Pre-Existing Tumoral B Cell Infiltration and Impaired Genome Maintenance Correlate with Response to Chemoradiotherapy in Locally Advanced Rectal Cancer

 , , ,
, , ,  , , add
Show full author list
, , add
Show full author list
Abstract
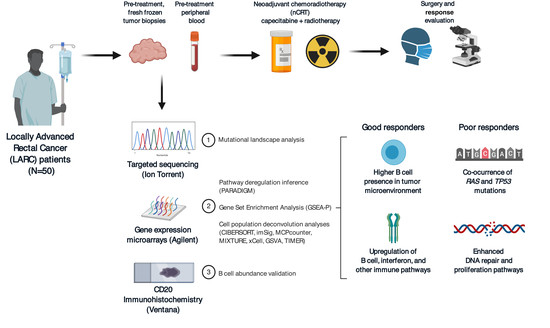
1. Introduction
2. Results
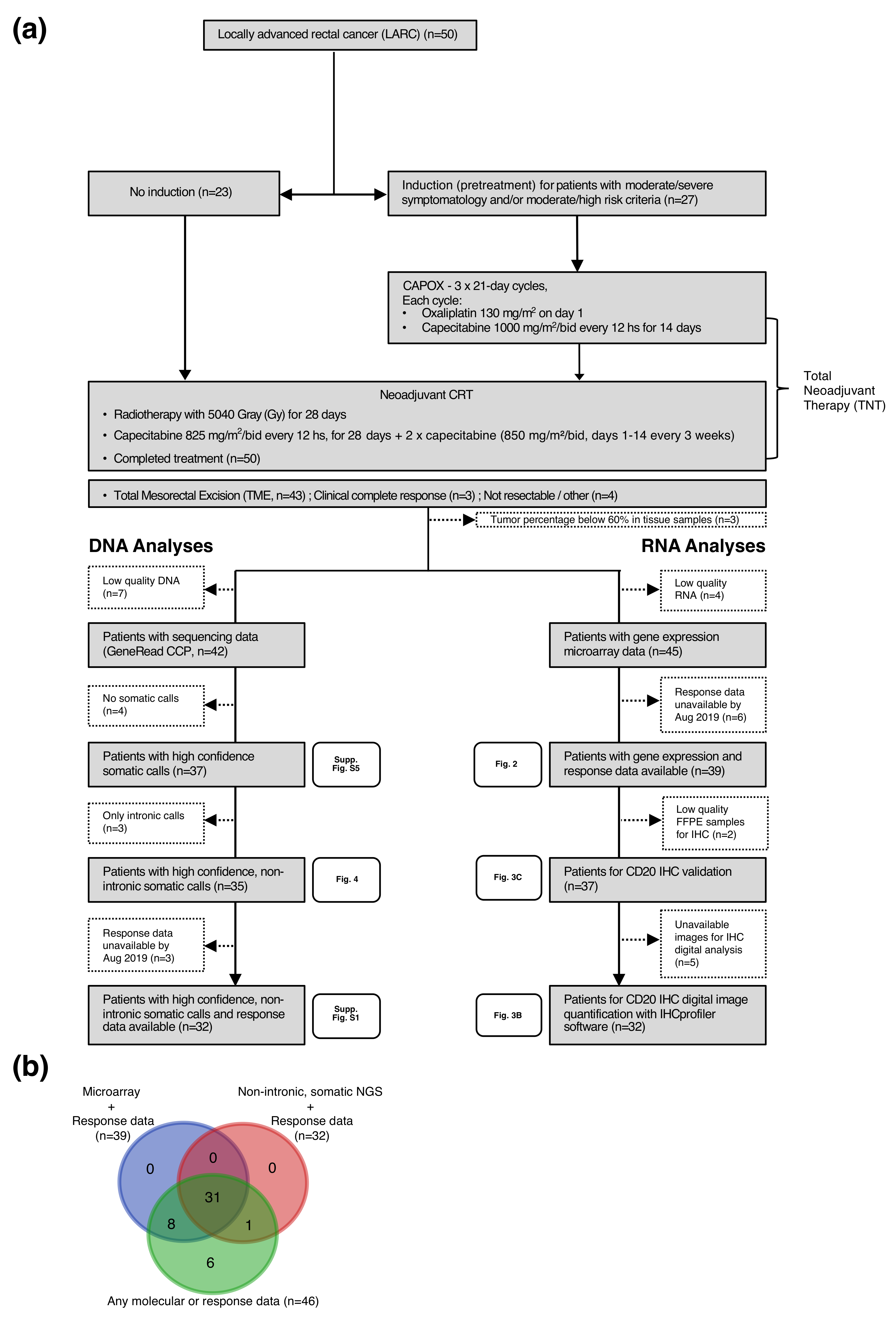
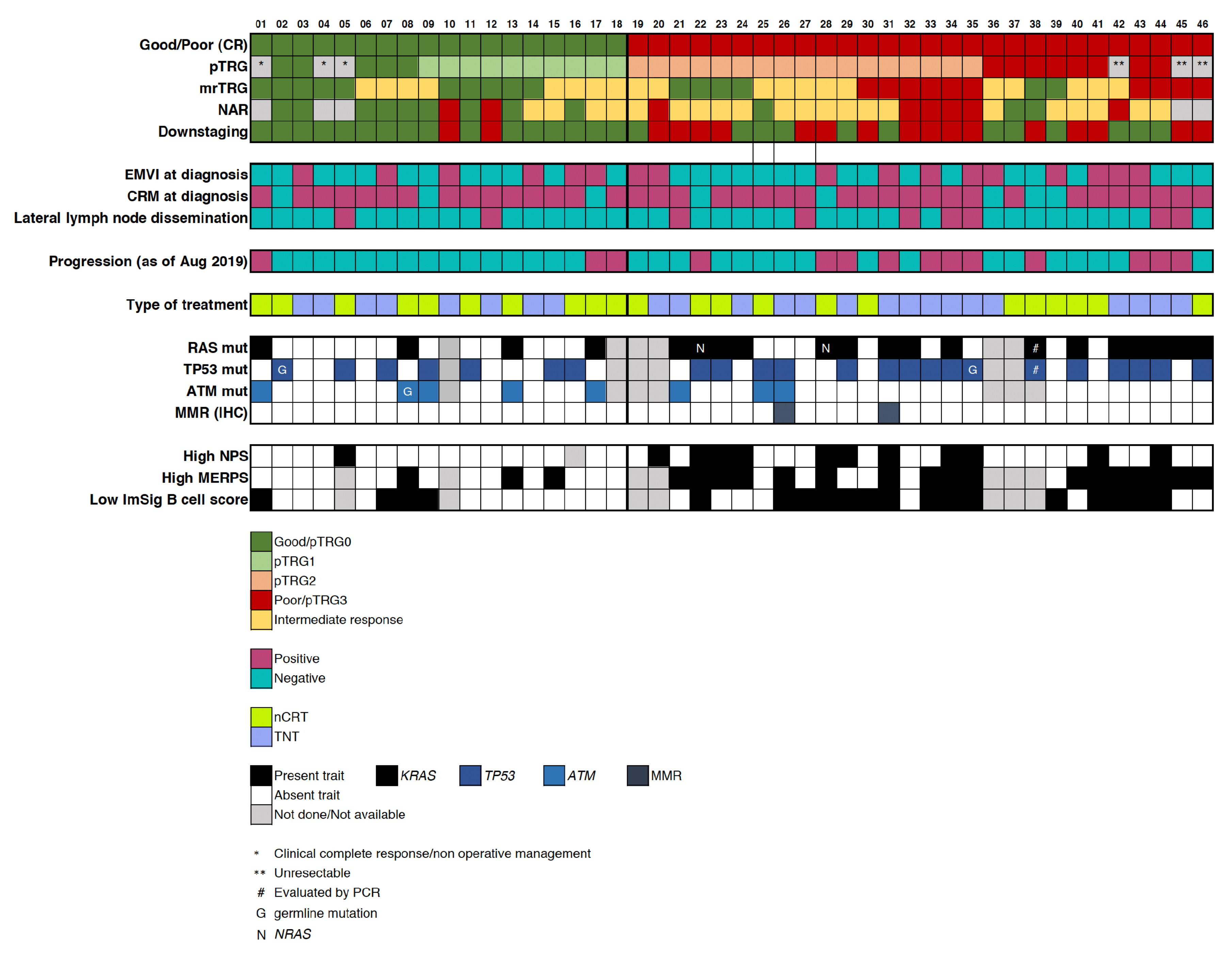
2.1. Patient Population and Response Assessment
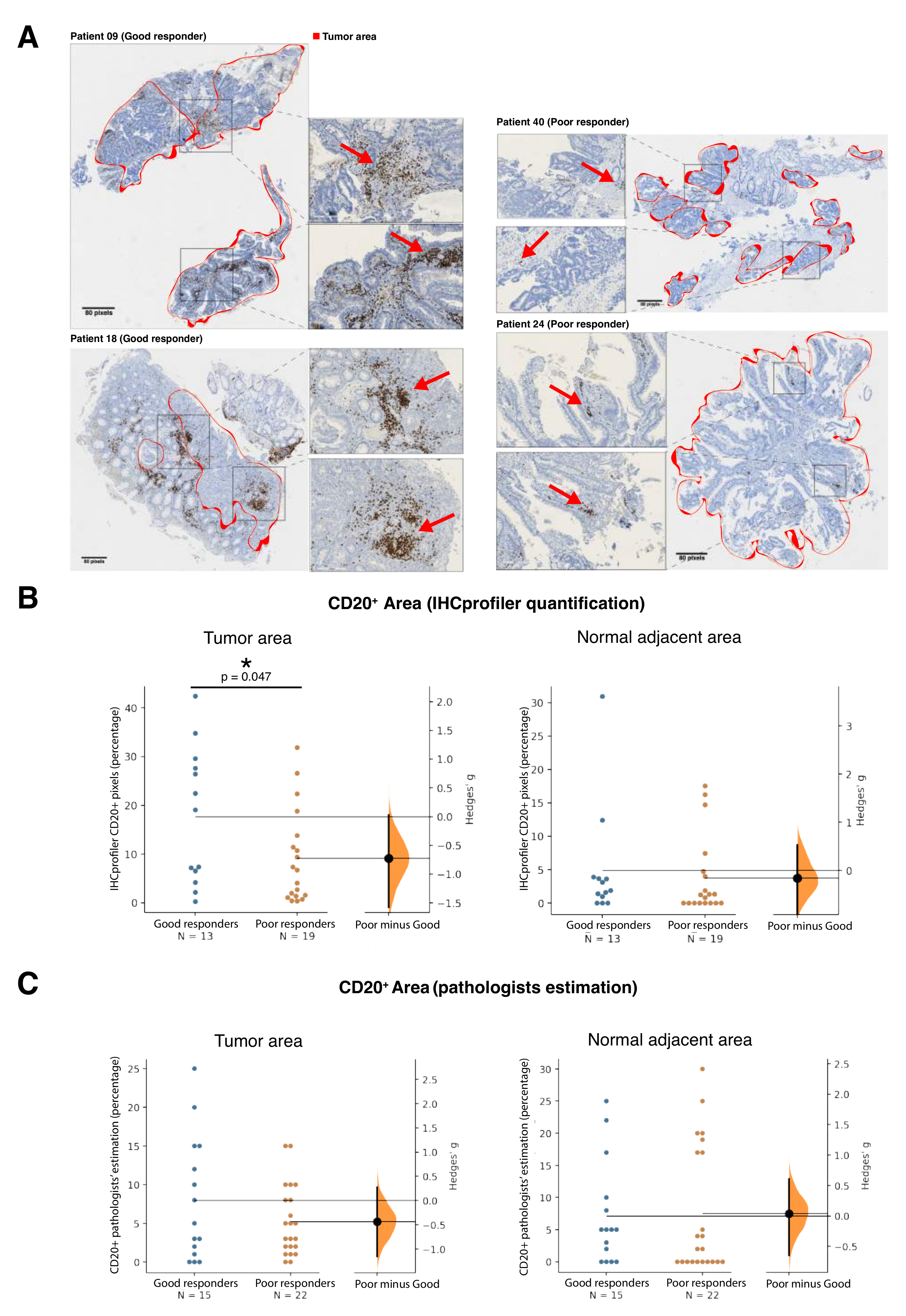
2.2. Gene Expression Analysis and Validation
2.3. Mutational Profile
2.4. Integration of Clinical and Multi-Omic Features for Response Prediction
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Quality Control
4.2. Targeted Sequencing
4.3. Gene Expression Analysis
4.4. CD20 Immunohistochemistry Validation
4.5. Statistical Analysis
4.6. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LARC | Locally Advanced Rectal Cancer |
| nCRT | neoadjuvant chemoradiotherapy |
| TME | Total Mesorectal Excision |
| 3D-LCRT | Three-dimensional Long Course Radiotherapy |
| CAPOX | Capecitabine + Oxaliplatin |
| TNT | Total Neoadjuvant Therapy |
| CRT | Chemoradiotherapy |
| pCR | Complete Pathological Response |
| AJCC | American Joint Committee on Cancer |
| pTRG | Pathological Tumor Regression Grade |
| mrTRG | MRI Tumor Regression Grade |
| NAR | NeoAdjuvant Rectal score |
| cCR | Complete Clinical Response |
| CMS | Colorectal Consensus Molecular Subtype |
| dMMR | Mismatch Repair Defficiency |
| IHC | Immunohistochemistry |
| NPS | Neutrophil Platelet Score |
| EMVI | Extramural Vascular Invasion |
| MRI | Magnetic Resonance Imaging |
| CRM | Circumferential Radial Margin |
| MERPS | Median Repair Pathway Score |
| OR | Odds Ratio |
| KP | KRAS-TP53 |
| MMR | Mismatch Repair |
| DAMPs | Damage Associated Molecular Patterns |
| MHC | Major Histocompatibility Complex |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| CT | Computed Tomography |
| LLND | Lateral Lymph Node Dissemination |
| DRE | Digital Rectal Examination |
| DW-MRI | Diffusion-Weighted Magnetic Resonance Imaging |
| CR | Consensus Response |
Appendix A. By-Subject Selected Clinical and Molecular Characteristics of Good and Poor Ncrt Responders

References
