Dual Inhibitory Action of a Novel AKR1C3 Inhibitor on Both Full-Length AR and the Variant AR-V7 in Enzalutamide Resistant Metastatic Castration Resistant Prostate Cancer

 , , , , , ,
, , , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
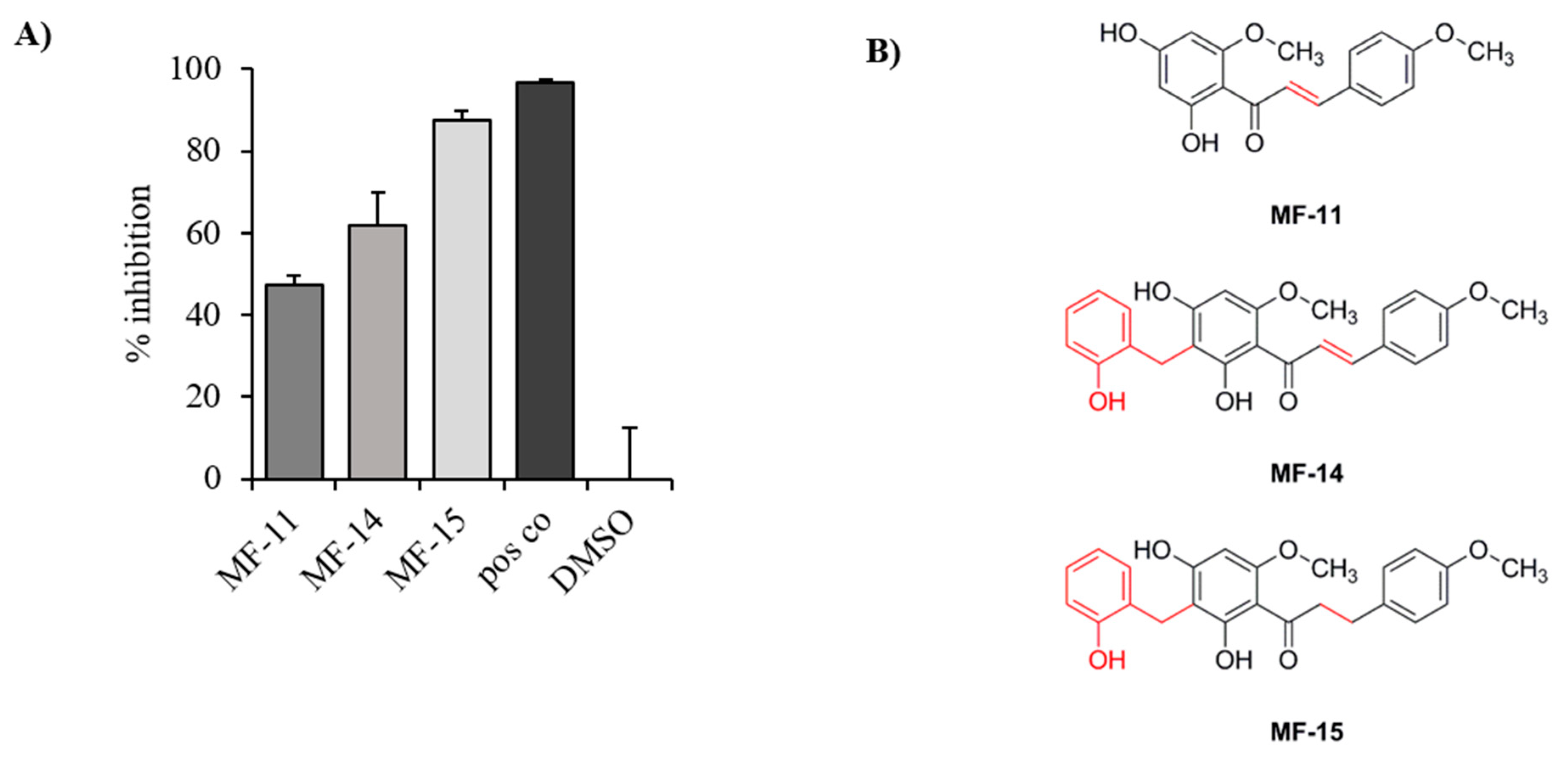
