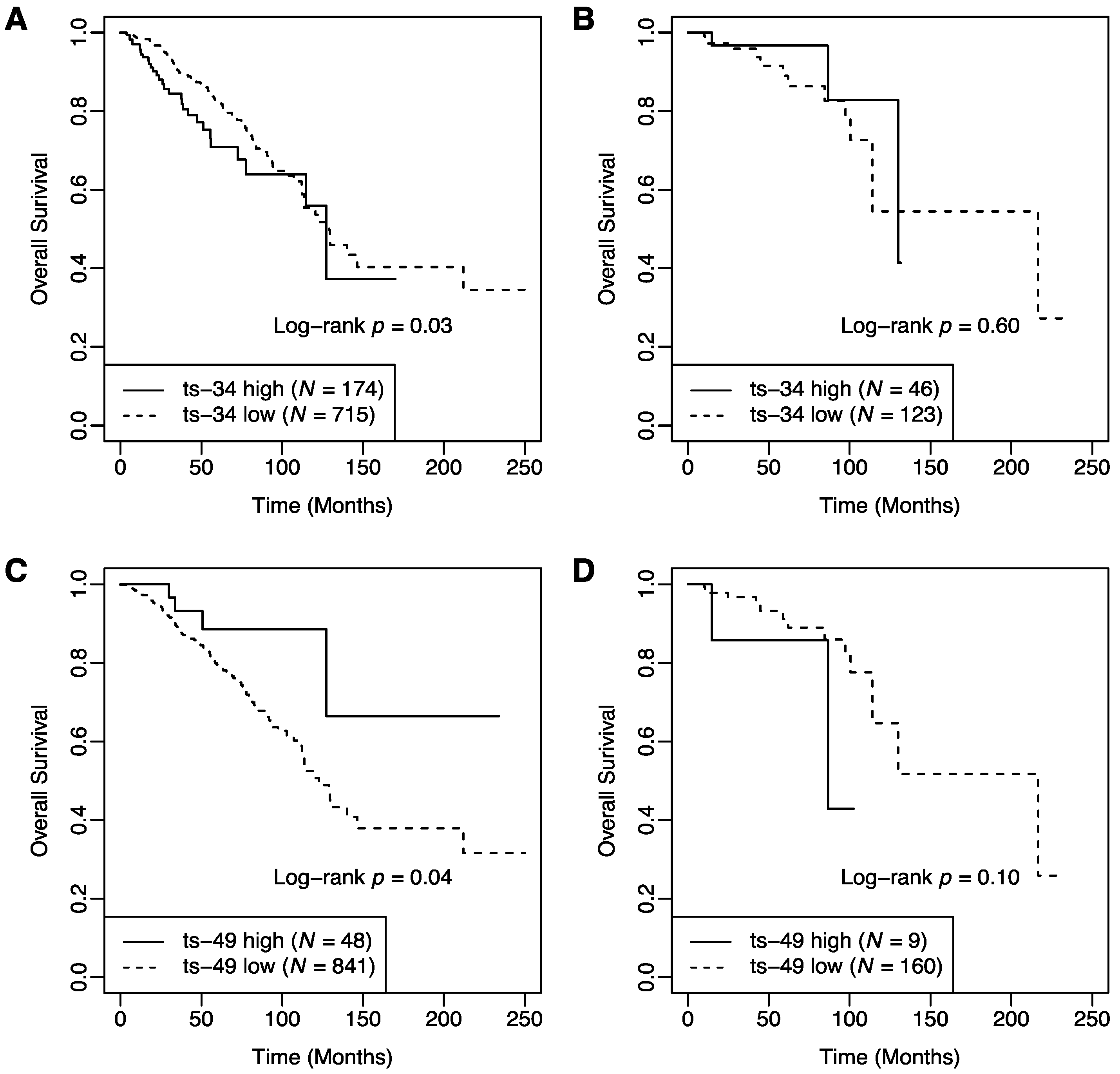
2.1. Association of tRFs with Patient Survival
2.2. Interaction between T Cell Activation Status and tRFs in Patient Survival
2.3. Correlation between tRFs and Clinical Pathological Variables
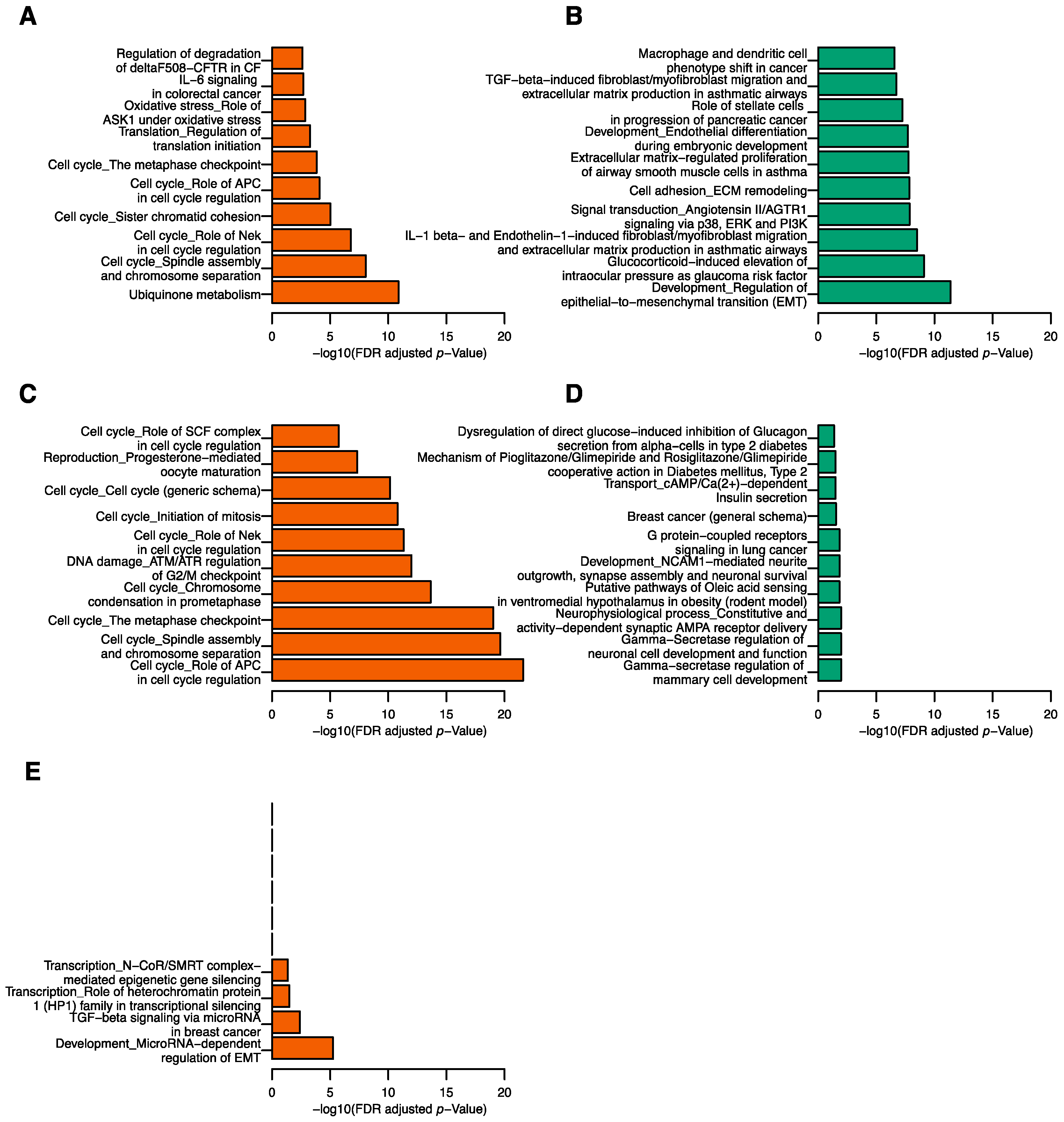
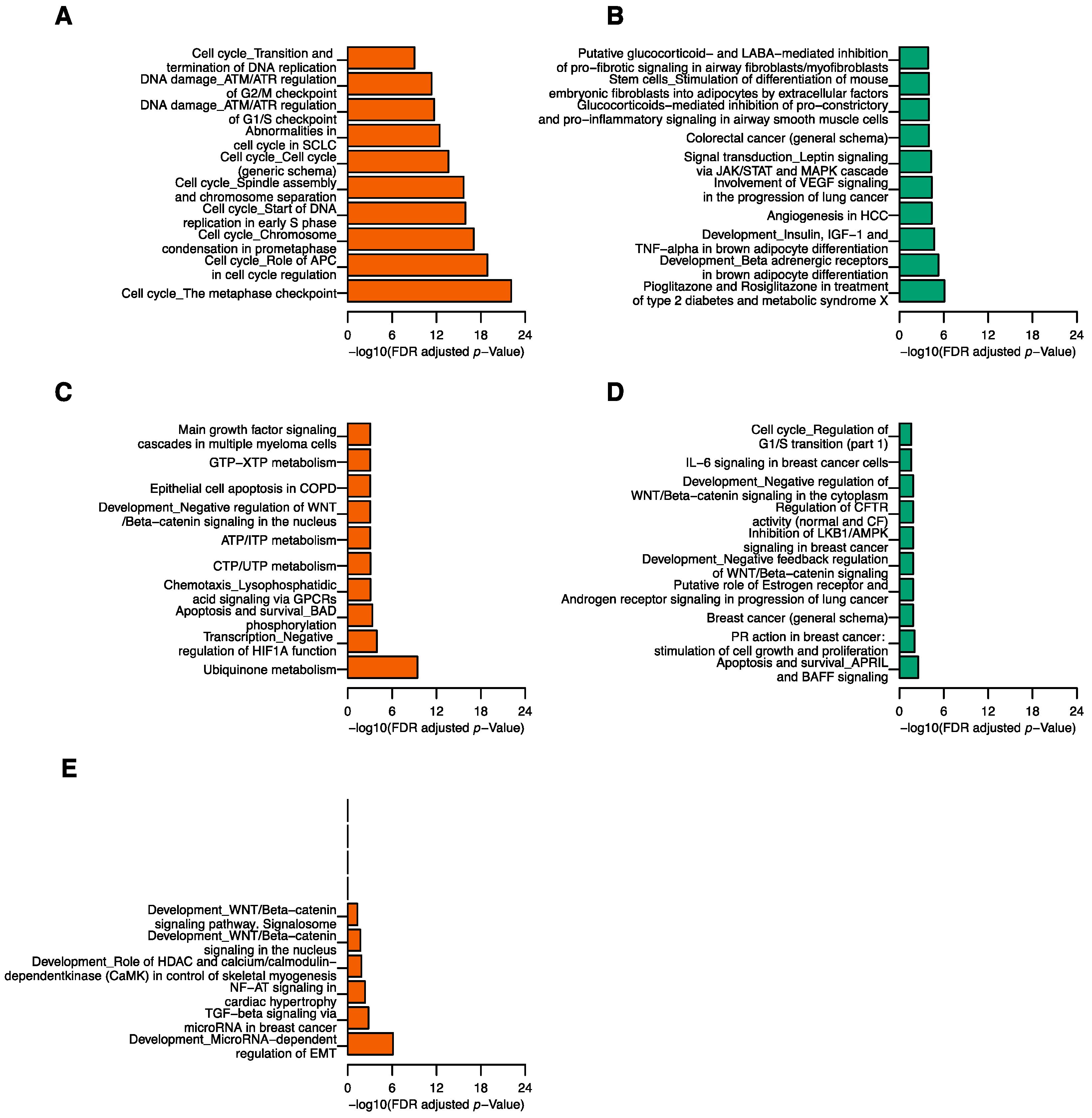
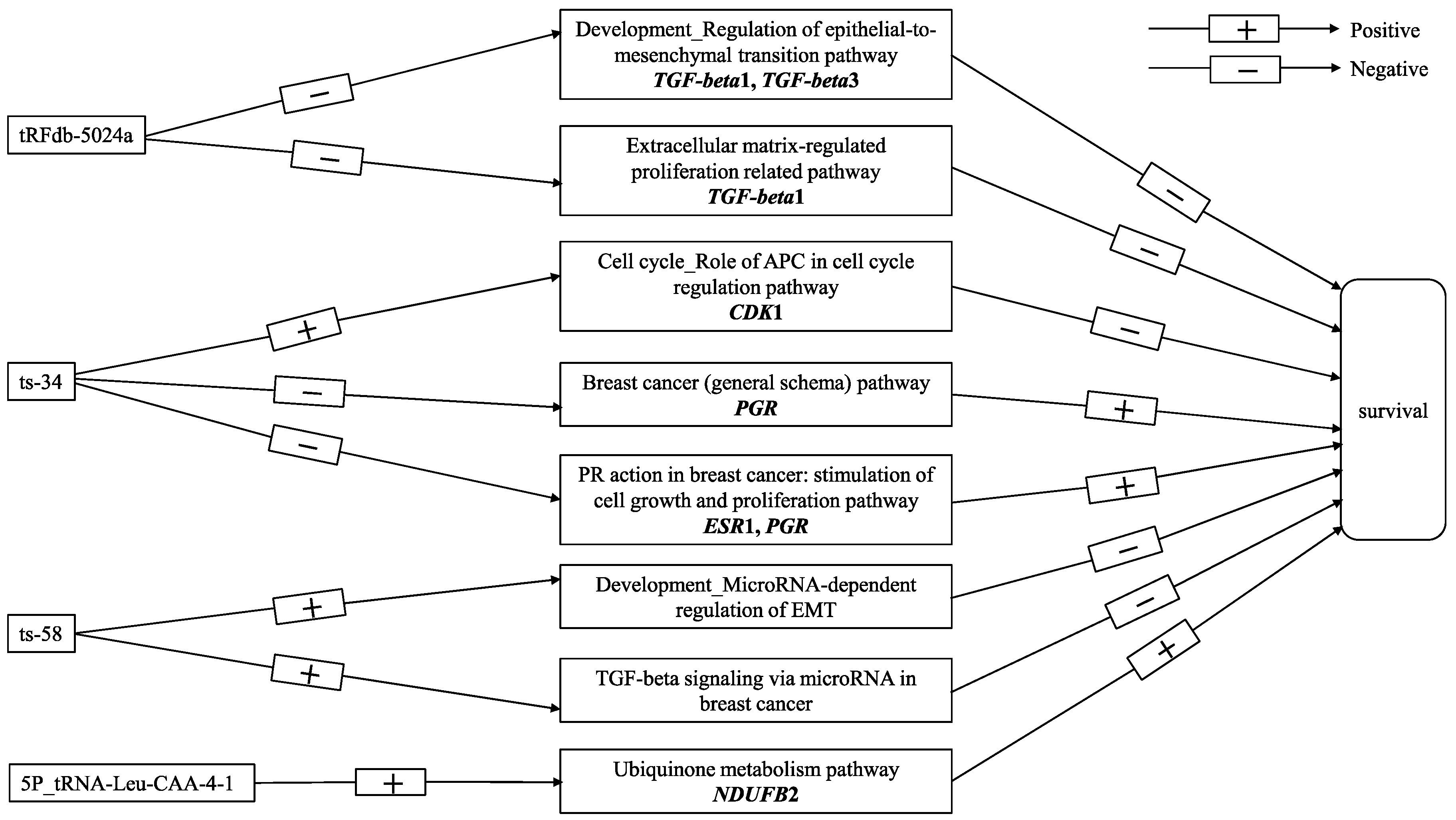
2.4. Correlation between tRFs and mRNA Transcripts
2.5. Correlation between tRFs and Gene Modules
3. Discussion
4. Materials and Methods
4.1. Study subjects and Data Sources
4.2. Statistical Analyses
4.3. Functional Pathway Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pekarsky, Y.; Balatti, V.; Palamarchuk, A.; Rizzotto, L.; Veneziano, D.; Nigita, G.; Rassenti, L.Z.; Pass, H.I.; Kipps, T.J.; Liu, C.G.; et al. Dysregulation of a family of short noncoding RNAs, tsRNAs, in human cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5071–5076. [Google Scholar] [CrossRef] [PubMed]
- Balatti, V.; Nigita, G.; Veneziano, D.; Drusco, A.; Stein, G.S.; Messier, T.L.; Farina, N.H.; Lian, J.B.; Tomasello, L.; Liu, C.G.; et al. tsRNA signatures in cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 8071–8076. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J. Tackling Tumors with Small RNAs Derived from Transfer RNA. N. Engl. J. Med. 2018, 378, 1842–1843. [Google Scholar] [CrossRef] [PubMed]