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.; Becker, H.; Hohenberger, W.; Rödel, C.; Wittekind, C.; Fietkau, R.; Martus, P.; Tschmelitsch, J.; Hager, E.; Hess, C.F.; et al. Preoperative versus postoperative chemoradiotherapy for rectal cancer. New Engl. J. Med. 2004, 351, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Atkin, W.; Lenz, H.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal cancer. Lancet 2010, 375, 1030–1047. [Google Scholar] [CrossRef]
- Cercek, A.; Roxburgh, C.S.; Strombom, P.; Smith, J.J.; Temple, L.K.; Nash, G.M.; Guillem, J.G.; Paty, P.B.; Yaeger, R.; Stadler, Z.K.; et al. Adoption of total neoadjuvant therapy for locally advanced rectal cancer. JAMA Oncol. 2018, 4, e180071. [Google Scholar] [CrossRef]
- Chau, I.; Brown, G.; Cunningham, D.; Tait, D.; Wotherspoon, A.; Norman, A.R.; Tebbutt, N.; Hill, M.; Ross, P.J.; Massey, A.; et al. Neoadjuvant capecitabine and oxaliplatin followed by synchronous chemoradiation and total mesorectal excision in magnetic resonance imaging–defined poor-risk rectal cancer. J. Clin. Oncol. 2006, 24, 668–674. [Google Scholar] [CrossRef]
- Fernandez-Martos, C.; Garcia-Albeniz, X.; Pericay, C.; Maurel, J.; Aparicio, J.; Montagut, C.; Safont, M.; Salud, A.; Vera, R.; Massuti, B.; et al. Chemoradiation, surgery and adjuvant chemotherapy versus induction chemotherapy followed by chemoradiation and surgery: Long-term results of the Spanish GCR-3 phase II randomized trial. Ann. Oncol. 2015, 26, 1722–1728. [Google Scholar] [CrossRef]
- Minsky, B.D.; Cohen, A.M.; Kemeny, N.; Enker, W.E.; Kelsen, D.P.; Reichman, B.; Saltz, L.; Sigurdson, E.R.; Frankel, J. Enhancement of radiation-induced downstaging of rectal cancer by fluorouracil and high-dose leucovorin chemotherapy. J. Clin. Oncol. 1992, 10, 79–84. [Google Scholar] [CrossRef]
- Mohiuddin, M.; Hayne, M.; Regine, W.F.; Hanna, N.; Hagihara, P.F.; McGrath, P.; Marks, G.M. Prognostic significance of postchemoradiation stage following preoperative chemotherapy and radiation for advanced/recurrent rectal cancers. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 1075–1080. [Google Scholar] [CrossRef]
- Sun, Z.; Adam, M.A.; Kim, J.; Shenoi, M.; Migaly, J.; Mantyh, C.R. Optimal timing to surgery after neoadjuvant chemoradiotherapy for locally advanced rectal cancer. J. Am. Coll. Surg. 2016, 222, 367–374. [Google Scholar] [CrossRef]
