TrkB Inhibits the BMP Signaling-Mediated Growth Inhibition of Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
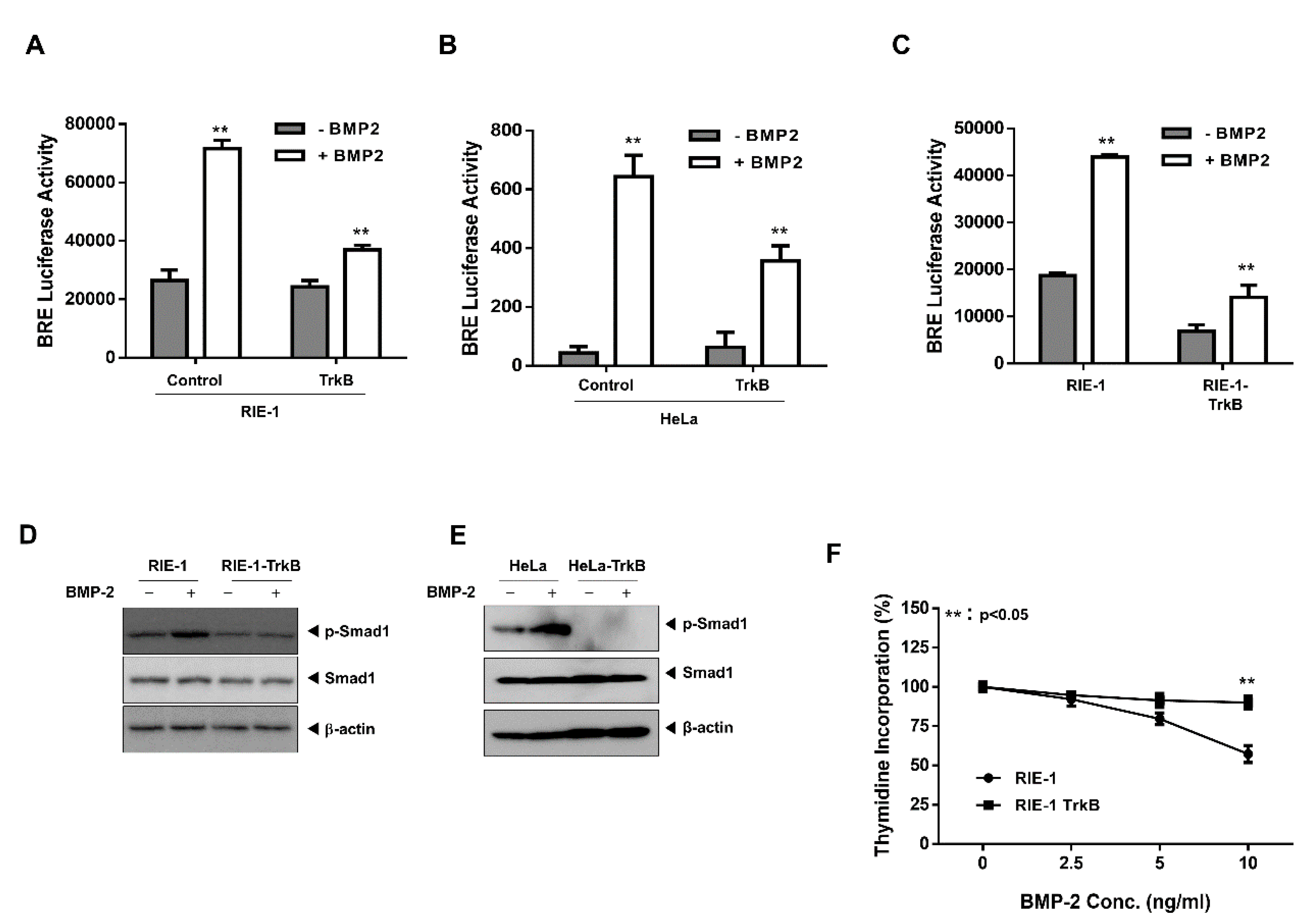
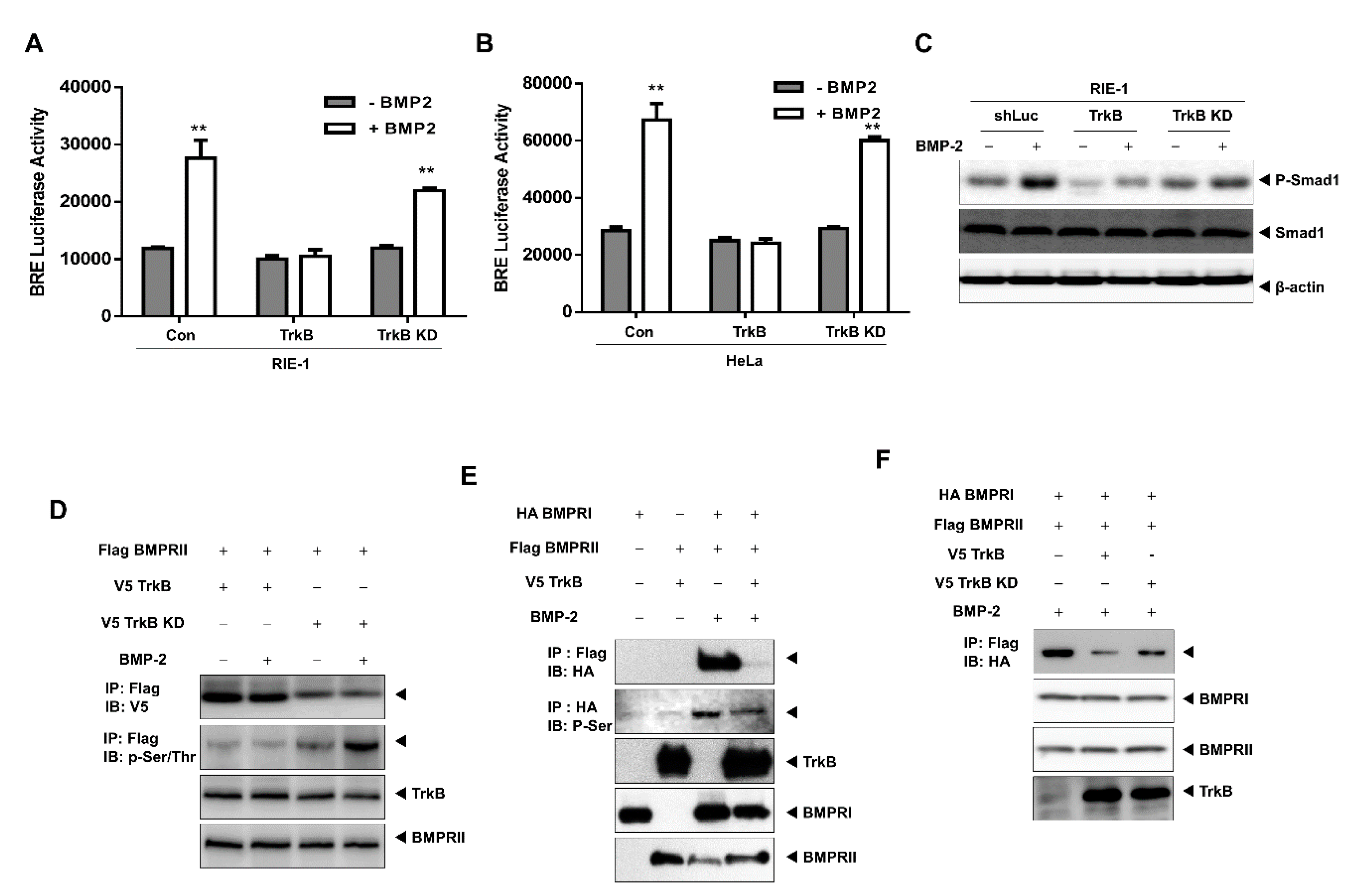
2.1. TrkB Expression Inhibits BMP Signaling Cascade
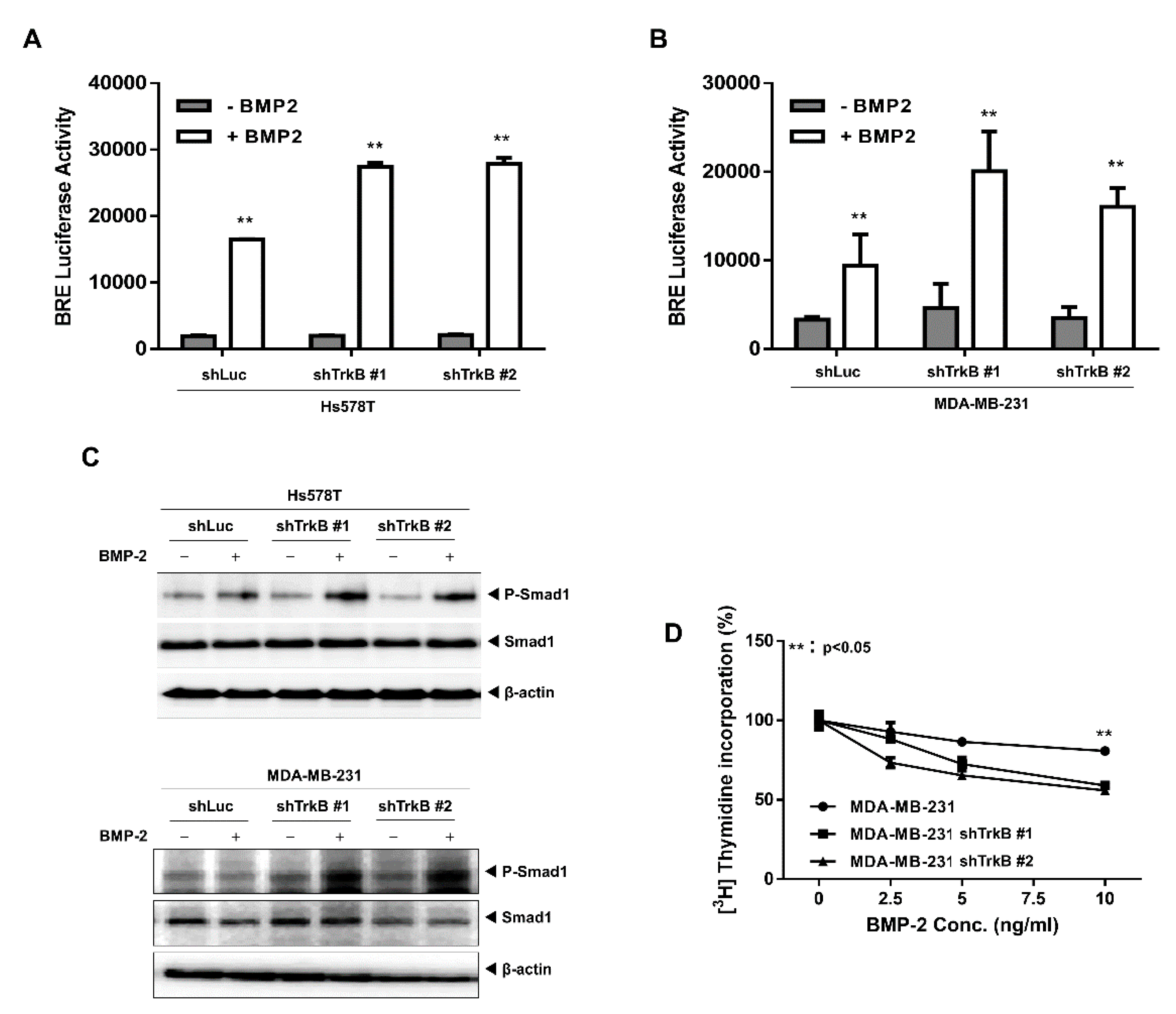
2.2. The Loss of TrkB Restores BMP-Mediated Tumor-Suppressive Activities
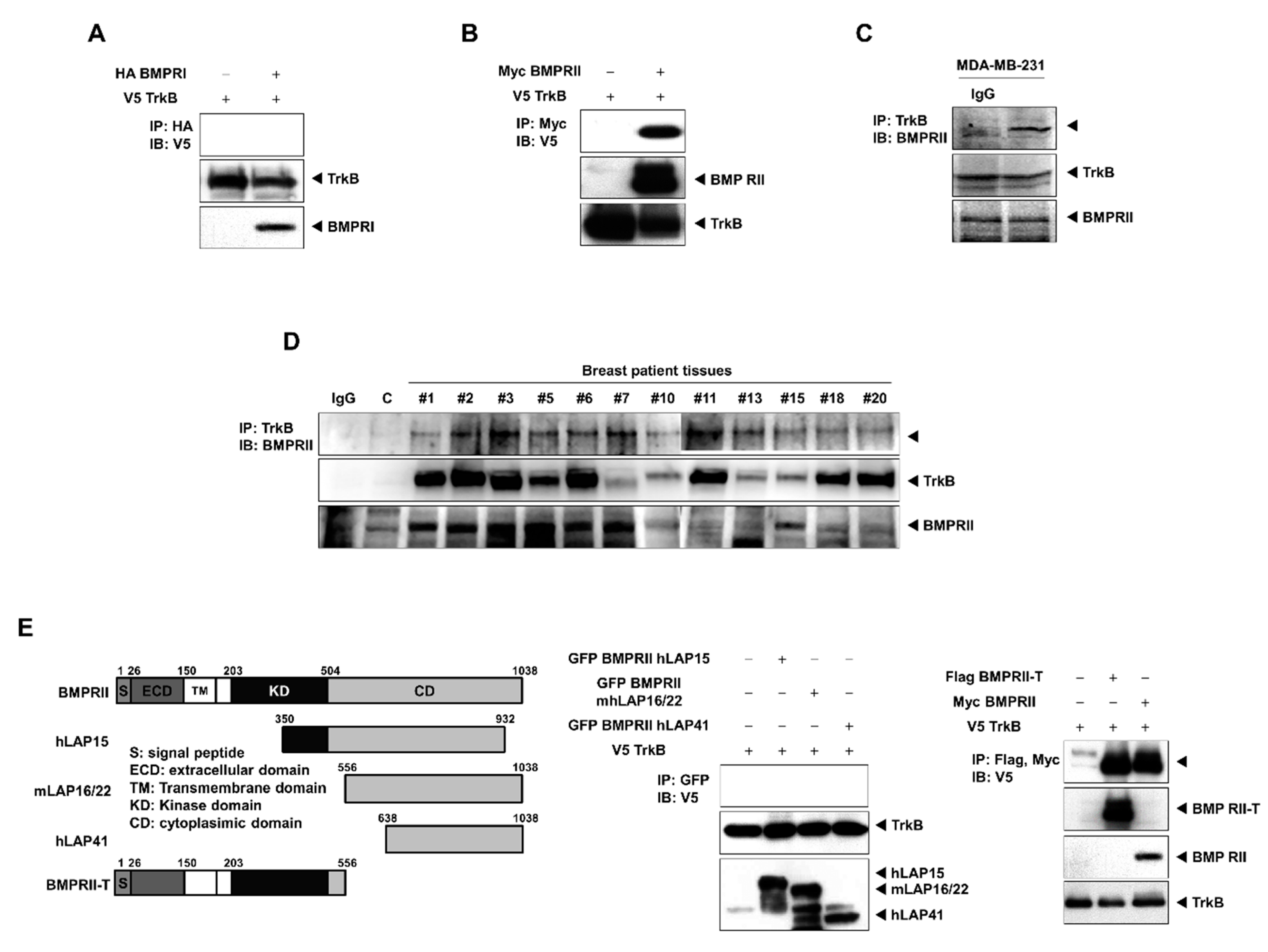
2.3. TrkB Directly Interacts with BMP Type II Receptors to Inhibit BMP Signaling
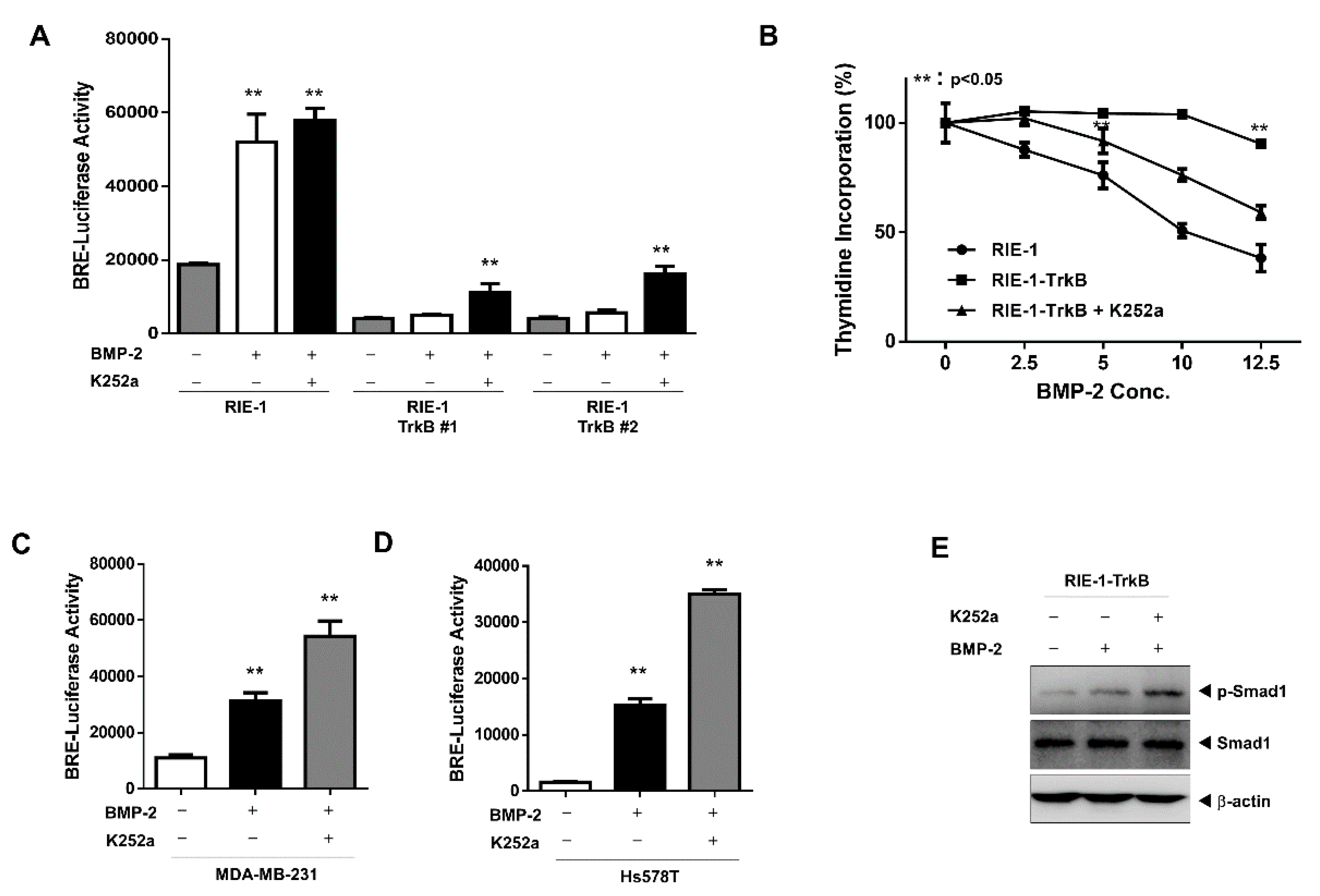
2.4. The Tyrosine Kinase Activity of TrkB is Required for the Inhibition of BMP-2 Signaling
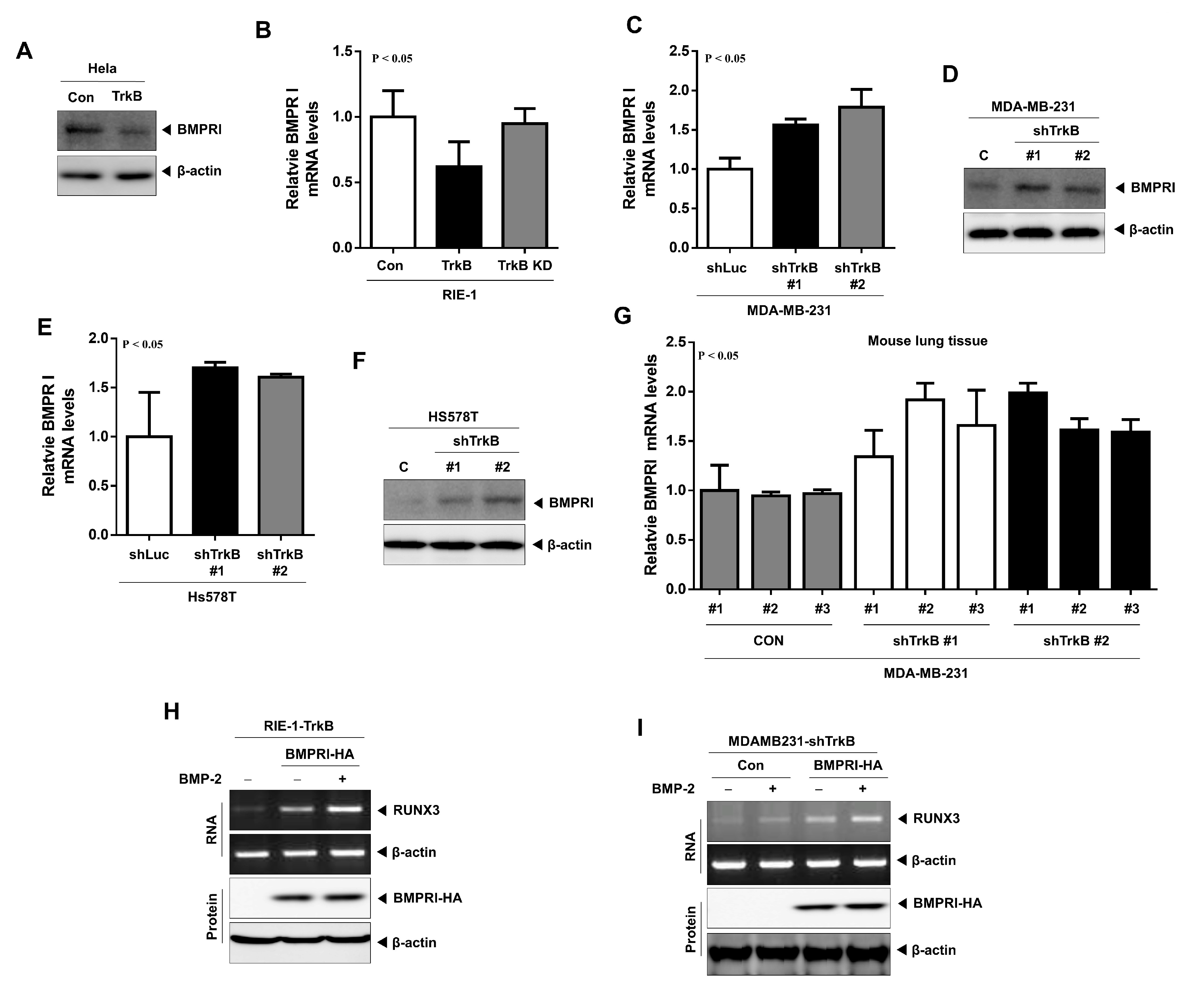
2.5. TrkB Regulates BMP Type I Receptor Expression to Inhibit BMP Signaling
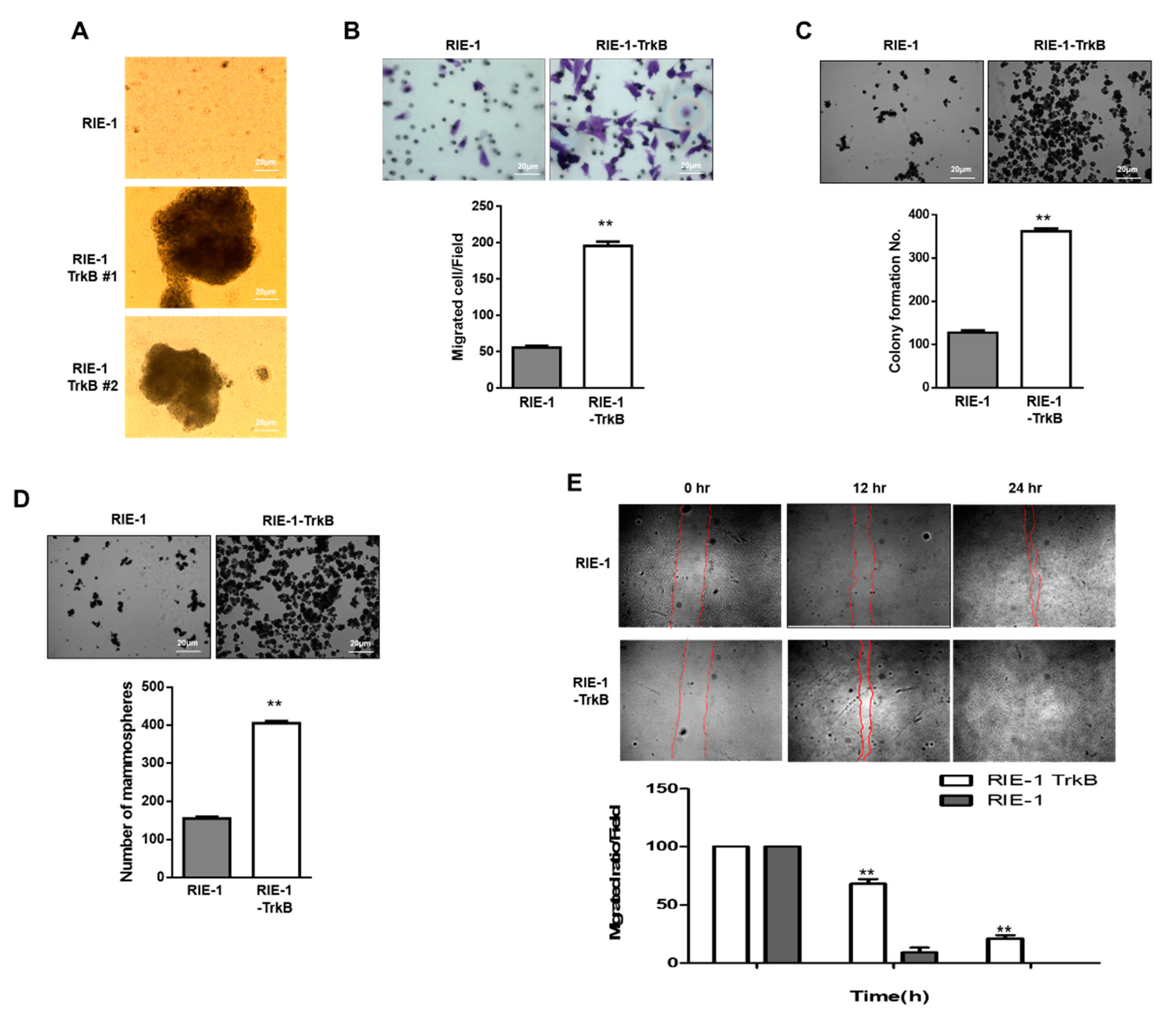
2.6. TrkB Promotes Invasion Ability of RIE-1 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Culture Conditions and Chemical Inhibitors
4.2. Plasmids and Viral Production
4.3. Human Breast Tumor Samples
4.4. Antibodies, Western Blotting, Immunoprecipitation, and Immunofluorescence
4.5. Invasion, Anoikis, Wound Healing, Anchorage-Independent Cell Growth, and Mammosphere Assays
4.6. Luciferase Reporter Assay
4.7. RNA Preparation and RT-PCR Analysis