- Dhahbi, J.M.; Spindler, S.R.; Atamna, H.; Boffelli, D.; Martin, D.I. Deep Sequencing of Serum Small RNAs Identifies Patterns of 5′ tRNA Half and YRNA Fragment Expression Associated with Breast Cancer. Biomark. Cancer 2014, 6, 37–47. [Google Scholar] [CrossRef]
- Goodarzi, H.; Liu, X.; Nguyen, H.C.; Zhang, S.; Fish, L.; Tavazoie, S.F. Endogenous tRNA-Derived Fragments Suppress Breast Cancer Progression via YBX1 Displacement. Cell 2015, 161, 790–802. [Google Scholar] [CrossRef]
- Honda, S.; Loher, P.; Shigematsu, M.; Palazzo, J.P.; Suzuki, R.; Imoto, I.; Rigoutsos, I.; Kirino, Y. Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proc. Natl. Acad. Sci. USA 2015, 112, E3816–E3825. [Google Scholar] [CrossRef]
- Sun, C.; Yang, F.; Zhang, Y.; Chu, J.; Wang, J.; Wang, Y.; Zhang, Y.; Li, J.; Li, Y.; Fan, R.; et al. tRNA-Derived Fragments as Novel Predictive Biomarkers for Trastuzumab-Resistant Breast Cancer. Cell. Physiol. Biochem. 2018, 49, 419–431. [Google Scholar] [CrossRef]
- Feng, W.; Li, Y.; Chu, J.; Li, J.; Zhang, Y.; Ding, X.; Fu, Z.; Li, W.; Huang, X.; Yin, Y. Identification of tRNA-derived small noncoding RNAs as potential biomarkers for prediction of recurrence in triple-negative breast cancer. Cancer Med. 2018, 7, 5130–5144. [Google Scholar] [CrossRef]