- Rodel, C.; Martus, P.; Papadoupolos, T.; Füzesi, L.; Klimpfinger, M.; Fietkau, R.; Liersch, T.; Hohenberger, W.; Raab, R.; Sauer, R.; et al. Prognostic significance of tumor regression after preoperative chemoradiotherapy for rectal cancer. J. Clin. Oncol. 2005, 23, 8688–8696. [Google Scholar] [CrossRef]
- Mace, A.G.; Pai, R.K.; Stocchi, L.; Kalady, M.F. American Joint Committee on Cancer and College of American Pathologists regression grade: A new prognostic factor in rectal cancer. Dis. Colon Rectum 2015, 58, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Edge, S.B. AJCC Cancer Staging Manual; Springer: New York, NY, USA, 2017. [Google Scholar]
- Trakarnsanga, A.; Gönen, M.; Shia, J.; Nash, G.M.; Temple, L.K.; Guillem, J.G.; Paty, P.B.; Goodman, K.A.; Wu, A.; Gollub, M.; et al. Comparison of tumor regression grade systems for locally advanced rectal cancer after multimodality treatment. JNCI J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Patel, U.B.; Brown, G.; Rutten, H.; West, N.; Sebag-Montefiore, D.; Glynne-Jones, R.; Rullier, E.; Peeters, M.; Van Cutsem, E.; Ricci, S.; et al. Comparison of magnetic resonance imaging and histopathological response to chemoradiotherapy in locally advanced rectal cancer. Ann. Surg. Oncol. 2012, 19, 2842–2852. [Google Scholar] [CrossRef]
- Sclafani, F.; Brown, G.; Cunningham, D.; Wotherspoon, A.; Mendes, L.S.T.; Balyasnikova, S.; Evans, J.; Peckitt, C.; Begum, R.; Tait, D.; et al. Comparison between MRI and pathology in the assessment of tumour regression grade in rectal cancer. Br. J. Cancer 2017, 117, 1478–1485. [Google Scholar] [CrossRef]
- George, T.J.; Allegra, C.J.; Yothers, G. Neoadjuvant rectal (NAR) score: A new surrogate endpoint in rectal cancer clinical trials. Curr. Color. Cancer Rep. 2015, 11, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Van der Valk, M.J.; Vuijk, F.A.; Putter, H.; van de Velde, C.J.; Beets, G.L.; Hilling, D.E. Disqualification of Neoadjuvant Rectal Score Based on Data of 6596 Patients From the Netherlands Cancer Registry. Clin. Color. Cancer 2019, 18, e231–e236. [Google Scholar] [CrossRef]
