Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies

 , , ,
, , ,
Abstract
1. An Aging Population and Cancer
2. Biological Changes of Aging and Senescence
3. Regulatory Pathway of Senescence
3.1. Replicative Senescence
3.2. Oncogene-Induced Senescence
3.3. Stress-Induced Senescence
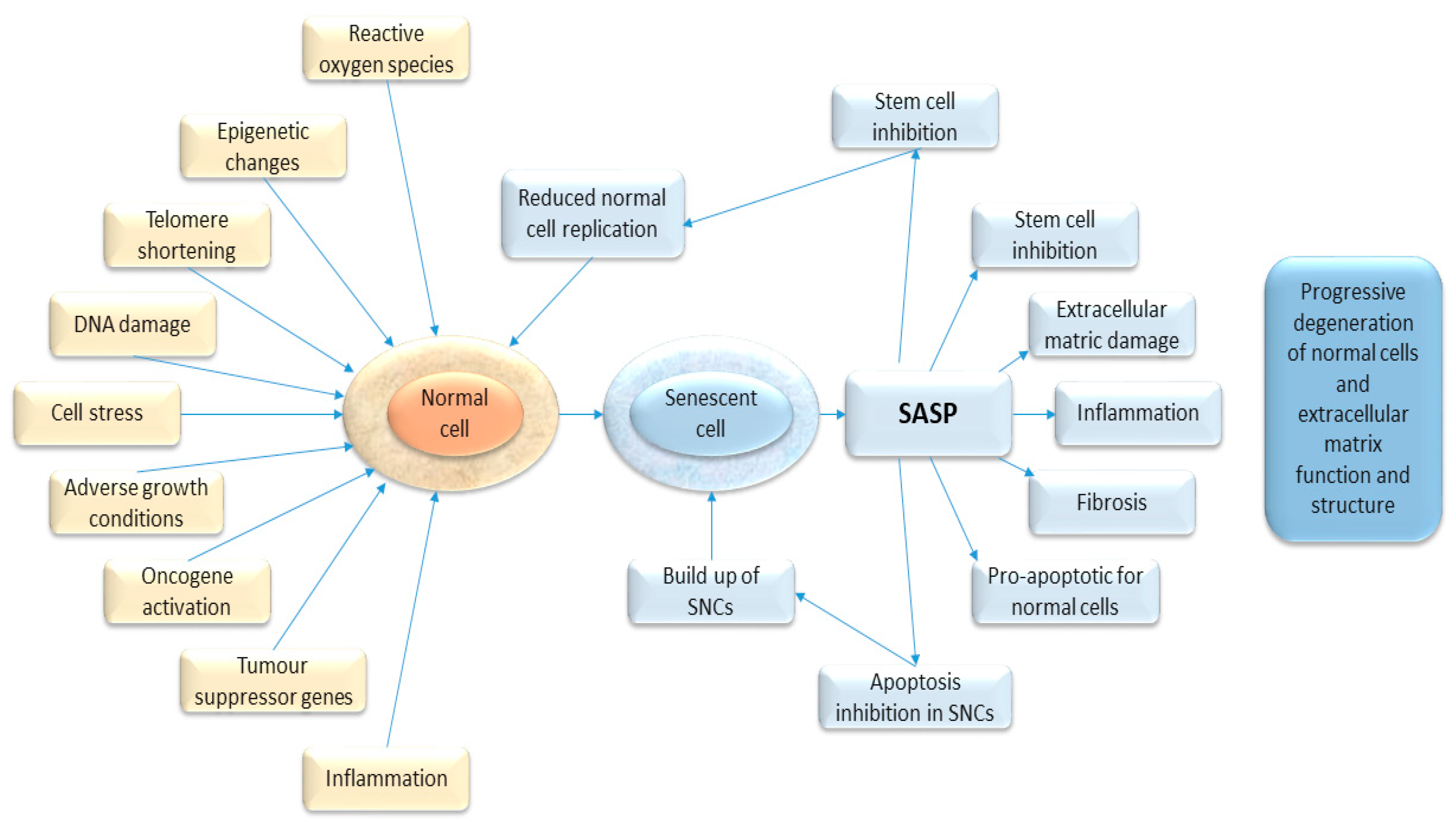
4. Senescence-Associated Secretory Phenotype, SASP
5. Role of Senescence in the Development of Cancer
6. Role of Senescence as a Prognostic Marker for Cancer
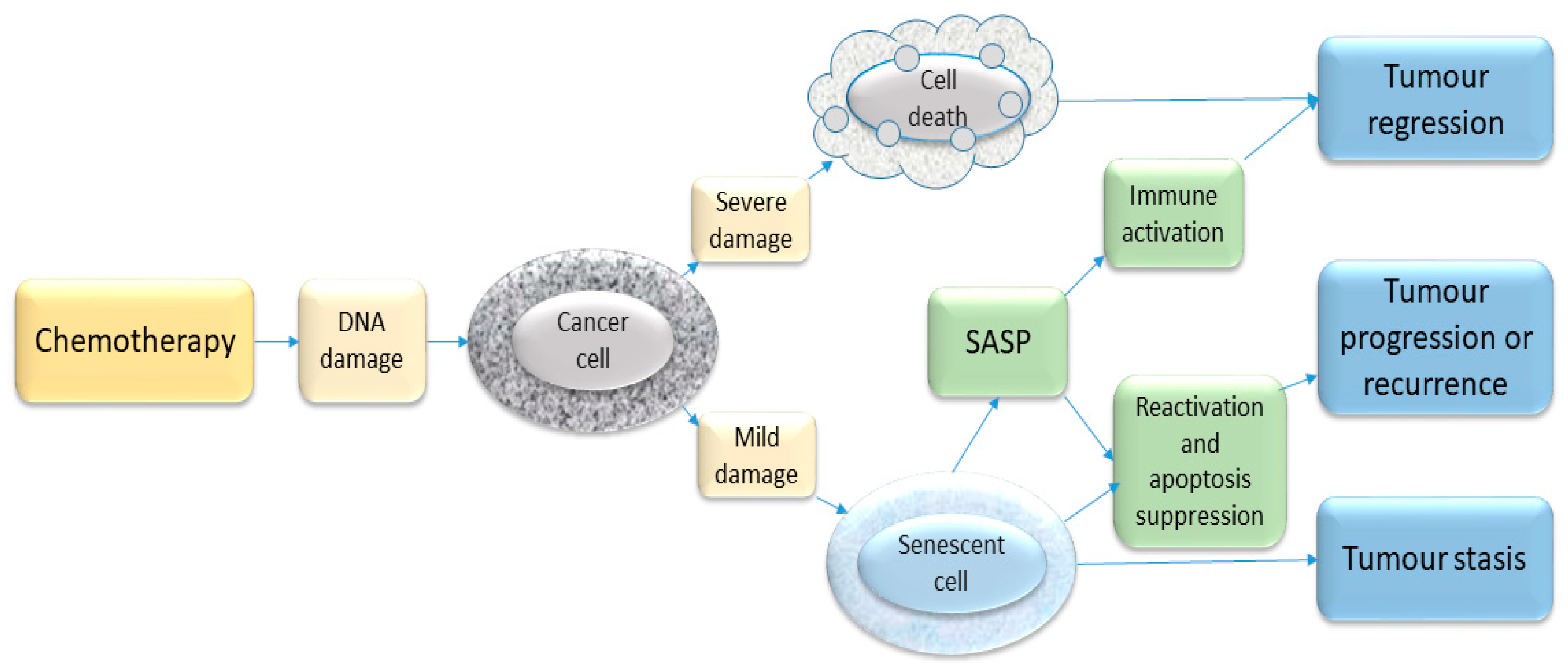
7. Role of Senescence in Cancer Treatment Response
Chemotherapy
8. Role of Senescence in Radiotherapy
9. Role of Senescence in Response to Hormonal Therapies (Anti-Oestrogens, Anti-Androgens)
10. Role of Senescence in Surgery
11. Frailty and Senescence Reducing Resilience to Cancer Therapies
12. Senotherapies
12.1. Navitoclax
12.2. Dasatinib Plus Quercetin
12.3. Fisetin
12.4. Metformin
12.5. Other Agents
13. Impact of Frailty on Cancer Treatment and Outcomes
14. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Oeppen, J.; Vaupel, J.W. Demography. Broken limits to life expectancy. Science 2002, 296, 1029–1031. [Google Scholar] [CrossRef]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef]
- Storey, A. Living Longer: How Our Population Is Changing and Why It Matters; Office for National Statistics: London, UK, 2018. [Google Scholar]
- Crimmins, E.M. Lifespan and Healthspan: Past, Present, and Promise. Gerontologist 2015, 55, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Pilleron, S.; Sarfati, D.; Janssen-Heijnen, M.; Vignat, J.; Ferlay, J.; Bray, F.; Soerjomataram, I. Global cancer incidence in older adults, 2012 and 2035: A population-based study. Int. J. Cancer 2019, 144, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing up to the global challenges of ageing. Nature 2018, 561, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Deelen, J.; Evans, D.S.; Arking, D.E.; Tesi, N.; Nygaard, M.; Liu, X.; Wojczynski, M.K.; Biggs, M.L.; van der Spek, A.; Atzmon, G.; et al. A meta-analysis of genome-wide association studies identifies multiple longevity genes. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef]
- Davaapil, H.; Brockes, J.P.; Yun, M.H. Conserved and novel functions of programmed cellular senescence during vertebrate development. Development 2017, 144, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Kauser, K.; Campisi, J.; Beausejour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Campisi, J.; Andersen, J.K.; Kapahi, P.; Melov, S. Cellular senescence: A link between cancer and age-related degenerative disease? Semin. Cancer Biol. 2011, 21, 354–359. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef]
- Bharadwaj, D.; Mandal, M. Senescence in polyploid giant cancer cells: A road that leads to chemoresistance. Cytokine Growth Factor Rev. 2020, 52, 68–75. [Google Scholar] [CrossRef]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Matjusaitis, M.; Chin, G.; Sarnoski, E.A.; Stolzing, A. Biomarkers to identify and isolate senescent cells. Ageing Res. Rev. 2016, 29, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Scriven, P.; Brown, N.J.; Pockley, A.G.; Wyld, L. The unfolded protein response and cancer: A brighter future unfolding? J. Mol. Med. 2007, 85, 331–341. [Google Scholar] [CrossRef]