4.8. Cell Proliferation Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.H.; Park, H.J.; Lee, S.K. The dual role of bone morphogenetic proteins in cancer. Mol. Ther. Oncolytics 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Alarmo, E.L.; Kallioniemi, A. Bone morphogenetic proteins in breast cancer: Dual role in tumourigenesis? Endocr. Relat. Cancer 2010, 17, R123–R139. [Google Scholar] [CrossRef]
- Hogan, B.L. Bone morphogenetic proteins: Multifunctional regulators of vertebrate development. Genes Dev. 1996, 10, 1580–1594. [Google Scholar] [CrossRef]
- Kim, I.Y.; Lee, D.H.; Ahn, H.J.; Tokunaga, H.; Song, W.; Devereaux, L.M.; Jin, D.; Sampath, T.K.; Morton, R.A. Expression of bone morphogenetic protein receptors type-IA, -IB and -II correlates with tumor grade in human prostate cancer tissues. Cancer Res. 2000, 60, 2840–2844. [Google Scholar] [PubMed]
- Voorneveld, P.W.; Stache, V.; Jacobs, R.J.; Smolders, E.; Sitters, A.I.; Liesker, A.; Korkmaz, K.S.; Lam, S.M.; De Miranda, N.F.; Morreau, H.; et al. Reduced expression of bone morphogenetic protein receptor IA in pancreatic cancer is associated with a poor prognosis. Br. J. Cancer 2013, 109, 1805–1812. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Neckmann, U.; Wolowczyk, C.; Hall, M.; Almaas, E.; Ren, J.; Zhao, S.; Johannessen, B.; Skotheim, R.I.; Bjorkoy, G.; Dijke, P.T.; et al. GREM1 is associated with metastasis and predicts poor prognosis in ER-negative breast cancer patients. Cell Commun. Signal. 2019, 17, 140. [Google Scholar] [CrossRef]