- Dhahbi, J.M. 5′ tRNA Halves: The Next Generation of Immune Signaling Molecules. Front. Immunol. 2015, 6, 74. [Google Scholar] [CrossRef]
- Li, S.; Xu, Z.; Sheng, J. tRNA-Derived Small RNA: A Novel Regulatory Small Non-Coding RNA. Genes 2018, 9, 246. [Google Scholar] [CrossRef]
- Wang, Z.; Xiang, L.; Shao, J.; Yuan, Z. The 3′ CCACCA sequence of tRNAAla(UGC) is the motif that is important in inducing Th1-like immune response, and this motif can be recognized by Toll-like receptor 3. Clin. Vaccine Immunol. 2006, 13, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Bai, Y.; Wang, Z. Elevated T cell activation score is associated with improved survival of breast cancer. Breast Cancer Res. Treat. 2017, 164, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Huang, H.; Zhou, J.; Ma, W.; Mackay, S.; Wang, Z. BRCA1 mRNA expression modifies the effect of T cell activation score on patient survival in breast cancer. BMC Cancer 2019, 19, 387. [Google Scholar] [CrossRef] [PubMed]
- Li, L.D.; Sun, H.F.; Liu, X.X.; Gao, S.P.; Jiang, H.L.; Hu, X.; Jin, W. Down-Regulation of NDUFB9 Promotes Breast Cancer Cell Proliferation, Metastasis by Mediating Mitochondrial Metabolism. PLoS ONE 2015, 10, e0144441. [Google Scholar] [CrossRef] [PubMed]