- Sarkisian, C.J.; Keister, B.A.; Stairs, D.B.; Boxer, R.B.; Moody, S.E.; Chodosh, L.A. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell Biol. 2007, 9, 493–505. [Google Scholar] [CrossRef]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Haferkamp, S.; Tran, S.L.; Becker, T.M.; Scurr, L.L.; Kefford, R.F.; Rizos, H. The relative contributions of the p53 and pRb pathways in oncogene-induced melanocyte senescence. Aging 2009, 1, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006, 66, 794–802. [Google Scholar] [CrossRef]
- Falci, C.; Gianesin, K.; Sergi, G.; Giunco, S.; De Ronch, I.; Valpione, S.; Solda, C.; Fiduccia, P.; Lonardi, S.; Zanchetta, M.; et al. Immune senescence and cancer in elderly patients: Results from an exploratory study. Exp. Gerontol. 2013, 48, 1436–1442. [Google Scholar] [CrossRef]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Jeyapalan, J.C.; Ferreira, M.; Sedivy, J.M.; Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007, 128, 36–44. [Google Scholar] [CrossRef]
- Waaijer, M.E.; Parish, W.E.; Strongitharm, B.H.; van Heemst, D.; Slagboom, P.E.; de Craen, A.J.; Sedivy, J.M.; Westendorp, R.G.; Gunn, D.A.; Maier, A.B. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, 11, 722–725. [Google Scholar] [CrossRef]
- Pare, R.; Soon, P.S.; Shah, A.; Lee, C.S. Differential expression of senescence tumour markers and its implications on survival outcomes of breast cancer patients. PLoS ONE 2019, 14, e0214604. [Google Scholar] [CrossRef]
- Shin, E.; Jung, W.H.; Koo, J.S. Expression of p16 and pRB in invasive breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8209–8217. [Google Scholar] [PubMed]
- Pare, R.; Shin, J.S.; Lee, C.S. Increased expression of senescence markers p14(ARF) and p16(INK4a) in breast cancer is associated with an increased risk of disease recurrence and poor survival outcome. Histopathology 2016, 69, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.; Schrauder, M.; Oswald, U.; Knoll, C.; Sellberg, P.; Palmqvist, R.; Niedobitek, G.; Brabletz, T.; Kirchner, T. The invasion front of human colorectal adenocarcinomas shows co-localization of nuclear beta-catenin, cyclin D1, and p16INK4A and is a region of low proliferation. Am. J. Pathol. 2001, 159, 1613–1617. [Google Scholar] [CrossRef]
- Horree, N.; van Diest, P.J.; Sie-Go, D.M.; Heintz, A.P. The invasive front in endometrial carcinoma: Higher proliferation and associated derailment of cell cycle regulators. Hum. Pathol. 2007, 38, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.H.; Yang, L.; Zhang, X.; Ma, X.H.; Miao, R.C.; Gu, J.X.; Fu, Y.N.; Yao, Q.; Zhang, J.Y.; Liu, C.; et al. Seven-senescence-associated gene signature predicts overall survival for Asian patients with hepatocellular carcinoma. World J. Gastroenterol. WJG 2019, 25, 1715–1728. [Google Scholar] [CrossRef]
- Schenker, H.; Buttner-Herold, M.; Fietkau, R.; Distel, L.V. Cell-in-cell structures are more potent predictors of outcome than senescence or apoptosis in head and neck squamous cell carcinomas. Radiat. Oncol. 2017, 12, 21. [Google Scholar] [CrossRef]
- Roxburgh, C.S.; Richards, C.H.; Macdonald, A.I.; Powell, A.G.; McGlynn, L.M.; McMillan, D.C.; Horgan, P.G.; Edwards, J.; Shiels, P.G. The in situ local immune response, tumour senescence and proliferation in colorectal cancer. Br. J. Cancer 2013, 109, 2207–2216. [Google Scholar] [CrossRef]