- Akiyoshi, T.; Tanaka, N.; Kiyotani, K.; Gotoh, O.; Yamamoto, N.; Oba, K.; Fukunaga, Y.; Ueno, M.; Mori, S. Immunogenomic profiles associated with response to neoadjuvant chemoradiotherapy in patients with rectal cancer. Br. J. Surg. 2019, 106, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Gim, J.; Cho, Y.B.; Hong, H.K.; Kim, H.C.; Yun, S.H.; Wu, H.G.; Jeong, S.Y.; Joung, J.G.; Park, T.; Park, W.Y.; et al. Predicting multi-class responses to preoperative chemoradiotherapy in rectal cancer patients. Radiat. Oncol. 2016, 11, 50. [Google Scholar] [CrossRef]
- Palma, P.; Cano, C.; Conde-Muiño, R.; Comino, A.; Bueno, P.; Ferrón, J.A.; Cuadros, M. Expression profiling of rectal tumors defines response to neoadjuvant treatment related genes. PLoS ONE 2014, 9, e112189. [Google Scholar] [CrossRef]
- Brettingham-Moore, K.H.; Duong, C.P.; Greenawalt, D.M.; Heriot, A.G.; Ellul, J.; Dow, C.A.; Murray, W.K.; Hicks, R.J.; Tjandra, J.; Chao, M.; et al. Pretreatment transcriptional profiling for predicting response to neoadjuvant chemoradiotherapy in rectal adenocarcinoma. Clin. Cancer Res. 2011, 17, 3039–3047. [Google Scholar] [CrossRef]
- Watanabe, T.; Komuro, Y.; Kiyomatsu, T.; Kanazawa, T.; Kazama, Y.; Tanaka, J.; Tanaka, T.; Yamamoto, Y.; Shirane, M.; Muto, T.; et al. Prediction of sensitivity of rectal cancer cells in response to preoperative radiotherapy by DNA microarray analysis of gene expression profiles. Cancer Res. 2006, 66, 3370–3374. [Google Scholar] [CrossRef]
- Kamran, S.C.; Lennerz, J.K.; Margolis, C.A.; Liu, D.; Reardon, B.; Wankowicz, S.A.; Van Seventer, E.E.; Tracy, A.; Wo, J.Y.; Carter, S.L.; et al. Integrative molecular characterization of resistance to neoadjuvant chemoradiation in rectal cancer. Clin. Cancer Res. 2019, 25, 5561–5571. [Google Scholar] [CrossRef] [PubMed]
- Millino, C.; Maretto, I.; Pacchioni, B.; Digito, M.; De Paoli, A.; Canzonieri, V.; D’Angelo, E.; Agostini, M.; Rizzolio, F.; Giordano, A.; et al. Gene and microRNA expression are predictive of tumor response in rectal adenocarcinoma patients treated with preoperative chemoradiotherapy. J. Cell. Physiol. 2017, 232, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Zangrando, A.; Pastrello, C.; D’Angelo, E.; Romano, G.; Giovannoni, R.; Giordan, M.; Maretto, I.; Bedin, C.; Zanon, C.; et al. A functional biological network centered on XRCC3: A new possible marker of chemoradiotherapy resistance in rectal cancer patients. Cancer Biol. Ther. 2015, 16, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Hur, H.; Tulina, I.; Cho, M.S.; Min, B.S.; Koom, W.S.; Lim, J.S.; Ahn, J.B.; Kim, N.K. Biomarker-based scoring system for prediction of tumor response after preoperative chemoradiotherapy in rectal cancer by reverse transcriptase polymerase chain reaction analysis. Dis. Colon Rectum 2016, 59, 1174–1182. [Google Scholar] [CrossRef]