- Olvedy, M.; Scaravilli, M.; Hoogstrate, Y.; Visakorpi, T.; Jenster, G.; Martens-Uzunova, E.S. A comprehensive repertoire of tRNA-derived fragments in prostate cancer. Oncotarget 2016, 7, 24766–24777. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, H.; Cheng, X.; Wang, D.; Fu, S.; Shen, W.; Zhang, Q.; Zhang, L.; Xue, Z.; Li, Y.; et al. tRF/miR-1280 Suppresses Stem Cell-like Cells and Metastasis in Colorectal Cancer. Cancer Res. 2017, 77, 3194–3206. [Google Scholar] [CrossRef]
- Shao, Y.; Sun, Q.; Liu, X.; Wang, P.; Wu, R.; Ma, Z. tRF-Leu-CAG promotes cell proliferation and cell cycle in non-small cell lung cancer. Chem. Biol. Drug Des. 2017, 90, 730–738. [Google Scholar] [CrossRef]
- Telonis, A.G.; Rigoutsos, I. Race Disparities in the Contribution of miRNA Isoforms and tRNA-Derived Fragments to Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 1140–1154. [Google Scholar] [CrossRef]
- Weidenfeld, K.; Barkan, D. EMT and Stemness in Tumor Dormancy and Outgrowth: Are They Intertwined Processes? Front. Oncol. 2018, 8, 381. [Google Scholar] [CrossRef]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef]
- Lu, L.; Zeng, H.; Gu, X.; Ma, W. Circulating tumor cell clusters-associated gene plakoglobin and breast cancer survival. Breast Cancer Res. Treat. 