- Calio, A.; Zamo, A.; Ponzoni, M.; Zanolin, M.E.; Ferreri, A.J.; Pedron, S.; Montagna, L.; Parolini, C.; Fraifeld, V.E.; Wolfson, M.; et al. Cellular Senescence Markers p16INK4a and p21CIP1/WAF Are Predictors of Hodgkin Lymphoma Outcome. Clin. Cancer Res. 2015, 21, 5164–5172. [Google Scholar] [CrossRef]
- Mo, Z.; Zheng, S.; Lv, Z.; Zhuang, Y.; Lan, X.; Wang, F.; Lu, X.; Zhao, Y.; Zhou, S. Senescence marker protein 30 (SMP30) serves as a potential prognostic indicator in hepatocellular carcinoma. Sci. Rep. 2016, 6, 39376. [Google Scholar] [CrossRef]
- Macher-Goeppinger, S.; Bermejo, J.L.; Schirmacher, P.; Pahernik, S.; Hohenfellner, M.; Roth, W. Senescence-associated protein p400 is a prognostic marker in renal cell carcinoma. Oncol. Rep. 2013, 30, 2245–2253. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar] [PubMed]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar] [PubMed]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008, 68, 7864–7871. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef]
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.A.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Munoz, D.P.; Yannone, S.M.; Daemen, A.; Sun, Y.; Vakar-Lopez, F.; Kawahara, M.; Freund, A.M.; Rodier, F.; Wu, J.D.; Desprez, P.Y.; et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- You, R.; Dai, J.; Zhang, P.; Barding, G.A., Jr.; Raftery, D. Dynamic Metabolic Response to Adriamycin-Induced Senescence in Breast Cancer Cells. Metabolites 2018, 8, 95. [Google Scholar] [CrossRef]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.Y.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.; Schlegelberger, B.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.C.; Ghadaouia, S.; Martinez, A.; Rodier, F. Premature aging/senescence in cancer cells facing therapy: Good or bad? Biogerontology 2016, 17, 71–87. [Google Scholar] [CrossRef]
- Zhang, B.; Lam, E.W.; Sun, Y. Senescent cells: A new Achilles’ heel to exploit for cancer medicine? Aging Cell 2019, 18, e12875. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef]
- Tsolou, A.; Lamprou, I.; Fortosi, A.O.; Liousia, M.; Giatromanolaki, A.; Koukourakis, M.I. ‘Stemness’ and ‘senescence’ related escape pathways are dose dependent in lung cancer cells surviving post irradiation. Life Sci. 2019, 232, 116562. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Kaur, E.; Rajendra, J.; Jadhav, S.; Shridhar, E.; Goda, J.S.; Moiyadi, A.; Dutt, S. Radiation-induced homotypic cell fusions of innately resistant glioblastoma cells mediate their sustained survival and recurrence. Carcinogenesis 2015, 36, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, E.; Vanpouille-Box, C.; Bornstein, S.; Yamazaki, T.; Demaria, S.; Galluzzi, L. Immune recognition of irradiated cancer cells. Immunol. Rev. 2017, 280, 220–230. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Thummuri, D.; Zheng, G.; Okunieff, P.; Citrin, D.E.; Vujaskovic, Z.; Zhou, D. Cellular senescence and radiation-induced pulmonary fibrosis. Transl. Res. 2019, 209, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Z.; Huang, Y.; Zhou, Y.; Sheng, X.; Jiang, Q.; Wang, Y.; Luo, P.; Luo, M.; Shi, C. Senolytics (DQ) Mitigates Radiation Ulcers by Removing Senescent Cells. Front. Oncol. 2019, 9, 1576. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kang, B.S.; Bae, Y.S. Premature senescence in human breast cancer and colon cancer cells by tamoxifen-mediated reactive oxygen species generation. Life Sci. 2014, 97, 116–122. [Google Scholar] [CrossRef]