- Ren, J.; Smid, M.; Iaria, J.; Salvatori, D.C.F.; van Dam, H.; Zhu, H.J.; Martens, J.W.M.; Ten Dijke, P. Cancer-associated fibroblast-derived Gremlin 1 promotes breast cancer progression. Breast Cancer Res. 2019, 21, 109. [Google Scholar] [CrossRef]
- Takahashi, M.; Otsuka, F.; Miyoshi, T.; Otani, H.; Goto, J.; Yamashita, M.; Ogura, T.; Makino, H.; Doihara, H. Bone morphogenetic protein 6 (BMP6) and BMP7 inhibit estrogen-induced proliferation of breast cancer cells by suppressing p38 mitogen-activated protein kinase activation. J. Endocrinol. 2008, 199, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.Y.; Lee, D.H.; Lee, D.K.; Kim, B.C.; Kim, H.T.; Leach, F.S.; Linehan, W.M.; Morton, R.A.; Kim, S.J. Decreased expression of bone morphogenetic protein (BMP) receptor type II correlates with insensitivity to BMP-6 in human renal cell carcinoma cells. Clin. Cancer Res. 2003, 9, 6046–6051. [Google Scholar]
- Kim, I.Y.; Lee, D.H.; Lee, D.K.; Kim, W.J.; Kim, M.M.; Morton, R.A.; Lerner, S.P.; Kim, S.J. Restoration of bone morphogenetic protein receptor type II expression leads to a decreased rate of tumor growth in bladder transitional cell carcinoma cell line TSU-Pr1. Cancer Res. 2004, 64, 7355–7360. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Raja, E.; Morikawa, M.; Nishida, J.; Tanabe, R.; Takahashi, K.; Seeherman, H.J.; Saito, N.; Todo, T.; Miyazono, K. Tyrosine kinase Eph receptor A6 sensitizes glioma-initiating cells towards bone morphogenetic protein-induced apoptosis. Cancer Sci. 2019, 110, 3486–3496. [Google Scholar] [CrossRef]