2015, 151, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Todorović, V.; Desai, B.V.; Patterson, M.J.; Amargo, E.V.; Dubash, A.D.; Yin, T.; Jones, J.C.; Green, K.J. Plakoglobin regulates cell motility through Rho- and fibronectin-dependent Src signaling. J. Cell Sci. 2010, 123, 3576–3586. [Google Scholar] [CrossRef] [PubMed]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. In Cell Cycle Control; Humana Press: New York, NY, USA, 2014; pp. 29–40. [Google Scholar]
- Lu, Y.; Yang, G.; Xiao, Y.; Zhang, T.; Su, F.; Chang, R.; Ling, X.; Bai, Y. Upregulated cyclins may be novel genes for triple-negative breast cancer based on bioinformatic analysis. Breast Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Purdie, C.A.; Quinlan, P.; Jordan, L.B.; Ashfield, A.; Ogston, S.; Dewar, J.A.; Thompson, A.M. Progesterone receptor expression is an independent prognostic variable in early breast cancer: A population-based study. Br. J. Cancer 2014, 110, 565–572. [Google Scholar] [CrossRef]
- Dunnwald, L.K.; Rossing, M.A.; Li, C.I. Hormone receptor status, tumor characteristics, and prognosis: A prospective cohort of breast cancer patients. Breast Cancer Res. 2007, 9, R6. [Google Scholar] [CrossRef]
- Qian, B.; Katsaros, D.; Lu, L.; Preti, M.; Durando, A.; Arisio, R.; Mu, L.; Yu, H. High miR-21 expression in breast cancer associated with poor disease-free survival in early stage disease and high TGF-beta1. Breast Cancer Res. Treat. 2009, 117, 131–140. [Google Scholar] [CrossRef]