- Barnell, E.K.; Ronning, P.; Campbell, K.M.; Krysiak, K.; Ainscough, B.J.; Sheta, L.M.; Pema, S.P.; Schmidt, A.D.; Richters, M.; Cotto, K.C.; et al. Standard operating procedure for somatic variant refinement of sequencing data with paired tumor and normal samples. Genet. Med. 2019, 21, 972–981. [Google Scholar] [CrossRef]
- Park, I.J.; Yu, Y.S.; Mustafa, B.; Park, J.Y.; Seo, Y.B.; Kim, G.D.; Kim, J.; Kim, C.M.; Noh, H.D.; Hong, S.M.; et al. A Nine-Gene Signature for Predicting the Response to Preoperative Chemoradiotherapy in Patients with Locally Advanced Rectal Cancer. Cancers 2020, 12, 800. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Watt, D.G.; Proctor, M.J.; Park, J.H.; Horgan, P.G.; McMillan, D.C. The neutrophil-platelet score (NPS) predicts survival in primary operable colorectal cancer and a variety of common cancers. PLoS ONE 2015, 10, e0142159. [Google Scholar] [CrossRef]
- Gonzalez, G.; Fresno, C.; Merino, G.; Llera, A.S.; Podhajcer, O.; Fernández, E.A. Agi4x44.2c: A two-colour Agilent 4x44 Quality Control R library for large microarray projects. In Proceedings of the Memorias del 3er Congreso Argentino de Bioinformática y Biología Computacional, Entre Rios, Argentina, 26–28 September 2012; Available online: https://www.academia.edu/31181814/Software_integration_to_bioimages_management_processing_and_analysis?source=swp_share (accessed on 3 July 2020).
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Subramanian, A.; Kuehn, H.; Gould, J.; Tamayo, P.; Mesirov, J.P. GSEA-P: A desktop application for Gene Set Enrichment Analysis. Bioinformatics 2007, 23, 3251–3253. [Google Scholar] [CrossRef] [PubMed]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Rubio-Perez, C.; Muiños, F.; Sabarinathan, R.; Piulats, J.M.; Muntasell, A.; Dienstmann, R.; Lopez-Bigas, N.; Gonzalez-Perez, A. A pan-cancer landscape of interactions between solid tumors and infiltrating immune cell populations. Clin. Cancer Res. 2018, 24, 3717–3728. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Nirmal, A.J.; Regan, T.; Shih, B.B.; Hume, D.A.; Sims, A.H.; Freeman, T.C. Immune cell gene signatures for profiling the microenvironment of solid tumors. Cancer Immunol. Res. 2018, 6, 1388–1400. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Krombach, J.; Hennel, R.; Brix, N.; Orth, M.; Schoetz, U.; Ernst, A.; Schuster, J.; Zuchtriegel, G.; Reichel, C.A.; Bierschenk, S.; et al. Priming anti-tumor immunity by radiotherapy: Dying tumor cell-derived DAMPs trigger endothelial cell activation and recruitment of myeloid cells. Oncoimmunology 2019, 8, e1523097. [Google Scholar] [CrossRef]
- Howe, E.; Holton, K.; Nair, S.; Schlauch, D.; Sinha, R.; Quackenbush, J. Mev: Multiexperiment viewer. In Biomedical Informatics for Cancer Research; Springer: Boston, MA, USA, 2010; pp. 267–277. [Google Scholar]
- Varghese, F.; Bukhari, A.B.; Malhotra, R.; De, A. IHC Profiler: An open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS ONE 2014, 9, e96801. [Google Scholar] [CrossRef]
- Ho, J.; Tumkaya, T.; Aryal, S.; Choi, H.; Claridge-Chang, A. Moving beyond P values: Data analysis with estimation graphics. Nat. Methods 2019, 16, 565–566. [Google Scholar] [CrossRef]
- Timmerman, C.; Taveras, L.R.; Huerta, S. Clinical and molecular diagnosis of pathologic complete response in rectal cancer: An update. Expert Rev. Mol. Diagn. 2018, 18, 887–896. [Google Scholar] [CrossRef]
- Glimelius, B.; Tiret, E.; Cervantes, A.; Arnold, D. Rectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24, vi81–vi88. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Edmonson, M.N.; Patel, A.N.; Hedges, D.J.; Wang, Z.; Rampersaud, E.; Kesserwan, C.A.; Zhou, X.; Liu, Y.; Newman, S.; Rusch, M.C.; et al. Pediatric Cancer Variant Pathogenicity Information Exchange (PeCanPIE): A cloud-based platform for curating and classifying germline variants. Genome Res. 2019, 29, 1555–1565. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Fernández, E.A.; Mahmoud, Y.D.; Veigas, F.; Rocha, D.; Balzarini, M.; Lujan, H.D.; Rabinovich, G.A.; Girotti, M.R. MIXTURE: An improved algorithm for immune tumor microenvironment estimation based on gene expression data. bioRxiv 2019. bioRxiv:726562. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: http://www.r-project.org (accessed on 19 May 2020).