- Langenfeld, E.; Hong, C.C.; Lanke, G.; Langenfeld, J. Bone morphogenetic protein type I receptor antagonists decrease growth and induce cell death of lung cancer cell lines. PLoS ONE 2013, 8, e61256. [Google Scholar] [CrossRef] [PubMed]
- Dai, K.; Qin, F.X.; Zhang, H.K.; Liu, X.L.; Guo, C.X.; Zhang, M.; Gu, F.; Fu, L.; Ma, Y.J. Low expression of BMPRIB indicates poor prognosis of breast cancer and is insensitive to taxane-anthracycline chemotherapy. Oncotarget 2016, 7, 4770–4784. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, W.S.; Jin, W. TrkB promotes breast cancer metastasis via suppression of Runx3 and Keap1 expression. Mol. Cells 2016, 39, 258–265. [Google Scholar] [CrossRef]
- Shen, T.; Cheng, X.S.; Xia, C.F.; Li, Q.; Gao, Y.; Pan, D.G.; Zhang, X.; Zhang, C.; Li, Y.F. Erlotinib inhibits colon cancer metastasis through inactivation of TrkB-dependent ERK signaling pathway. J. Cell. Biochem. 2019, 120, 11248–11255. [Google Scholar] [CrossRef]
- Sinkevicius, K.W.; Kriegel, C.; Bellaria, K.J.; Lee, J.; Lau, A.N.; Leeman, K.T.; Zhou, P.C.; Beede, A.M.; Fillmore, C.M.; Caswell, D.; et al. Neurotrophin receptor TrkB promotes lung adenocarcinoma metastasis. Proc. Natl. Acad. Sci. USA 2014, 111, 10299–10304. [Google Scholar] [CrossRef]