- Mu, L.; Katsaros, D.; Lu, L.; Preti, M.; Durando, A.; Arisio, R.; Yu, H. TGF-beta1 genotype and phenotype in breast cancer and their associations with IGFs and patient survival. Br. J. Cancer 2008, 99, 1357–1363. [Google Scholar] [CrossRef]
- Xu, Q.; Wang, L.; Li, H.; Han, Q.; Li, J.; Qu, X.; Huang, S.; Zhao, R.C. Mesenchymal stem cells play a potential role in regulating the establishment and maintenance of epithelial-mesenchymal transition in MCF7 human breast cancer cells by paracrine and induced autocrine TGF-β. Int. J. Oncol. 2012, 41, 959–968. [Google Scholar] [CrossRef]
- cBioPortal. Available online: http://www.cbioportal.org/ (accessed on 10 July 2018).
- Genomic Data Commons Data Portal. Available online: https://portal.gdc.cancer.gov/ (accessed on 10 November 2018).
- GtRNAdb. Available online: http://gtrnadb.ucsc.edu/ (accessed on 3 December 2019).
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef]
- tRFexplorer. Available online: https://trfexplorer.cloud/ (accessed on 3 December 2019).
- La Ferlita, A.; Alaimo, S.; Veneziano, D.; Nigita, G.; Balatti, V.; Croce, C.M.; Ferro, A.; Pulvirenti, A. Identification of tRNA-derived ncRNAs in TCGA and NCI-60 panel cell lines and development of the public database tRFexplorer. Database 2019, 2019. [Google Scholar] [CrossRef]
- The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 19 January 2019).