- Pernicova, Z.; Slabakova, E.; Kharaishvili, G.; Bouchal, J.; Kral, M.; Kunicka, Z.; Machala, M.; Kozubik, A.; Soucek, K. Androgen depletion induces senescence in prostate cancer cells through down-regulation of Skp2. Neoplasia 2011, 13, 526–536. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef]
- Wilkinson, H.N.; Clowes, C.; Banyard, K.L.; Matteuci, P.; Mace, K.A.; Hardman, M.J. Elevated Local Senescence in Diabetic Wound Healing Is Linked to Pathological Repair via CXCR2. J. Investig. Dermatol. 2019, 139, 1171–1181.e1176. [Google Scholar] [CrossRef]
- Stanley, A.; Osler, T. Senescence and the healing rates of venous ulcers. J. Vasc. Surg. 2001, 33, 1206–1211. [Google Scholar] [CrossRef]
- Guo, S.; Dipietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Ikemoto, T.; Utsunomiya, T.; Yamada, S.; Morine, Y.; Imura, S.; Arakawa, Y.; Takasu, C.; Ishikawa, D.; Shimada, M. Senescence-related genes possibly responsible for poor liver regeneration after hepatectomy in elderly patients. J. Gastroenterol. Hepatol. 2014, 29, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Koppelstaetter, C.; Schratzberger, G.; Perco, P.; Hofer, J.; Mark, W.; Ollinger, R.; Oberbauer, R.; Schwarz, C.; Mitterbauer, C.; Kainz, A.; et al. Markers of cellular senescence in zero hour biopsies predict outcome in renal transplantation. Aging Cell 2008, 7, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Moug, S.J.; Stechman, M.; McCarthy, K.; Pearce, L.; Myint, P.K.; Hewitt, J. Frailty and cognitive impairment: Unique challenges in the older emergency surgical patient. Ann. R. Coll. Surg. Engl. 2016, 98, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Bratzke, L.C.; Koscik, R.L.; Schenning, K.J.; Clark, L.R.; Sager, M.A.; Johnson, S.C.; Hermann, B.P.; Hogan, K.J. Cognitive decline in the middle-aged after surgery and anaesthesia: Results from the Wisconsin Registry for Alzheimer’s Prevention cohort. Anaesthesia 2018, 73, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Adjogatse, D.; Thanopoulou, E.; Okines, A.; Thillai, K.; Tasker, F.; Johnston, S.R.; Harper-Wynne, C.; Torrisi, E.; Ring, A. Febrile neutropaenia and chemotherapy discontinuation in women aged 70 years or older receiving adjuvant chemotherapy for early breast cancer. Clin. Oncol. 2014, 26, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Zampino, M.; Ferrucci, L.; Semba, R.D. Biomarkers in the path from cellular senescence to frailty. Exp. Gerontol. 2020, 129, 110750. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gualda, E.; Paez-Ribes, M.; Lozano-Torres, B.; Macias, D.; Wilson, J.R., 3rd; Gonzalez-Lopez, C.; Ou, H.L.; Miron-Barroso, S.; Zhang, Z.; Lerida-Viso, A.; et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 2020, 19, e13142. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.; Malek, M.; Zha, J.; Yue, P.; Kassees, R.; Berry, L.; Fairbrother, W.J.; Sampath, D.; Belmont, L.D. Navitoclax enhances the efficacy of taxanes in non-small cell lung cancer models. Clin. Cancer Res. 2011, 17, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Stamelos, V.A.; Robinson, E.; Redman, C.W.; Richardson, A. Navitoclax augments the activity of carboplatin and paclitaxel combinations in ovarian cancer cells. Gynecol. Oncol. 2013, 128, 377–382. [Google Scholar] [CrossRef]