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Package ‘survminer’. Drawing Survival Curves Using ‘ggplot2’. (R Package Version 0.3.1); The Comprehensive R Archive Network. 2017. Available online: https://CRAN.R-project.org/package=survminer (accessed on 3 July 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | N = 50 |
|---|---|
| Demographic and clinical variables | |
| Age at diagnosis, median (range) | 61 (34–75) |
| <50 years, n (%) | 13 (26%) |
| Male, n (%) | 37 (74%) |
| Type of treatment, n (%) | |
| nCRT | 23 (46%) |
| TNT | 27 (54%) |
| Histological classification, n(%) | |
| Adenocarcinoma, well differentiated | 17 (34%) |
| Adenocarcinoma, moderately differentiated | 27 (54%) |
| Adenocarcinoma, poorly differentiated | 1 (2%) |
| Adenocarcinoma, differentiation data not available | 5 (10%) |
| Mucinous classification, n(%) | |
| Mucinous | 5 (10%) |
| Non-mucinous | 45 (95%) |
| Mismatch repair IHC assessment, n (%) | |
| MMR proficient by IHC | 47 (94%) |
| MMR deficient by IHC | 3 (6%) |
| Tumor localization, n(%) | |
| Low rectum (<7 cm) | 23 (46%) |
| Middle rectum (7–12 cm) | 20 (40%) |
| Upper rectum (>12 cm) | 7 (14%) |
| Clinical staging by MRI, n(%) | |
| T2-T3abN0, very low tumors, N0 | 3 (6%) |
| T3c-d/ T4, N0 | 18 (36%) |
| N1 | 19 (38%) |
| N2 | 10 (20%) |
| MRI risk factors beyond TNM, n(%) | |
| Initial EMVI | 19 (38%) |
| Initial CRM | 41 (82%) |
| Initial lateral lymph node dissemination | 10 (20%) |
| Post-nCRT outcomes, n(%) | |
| Clinical complete response (‘Watch and wait‘ strategy) | 3 (6%) |
| TME | 43 (86%) |
| Not resectable during surgery | 3 (6%) |
| Disease progression during treatment | 1 (2%) |
| Category | Dichotomic Value | Total | Good Response (CAP 0–1) * | Poor Response (CAP 2–3) ≠ | p-Value (Fisher’s Test) | OR | 95% CI |
|---|---|---|---|---|---|---|---|
| Age (years) | >50 | 35 | 15 | 20 | 0.487 | 2 | 0.434–7.793 |
| 50 | 11 | 3 | 8 | ||||
| Sex | Male | 33 | 15 | 18 | 0.197 | 2.778 | 0.673–10.54 |
| Female | 13 | 3 | 10 | ||||
| Treatment type | nCRT | 22 | 10 | 12 | 0.295 | 0.6 | 0.182–1.979 |
| TNT | 24 | 8 | 16 | ||||
| CEA | >3 ng/mL | 30 | 11 | 19 | 0.753 | 0.712 | 0.175–2.931 |
| 3 ng/mL | 16 | 7 | 9 | ||||
| Neutrophil/Platelet Score (NPS) | high (1 or 2) | 12 | 1 | 11 | 0.034 | 9.387 | 1.131–445.6 |
| low (0) | 34 | 16 | 18 | ||||
| MRI-assessed initial extramural vascular invasion (EMVI) | Yes | 17 | 6 | 11 | 0.761 | 0.773 | 0.237–2.454 |
| No | 29 | 12 | 17 | ||||
| MRI-assessed initial circumferential radial margin (CRM) | Positive | 38 | 15 | 23 | 1 | 1.087 | 0.223–4.578 |
| Negative | 8 | 3 | 5 | ||||
| Nodal status | N0 | 22 | 9 | 13 | 1 | 1.154 | 0.382–3.514 |
| N1 or higher | 24 | 9 | 15 | ||||
| Lateral lymph node dissemination | Yes | 9 | 2 | 7 | 0.447 | 0.4 | 0.077–1.854 |