- Dubovenko, A.; Nikolsky, Y.; Rakhmatulin, E.; Nikolskaya, T. Functional Analysis of OMICs Data and Small Molecule Compounds in an Integrated “Knowledge-Based” Platform. In Biological Networks and Pathway Analysis; Humana Press: New York, NY, USA, 2017; pp. 101–124. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.X.; Sieuwerts, A.M.; Zhang, Y.; Martens, J.W.; Smid, M.; Klijn, J.G.; Wang, Y.; Foekens, J.A. Pathway analysis of gene signatures predicting metastasis of node-negative primary breast cancer. BMC Cancer 2007, 7, 182. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Death | ||
|---|---|---|---|
| HR | 95% CI | p-Value | |
| T Cell Activation | |||
| Exhaustion | 1.00 | ||
| Activation | 0.48 | 0.27–0.83 | 0.009 |
| tRFdb-5024a | |||
| Low | 1.00 | ||
| High | 0.52 | 0.37–0.74 | p < 0.001 |
| 5P_tRNA-Leu-CAA-4-1 | |||
| Low | 1.00 | ||
| High | 0.55 | 0.35–0.87 | 0.011 |
| ts-34 | |||
| Low | 1.00 | ||
| High | 1.62 | 1.08–2.44 | 0.019 |
| ts-49 | |||
| Low | 1.00 | ||
| High | 0.40 | 0.17–0.93 | 0.032 |
| ts-58 | |||
| Low | 1.00 | ||
| High | 1.56 | 1.10–2.20 | 0.013 |
| tRFdb-1040 | |||
| Low | 1.00 | ||
| High | 1.49 | 0.93–2.38 | 0.096 |
| 5P_tRNA-Ala-AGC-8-2 | |||
| Low | 1.00 | ||
| High | 1.49 | 0.99–2.27 | 0.059 |
| ts-13 | |||
| Low | 1.00 | ||
| High | 1.38 | 0.94–2.03 | 0.103 |
| Age (per 5 years) | 1.21 | 1.13–1.29 | p < 0.001 |
| Disease Stage | |||
| Stage I | 1.00 | ||
| Stage II | 2.38 | 1.31–4.31 | 0.004 |
| Stage III or IV | 7.03 | 3.84–12.85 | p < 0.001 |
| Histological type | |||
| Ductal | 1.00 | ||
| Lobular | 0.55 | 0.34–0.87 | 0.011 |
| Mix | 0.59 | 0.28–1.24 | 0.161 |
| Other | 2.38 | 1.24–4.57 | 0.009 |
| Variables | Death | ||
|---|---|---|---|
| HR | 95% CI | p-Value | |
| T Cell Activation | |||
| Exhaustion | 1.00 | ||
| Activation | 0.60 | 0.32–1.12 | 0.110 |
| tRFdb-5024a | |||
| Low | 1.00 | ||
| High | 0.50 | 0.36–0.71 | p < 0.001 |
| 5P_tRNA-Leu-CAA-4-1 | |||
| Low | 1.00 | ||
| High | 0.58 | 0.37–0.92 | 0.021 |
| ts-34 | |||
| Low | 1.00 | ||
| High | 2.12 | 1.40–3.22 | p < 0.001 |
| ts-49 | |||
| Low | 1.00 | ||
| High | 0.27 | 0.10–0.74 | 0.011 |
| ts-58 | |||
| Low | 1.00 | ||
| High | 1.51 | 1.07–2.12 | 0.018 |
| T cell Activation × ts-34 | 0.22 | 0.05–0.94 | 0.040 |
| T cell Activation × ts-49 | 13.49 | 2.00–91.02 | 0.008 |
| Age (per 5 years) | 1.20 | 1.12–1.28 | p < 0.001 |
| Disease Stage | |||
| Stage I | 1.00 | ||
| Stage II | 2.18 | 1.21–3.94 | 0.010 |
| Stage III or IV | 6.35 | 3.50–11.52 | p < 0.001 |
| Histological type | |||
| Ductal | 1.00 | ||
| Lobular | 0.53 | 0.33–0.83 | 0.006 |
| Mix | 0.56 | 0.26–1.19 | 0.130 |
| Other | 2.60 | 1.34–5.02 | 0.005 |