- Miknyoczki, S.J.; Lang, D.; Huang, L.Y.; Klein-Szanto, A.J.P.; Dionne, C.A.; Ruggeri, B.A. Neurotrophins and Trk receptors in human pancreatic ductal adenocarcinoma: Expression patterns and effects on in vitro invasive behavior. Int. J. Cancer 1999, 81, 417–427. [Google Scholar] [CrossRef]
- Xu, Y.; Jiang, W.G.; Wang, H.C.; Martin, T.; Zeng, Y.X.; Zhang, J.; Qi, Y.S. BDNF activates TrkB/PLC gamma 1 signaling pathway to promote proliferation and invasion of ovarian cancer cells through inhibition of apoptosis. Eur. Rev. Med. Pharmaco. Sci. 2019, 23, 5093–5100. [Google Scholar]
- Antunes, L.C.M.; Cartell, A.; de Farias, C.B.; Bakos, R.M.; Roesler, R.; Schwartsmann, G. Tropomyosin-related kinase receptor and neurotrophin expression in cutaneous melanoma is associated with a poor prognosis and decreased survival. Oncol. Basel 2019, 97, 26–37. [Google Scholar] [CrossRef] [PubMed]
- de Moraes, J.K.; Wagner, V.P.; Fonseca, F.P.; do Amaral-Silva, G.K.; de Farias, C.B.; Pilar, E.F.S.; Gregianin, A.; Roesler, R.; Vargas, P.A.; Martins, M.D. Activation of BDNF/TrkB/Akt pathway is associated with aggressiveness and unfavorable survival in oral squamous cell carcinoma. Oral Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Thomaz, A.; Pinheiro, K.D.; Souza, B.K.; Gregianin, L.; Brunetto, A.L.; Brunetto, A.T.; de Farias, C.B.; Jaeger, M.D.; Ramaswamy, V.; Nor, C.; et al. Antitumor activities and cellular changes induced by TrkB inhibition in medulloblastoma. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, W.S.; Jeong, J.; Kim, S.J.; Jin, W. Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget 2015, 6, 40158–40171. [Google Scholar] [CrossRef]