| No | 36 | 15 | 21 | ||||
| Mismatch repair deficit | Yes | 2 | 0 | 2 | 0.513 | 0 | 0–3.348 |
| No | 44 | 18 | 26 | ||||
| RAS mutation | Yes | 20 | 4 | 16 | 0.023 | 0.167 | 0.049–0.663 |
| No | 20 | 12 | 8 | ||||
| TP53 mutation | Yes | 22 | 7 | 15 | 0.335 | 0.467 | 0.138–1.653 |
| No | 18 | 9 | 9 | ||||
| RAS/TP53 double mutation | Yes | 12 | 0 | 12 | 0.001 | 0 | 0–0.282 |
| No | 28 | 16 | 12 | ||||
| ATM mutation | Yes | 8 | 5 | 3 | 0.235 | 3.03 | 0.576–12.79 |
| No | 31 | 11 | 20 | ||||
| Consensus Molecular Subtype (CMS) | 2 | 35 | 14 | 21 | 1 | 0.674 | 0.044–10.31 |
| Other than 2 | 4 | 2 | 2 | ||||
| % CD20 in tumor (IHCProfiler) | high | 16 | 8 | 8 | 0.473 | 2.145 | 0.422–11.957 |
| low | 16 | 5 | 11 | ||||
| % CD20 in tumor (pathologists) | high | 19 | 8 | 11 | 1 | 1.038 | 0.227–4.794 |
| low | 17 | 7 | 10 | ||||
| B cell ImSig score (median) | High | 20 | 12 | 8 | 0.022 | 5.625 | 1.394–19.18 |
| Low | 19 | 4 | 15 | ||||
| Median Repair Pathway Score (MERPS) | Low | 19 | 13 | 6 | 0.001 | 12.28 | 2.548–47.65 |
| High | 20 | 3 | 17 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sendoya, J.M.; Iseas, S.; Coraglio, M.; Golubicki, M.; Robbio, J.; Salanova, R.; Kujaruk, M.; Mikolaitis, V.; Rizzolo, M.; Ruiz, G.; et al. Pre-Existing Tumoral B Cell Infiltration and Impaired Genome Maintenance Correlate with Response to Chemoradiotherapy in Locally Advanced Rectal Cancer. Cancers 2020, 12, 2227. https://doi.org/10.3390/cancers12082227
Sendoya JM, Iseas S, Coraglio M, Golubicki M, Robbio J, Salanova R, Kujaruk M, Mikolaitis V, Rizzolo M, Ruiz G, et al. Pre-Existing Tumoral B Cell Infiltration and Impaired Genome Maintenance Correlate with Response to Chemoradiotherapy in Locally Advanced Rectal Cancer. Cancers. 2020; 12(8):2227. https://doi.org/10.3390/cancers12082227
Chicago/Turabian StyleSendoya, Juan M., Soledad Iseas, Mariana Coraglio, Mariano Golubicki, Juan Robbio, Ruben Salanova, Mirta Kujaruk, Vanesa Mikolaitis, Mariana Rizzolo, Gonzalo Ruiz, and et al. 2020. "Pre-Existing Tumoral B Cell Infiltration and Impaired Genome Maintenance Correlate with Response to Chemoradiotherapy in Locally Advanced Rectal Cancer" Cancers 12, no. 8: 2227. https://doi.org/10.3390/cancers12082227
APA StyleSendoya, J. M., Iseas, S., Coraglio, M., Golubicki, M., Robbio, J., Salanova, R., Kujaruk, M., Mikolaitis, V., Rizzolo, M., Ruiz, G., Cabanne, A., Gualdrini, U., Mendez, G., Hirmas, S., Rotondaro, C., Viglino, J., Eleta, M., Fernandez, E., Abba, M., ... Llera, A. S. (2020). Pre-Existing Tumoral B Cell Infiltration and Impaired Genome Maintenance Correlate with Response to Chemoradiotherapy in Locally Advanced Rectal Cancer. Cancers, 12(8), 2227. https://doi.org/10.3390/cancers12082227