| Stratification Variable | Variables | Death | ||
|---|---|---|---|---|
| HR | 95% CI | p-Value | ||
| T cell Exhaustion group | tRFdb-5024a | |||
| Low | 1.00 | |||
| High | 0.51 | 0.35–0.73 | p < 0.001 | |
| 5P_tRNA-Leu-CAA-4-1 | ||||
| Low | 1.00 | |||
| High | 0.54 | 0.33–0.88 | 0.014 | |
| ts-34 | ||||
| Low | 1.00 | |||
| High | 2.13 | 1.40–3.23 | p < 0.001 | |
| ts-49 | ||||
| Low | 1.00 | |||
| High | 0.28 | 0.10–0.76 | 0.013 | |
| ts-58 | ||||
| Low | 1.00 | |||
| High | 1.58 | 1.10–2.26 | 0.013 | |
| Age (per 5 years) | 1.21 | 1.13–1.30 | p < 0.001 | |
| Disease Stage | ||||
| Stage I | 1.00 | |||
| Stage II | 2.60 | 1.35–4.99 | 0.004 | |
| Stage III or IV | 7.18 | 3.72–13.86 | p < 0.001 | |
| Histological type | ||||
| Ductal | 1.00 | |||
| Lobular | 0.49 | 0.30–0.80 | 0.004 | |
| Mix | 0.51 | 0.23–1.12 | 0.094 | |
| Other | 2.16 | 0.98–4.76 | 0.056 | |
| T cell Activation group | tRFdb-5024a | |||
| Low | 1.00 | |||
| High | 0.57 | 0.15–2.06 | 0.388 | |
| 5P_tRNA-Leu-CAA-4-1 | ||||
| Low | 1.00 | |||
| High | 0.55 | 0.12–2.49 | 0.442 | |
| ts-34 | ||||
| Low | 1.00 | |||
| High | 0.18 | 0.03-1.14 | 0.069 | |
| ts-49 | ||||
| Low | 1.00 | |||
| High | 3.91 | 0.61–24.95 | 0.150 | |
| ts-58 | ||||
| Low | 1.00 | |||
| High | 0.50 | 0.14–1.81 | 0.291 | |
| Age (per 5 years) | 1.16 | 0.94–1.43 | 0.157 | |
| Disease Stage | ||||
| Stage I | 1.00 | |||
| Stage II | 0.53 | 0.11–2.71 | 0.449 | |
| Stage III or IV | 4.37 | 0.74–25.68 | 0.103 | |
| Histological type | ||||
| Ductal | 1.00 | |||
| Lobular | 0.99 | 0.19–5.32 | 0.999 | |
| Mix | 3.65 | 0.33–40.56 | 0.292 | |
| Other | 6.21 | 1.39–27.71 | 0.017 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shan, N.; Li, N.; Dai, Q.; Hou, L.; Yan, X.; Amei, A.; Lu, L.; Wang, Z. Interplay of tRNA-Derived Fragments and T Cell Activation in Breast Cancer Patient Survival. Cancers 2020, 12, 2230. https://doi.org/10.3390/cancers12082230
Shan N, Li N, Dai Q, Hou L, Yan X, Amei A, Lu L, Wang Z. Interplay of tRNA-Derived Fragments and T Cell Activation in Breast Cancer Patient Survival. Cancers. 2020; 12(8):2230. https://doi.org/10.3390/cancers12082230
Chicago/Turabian StyleShan, Nayang, Ningshan Li, Qile Dai, Lin Hou, Xiting Yan, Amei Amei, Lingeng Lu, and Zuoheng Wang. 2020. "Interplay of tRNA-Derived Fragments and T Cell Activation in Breast Cancer Patient Survival" Cancers 12, no. 8: 2230. https://doi.org/10.3390/cancers12082230
APA StyleShan, N., Li, N., Dai, Q., Hou, L., Yan, X., Amei, A., Lu, L., & Wang, Z. (2020). Interplay of tRNA-Derived Fragments and T Cell Activation in Breast Cancer Patient Survival. Cancers, 12(8), 2230. https://doi.org/10.3390/cancers12082230