- Lee, C.W.; Ito, K.; Ito, Y. Role of RUNX3 in bone morphogenetic protein signaling in colorectal cancer. Cancer Res. 2010, 70, 4243–4252. [Google Scholar] [CrossRef]
- Yin, B.; Ma, Z.Y.; Zhou, Z.W.; Gao, W.C.; Du, Z.G.; Zhao, Z.H.; Li, Q.Q. The TrkB+ cancer stem cells contribute to post-chemotherapy recurrence of triple-negative breast cancers in an orthotopic mouse model. Oncogene 2015, 34, 761–770. [Google Scholar] [CrossRef]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Yan, K.; Wu, Q.; Yan, D.H.; Lee, C.H.; Rahim, N.; Tritschler, I.; DeVecchio, J.; Kalady, M.F.; Hjelmeland, A.B.; Rich, J.N. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014, 28, 1085–1100. [Google Scholar] [CrossRef]
- Davis, H.; Irshad, S.; Bansal, M.; Rafferty, H.; Boitsova, T.; Bardella, C.; Jaeger, E.; Lewis, A.; Freeman-Mills, L.; Giner, F.C.; et al. Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nat. Med. 2015, 21, 62–70. [Google Scholar] [CrossRef]
- Ito, K. RUNX3 in oncogenic and anti-oncogenic signaling in gastrointestinal cancers. J. Cell. Biochem. 2011, 112, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Liu, Q.; Salto-Tellez, M.; Yano, T.; Tada, K.; Ida, H.; Huang, C.; Shah, N.; Inoue, M.; Rajnakova, A.; et al. RUNX3, a novel tumor suppressor, is frequently inactivated in gastric cancer by protein mislocalization. Cancer Res. 2005, 65, 7743–7750. [Google Scholar] [CrossRef] [PubMed]