- Jeong, J.H.; Oh, J.M.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Combination Treatment with the BRAF(V600E) Inhibitor Vemurafenib and the BH3 Mimetic Navitoclax for BRAF-Mutant Thyroid Carcinoma. Thyroid 2019, 29, 540–548. [Google Scholar] [CrossRef]
- Nakajima, W.; Sharma, K.; Hicks, M.A.; Le, N.; Brown, R.; Krystal, G.W.; Harada, H. Combination with vorinostat overcomes ABT-263 (navitoclax) resistance of small cell lung cancer. Cancer Biol. Ther. 2016, 17, 27–35. [Google Scholar] [CrossRef]
- Ackler, S.; Mitten, M.J.; Chen, J.; Clarin, J.; Foster, K.; Jin, S.; Phillips, D.C.; Schlessinger, S.; Wang, B.; Leverson, J.D.; et al. Navitoclax (ABT-263) and bendamustine +/- rituximab induce enhanced killing of non-Hodgkin’s lymphoma tumours in vivo. Br. J. Pharmacol. 2012, 167, 881–891. [Google Scholar] [CrossRef]
- Tolcher, A.W.; LoRusso, P.; Arzt, J.; Busman, T.A.; Lian, G.; Rudersdorf, N.S.; Vanderwal, C.A.; Waring, J.F.; Yang, J.; Holen, K.D.; et al. Safety, efficacy, and pharmacokinetics of navitoclax (ABT-263) in combination with irinotecan: Results of an open-label, phase 1 study. Cancer Chemother. Pharmacol. 2015, 76, 1041–1049. [Google Scholar] [CrossRef]
- Kipps, T.J.; Eradat, H.; Grosicki, S.; Catalano, J.; Cosolo, W.; Dyagil, I.S.; Yalamanchili, S.; Chai, A.; Sahasranaman, S.; Punnoose, E.; et al. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk. Lymphoma 2015, 56, 2826–2833. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; Ribeiro de Oliveira, M.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Wissler Gerdes, E.O.; Zhu, Y.; Tchkonia, T.; Kirkland, J.L. Discovery, development, and future application of senolytics: Theories and predictions. FEBS J. 2020, 287, 2418–2427. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Kirkland, J.L. Aging, Cell Senescence, and Chronic Disease: Emerging Therapeutic Strategies. JAMA 2018, 320, 1319–1320. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY) 2017, 9, 955–963. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Maher, P. How fisetin reduces the impact of age and disease on CNS function. Front. Biosci. 2015, 7, 58–82. [Google Scholar] [CrossRef]
- Kashyap, D.; Sharma, A.; Sak, K.; Tuli, H.S.; Buttar, H.S.; Bishayee, A. Fisetin: A bioactive phytochemical with potential for cancer prevention and pharmacotherapy. Life Sci. 2018, 194, 75–87. [Google Scholar] [CrossRef]
- Li, J.; Gong, X.; Jiang, R.; Lin, D.; Zhou, T.; Zhang, A.; Li, H.; Zhang, X.; Wan, J.; Kuang, G.; et al. Fisetin Inhibited Growth and Metastasis of Triple-Negative Breast Cancer by Reversing Epithelial-to-Mesenchymal Transition via PTEN/Akt/GSK3beta Signal Pathway. Front. Pharmacol. 2018, 9, 772. [Google Scholar] [CrossRef]
- Xiao, X.; Zou, J.; Fang, Y.; Meng, Y.; Xiao, C.; Fu, J.; Liu, S.; Bai, P.; Yao, Y. Fisetin and polymeric micelles encapsulating fisetin exhibit potent cytotoxic effects towards ovarian cancer cells. BMC Complement. Altern. Med. 2018, 18, 91. [Google Scholar] [CrossRef]