- Yanada, M.; Yaoi, T.; Shimada, J.; Sakakura, C.; Nishimura, M.; Ito, K.; Terauchi, K.; Nishiyama, K.; Itoh, K.; Fushiki, S. Frequent hemizygous deletion at 1p36 and hypermethylation downregulate RUNX3 expression in human lung cancer cell lines. Oncol. Rep. 2005, 14, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.P.; Zhu, J.S.; Zhang, Q.; Wang, X.Y. A breakdown of the Hippo pathway in gastric cancer. Hepatogastroenterology 2011, 58, 1611–1617. [Google Scholar] [CrossRef]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, M.; Tan, T.Z.; Syed Sulaiman, N.B.; Lamar, J.M.; Bansal, P.; Cui, J.; Qiao, Y.; Ito, Y. RUNX1 and RUNX3 protect against YAP-mediated EMT, stem-ness and shorter survival outcomes in breast cancer. Oncotarget 2018, 9, 14175–14192. [Google Scholar] [CrossRef]
- Kim, S.M.; Ye, S.; Rah, S.Y.; Park, B.H.; Wang, H.; Kim, J.R.; Kim, S.H.; Jang, K.Y.; Lee, K.B. RhBMP-2 activates hippo signaling through RASSF1 in esophageal cancer cells. Sci. Rep. 2016, 6, 26821. [Google Scholar] [CrossRef]
- Geiger, T.R.; Song, J.Y.; Rosado, A.; Peeper, D.S. Functional characterization of human cancer-derived TRKB mutations. PLoS ONE 2011, 6, e16871. [Google Scholar] [CrossRef]
- Kim, M.S.; Jeong, J.; Seo, J.; Kim, H.S.; Kim, S.J.; Jin, W. Dysregulated JAK2 expression by TrkC promotes metastasis potential, and EMT program of metastatic breast cancer. Sci. Rep. UK 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, H.S.; Kim, Y.J.; Lee, D.Y.; Kang, S.G.; Jin, W. MEST induces Twist-1-mediated EMT through STAT3 activation in breast cancers. Cell Death Differ. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Kim, G.M.; Kim, M.S.; Lim, M.H.; Yun, C.; Jeong, J.; Nam, J.S.; Kim, S.J. TrkC plays an essential role in breast tumor growth and metastasis. Carcinogenesis 2010, 31, 1939–1947. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, M.S.; Suh, K.W.; Hong, S.; Jin, W. TrkC promotes colorectal cancer growth and metastasis. Oncotarget 2017, 8, 41319–41333. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.S.; Jin, W. TrkB Inhibits the BMP Signaling-Mediated Growth Inhibition of Cancer Cells. Cancers 2020, 12, 2095. https://doi.org/10.3390/cancers12082095
Kim MS, Jin W. TrkB Inhibits the BMP Signaling-Mediated Growth Inhibition of Cancer Cells. Cancers. 2020; 12(8):2095. https://doi.org/10.3390/cancers12082095
Chicago/Turabian StyleKim, Min Soo, and Wook Jin. 2020. "TrkB Inhibits the BMP Signaling-Mediated Growth Inhibition of Cancer Cells" Cancers 12, no. 8: 2095. https://doi.org/10.3390/cancers12082095
APA StyleKim, M. S., & Jin, W. (2020). TrkB Inhibits the BMP Signaling-Mediated Growth Inhibition of Cancer Cells. Cancers, 12(8), 2095. https://doi.org/10.3390/cancers12082095