- Jia, S.; Xu, X.; Zhou, S.; Chen, Y.; Ding, G.; Cao, L. Fisetin induces autophagy in pancreatic cancer cells via endoplasmic reticulum stress- and mitochondrial stress-dependent pathways. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Youns, M.; Abdel Halim Hegazy, W. The Natural Flavonoid Fisetin Inhibits Cellular Proliferation of Hepatic, Colorectal, and Pancreatic Cancer Cells through Modulation of Multiple Signaling Pathways. PLoS ONE 2017, 12, e0169335. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Chen, S.; Zhao, Y.; Ye, X. Fisetin inhibits the proliferation of gastric cancer cells and induces apoptosis through suppression of ERK 1/2 activation. Oncol. Lett. 2018, 15, 8442–8446. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.M.; Tseng, H.H.; Peng, C.W.; Chen, W.S.; Chiu, S.J. Dietary flavonoid fisetin targets caspase-3-deficient human breast cancer MCF-7 cells by induction of caspase-7-associated apoptosis and inhibition of autophagy. Int. J. Oncol. 2012, 40, 469–478. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lin, M.T.; Lin, C.L.; Lin, T.Y.; Cheng, C.W.; Yang, S.F.; Lin, C.L.; Wu, C.C.; Hsieh, Y.H.; Tsai, J.P. Synergistic effect of fisetin combined with sorafenib in human cervical cancer HeLa cells through activation of death receptor-5 mediated caspase-8/caspase-3 and the mitochondria-dependent apoptotic pathway. Tumour Biol. 2016, 37, 6987–6996. [Google Scholar] [CrossRef] [PubMed]
- Pal, H.C.; Baxter, R.D.; Hunt, K.M.; Agarwal, J.; Elmets, C.A.; Athar, M.; Afaq, F. Fisetin, a phytochemical, potentiates sorafenib-induced apoptosis and abrogates tumor growth in athymic nude mice implanted with BRAF-mutated melanoma cells. Oncotarget 2015, 6, 28296–28311. [Google Scholar] [CrossRef]
- Khan, N.; Jajeh, F.; Eberhardt, E.L.; Miller, D.D.; Albrecht, D.M.; Van Doorn, R.; Hruby, M.D.; Maresh, M.E.; Clipson, L.; Mukhtar, H.; et al. Fisetin and 5-fluorouracil: Effective combination for PIK3CA-mutant colorectal cancer. Int. J. Cancer 2019, 145, 3022–3032. [Google Scholar] [CrossRef]
- Zhuo, W.; Zhang, L.; Zhu, Y.; Zhu, B.; Chen, Z. Fisetin, a dietary bioflavonoid, reverses acquired Cisplatin-resistance of lung adenocarcinoma cells through MAPK/Survivin/Caspase pathway. Am. J. Transl. Res. 2015, 7, 2045–2052. [Google Scholar]
- Touil, Y.S.; Seguin, J.; Scherman, D.; Chabot, G.G. Improved antiangiogenic and antitumour activity of the combination of the natural flavonoid fisetin and cyclophosphamide in Lewis lung carcinoma-bearing mice. Cancer Chemother. Pharmacol. 2011, 68, 445–455. [Google Scholar] [CrossRef]
- Farsad-Naeimi, A.; Alizadeh, M.; Esfahani, A.; Darvish Aminabad, E. Effect of fisetin supplementation on inflammatory factors and matrix metalloproteinase enzymes in colorectal cancer patients. Food Funct. 2018, 9, 2025–2031. [Google Scholar] [CrossRef]
- Kirkland, J.L. Alleviation by Fisetin of Frailty, Inflammation, and Related Measures in Older Adults (AFFIRM-LITE.; NCT03675724). Available online: https://clinicaltrials.gov/ct2/show/NCT03675724 (accessed on 29 July 2020).
- Moiseeva, O.; Deschenes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Bellman, S.M.; Stephenson, M.D.; Lisy, K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: A systematic review and meta-analysis. Ageing Res. Rev. 2017, 40, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D. Hsp90 inhibitors as senolytic drugs to extend healthy aging. Cell Cycle 2018, 17, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M. Hematopoietic Stem Cell Transplant Survivors Study (HTSS Study; NCT02652052). Clinical TrialsGov; 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT02652052 (accessed on 29 July 2020).



{kind=link}
{kind=link}
| Characteristic | Marker |
|---|---|
| Proliferative arrest | Low expression of Ki-67 or bromodeoxyuridine BrdU |
| Persistent activation of the DNA damage response | Activation of tumour suppressors, such as p53, p16INK4A, cyclins, and cyclin-dependent kinases |
| Heterochromatic foci (DNA becomes denser than normal) | Senescence-associated heterochromatic foci on DNA staining with DAPI |
| Cells become flattened and enlarged | Light microscopy changes |
| Altered metabolism including increased β-galactosidase activity, which is part of carbohydrate metabolism | Measurement of β-galactosidase levels |
| Senescence-associated secretory phenotype | Interleukins-1, -6, and -8, matrix metalloproteinases, plasminogen activator inhibitor-1 |
| Model | Cancer Type | Prognostic Significance of Senescence | Reference |
|---|---|---|---|
| Ex vivo human tumours | Breast | Senescence indicates better survival | Althubiti et al. 2014 [29] |
| Ex vivo human tumours | Hepatocellular carcinoma | Panel of seven senescence associated genes has prognostic significance | Xiang et al. 2019 [46] |
| Ex vivo human tumours | Squamous head and neck cancer | Senescent cells associated with a non-significant trend to improved prognosis | Schenker et al. 2017 [47] |
| Ex vivo human tumours | Colorectal cancer | Lower levels of senescence associated with poorer survival | Roxburgh et al. 2013 [48] |
| Ex vivo human tumours | Lymph node tissue from Hodgkin Lymphoma | High levels of senescence marker expression linked to improved prognosis | Calio et al. 2015 [49] |
| Ex vivo human tumours | Hepatocellular carcinoma | Low levels of senescence linked to poor prognosis | Mo et al. 2016 [50] |
| Ex vivo human tumours | Renal cell cancer | Low levels of senescence linked to worse prognosis. | Macher-Goeppinger et al. 2013 [51] |
| Reference | Model | Drug | Cancer Type | Effect |
|---|---|---|---|---|
| Tan et al. 2011 [94] | In vitro | Naviticlax+Paclitaxel | Non-small cell lung cancer | More than additive cell killing with combination |
| Stamelos et al. 2013 [95] | In vitro | Naviticlax+Paclitaxel or Carboplatin | Ovarian cancer | More than additive cell killing with combination |
| Jeong et al. 2019 [96] | In vitro | Vemurafenib+Navitoclax | Papillary thyroid cancer | Enhanced growth arrest and increase apoptosis with combination |
| Nakajima et al. 2016 [97] | In vitro | Vorinostat+Navitoclax | Small cell lung cancer | Increased induction of apoptosis with combination |
| Gonzalez-Gualda et al. 2020 [93] | In vitro and ex vitro | Galactose conjugated Navitoclax+Cisplatin | Lung cancer | Increased cell killing with combination. Reduced platelet toxicity in ex vivo blood |
| Ackler et al. 2012 [98] | In vivo mouse model | Bendamustine+Navitoclax±Rituximab | Non-Hodgkins lymphoma | Enhanced efficacy in combination |
| Tolcher et al. 2015 [99] | Phase 1 clinical trial | Irinotecan+Navitoclax | Advanced solid tumours, n = 31 | 6% rate of partial response |
| Kipps et al. 2015 [100] | Phase 2 clinical trial | Rituximab+Navitoclax | Chronic lymphocytic lymphoma | Combination increase progression free survival and response rates |
| Rudin et al. 2012 [101] | Phase 2 clinical trial | Single agent navitoclax | Relapsed small cell lung cancer, n = 39 | 23% static disease, 2.6% partial response |
| Reference | Model | Drug | Cancer Type | Effect |
|---|---|---|---|---|
| Li et al., 2018 [110] | In vitro and in vivo mouse xenograft | Fisetin alone | Triple negative breast cancer | Inhibition of proliferation, migration and metastases |
| Xiao et al., 2018 [111] | In vitro and in vivo mouse xenograft | Fisetin and fisetin micelles | Ovarian cancer | Antiproliferative and proapoptotic effects |
| Jia et al., 2019 [112] | In vitro and in vivo mouse xenograft | Fisetin alone | Pancreatic cancer | Antiproliferative |
| Youns et al., 2017 [113] | In vitro | Fisetin alone | Hepatic, colorectal, and pancreatic | Growth arrest and apoptosis |
| Yan et al., 2018 [114] | In vitro | Fisetin alone | Gastric cancer | Antiproliferative and pro-apoptotic |
| Yang et al., 2012 [115] | In vitro | Fisetin alone | Breast cancer | Induction of apoptosis |
| Lin et al., 2015 [116] | In vitro and in vivo | Fisetin and sorafenib | Cervical cancer | Combination superior to either agent alone in anticancer efficacy |
| Pal et al., 2015 [117] | In vivo mouse model | Fisetin and sorafenib | BRAF mutated melanoma cells | Reduced proliferation, increased apoptosis and reduced metastases in combination |
| Khan et al., 2019 [118] | In vivo mouse model | Fisetin and 5FU | Colorectal cancer | Reduced incidence of colorectal cancer formation with Fisetin alone and in combination |
| Zhuo et al., 2015 [119] | In vitro | Fisetin and cisplatin | Lung adenocarcinoma | Increased apoptosis and decreased viability with combination |
| Touil et al., 2011 [120] | In vivo mouse model | Fisetin and cyclophosphamide | Lung cancer | 92% growth inhibition of combination compared to single agent |
| Farsad-Naemi et al., 2018 [121] | Randomised clinical trial of dietary supplement dose of Fisetin | Oxaliplatin and capecitabine chemotherapy ± Fisetin 100 mg daily for 7 weeks | Colorectal cancer | Reduced levels of inflammatory mediators (IL8, CRP and MMP7) in Fisetin group. Tumour response was not reported |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. https://doi.org/10.3390/cancers12082134
Wyld L, Bellantuono I, Tchkonia T, Morgan J, Turner O, Foss F, George J, Danson S, Kirkland JL. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers. 2020; 12(8):2134. https://doi.org/10.3390/cancers12082134
Chicago/Turabian StyleWyld, Lynda, Ilaria Bellantuono, Tamara Tchkonia, Jenna Morgan, Olivia Turner, Fiona Foss, Jayan George, Sarah Danson, and James L. Kirkland. 2020. "Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies" Cancers 12, no. 8: 2134. https://doi.org/10.3390/cancers12082134
APA StyleWyld, L., Bellantuono, I., Tchkonia, T., Morgan, J., Turner, O., Foss, F., George, J., Danson, S., & Kirkland, J. L. (2020). Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers, 12(8), 2134. https://doi.org/10.3390/cancers12082134