2.1. Identification of Three Natural Chalcones as Potential AKR1C3 Inhibitors
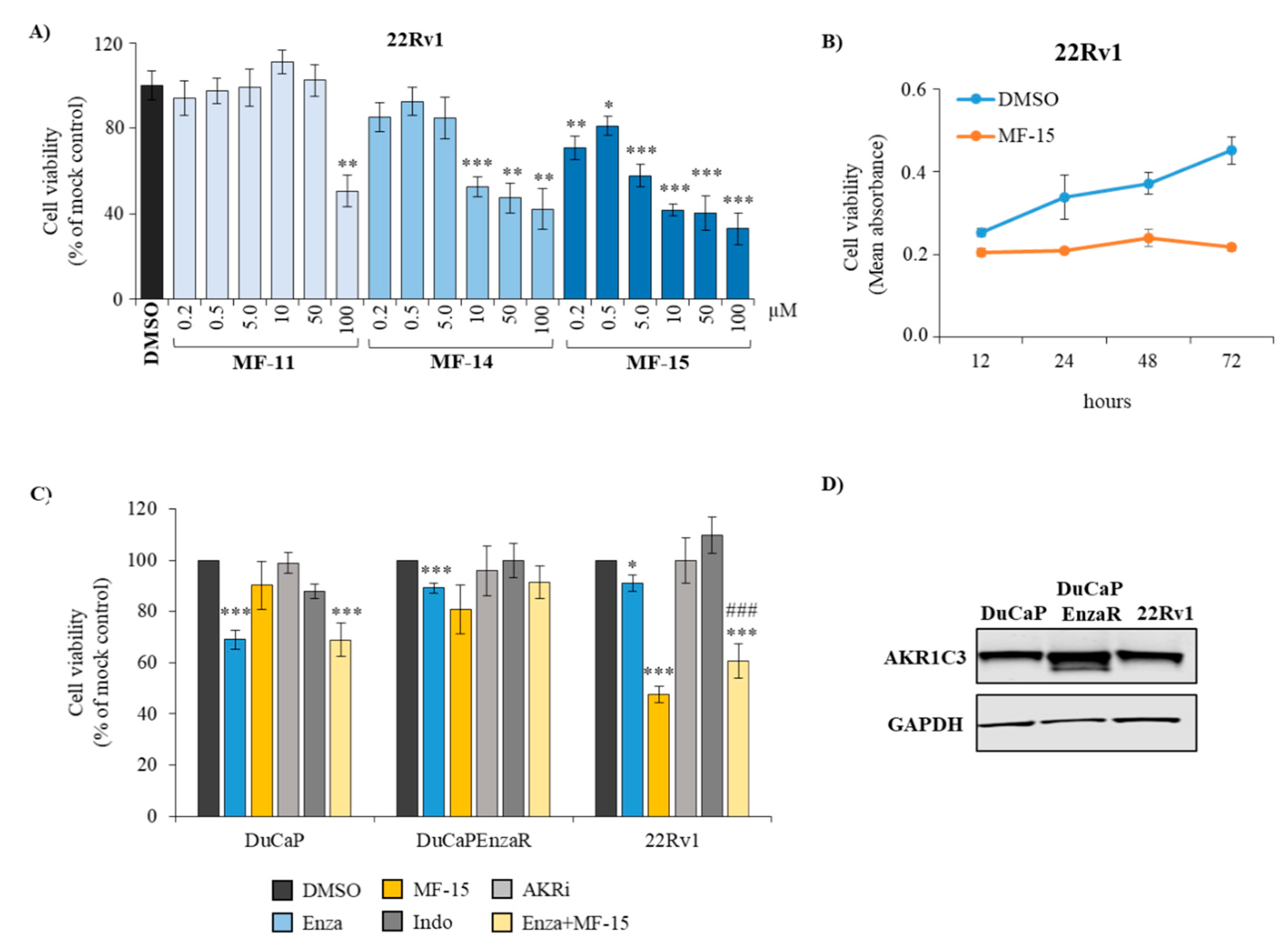
2.2. Amongst the Three Natural Compounds, MF-15 Displays the Most Potent Growth-Inhibitory Capacity in 22Rv1 Prostate Cancer Cells
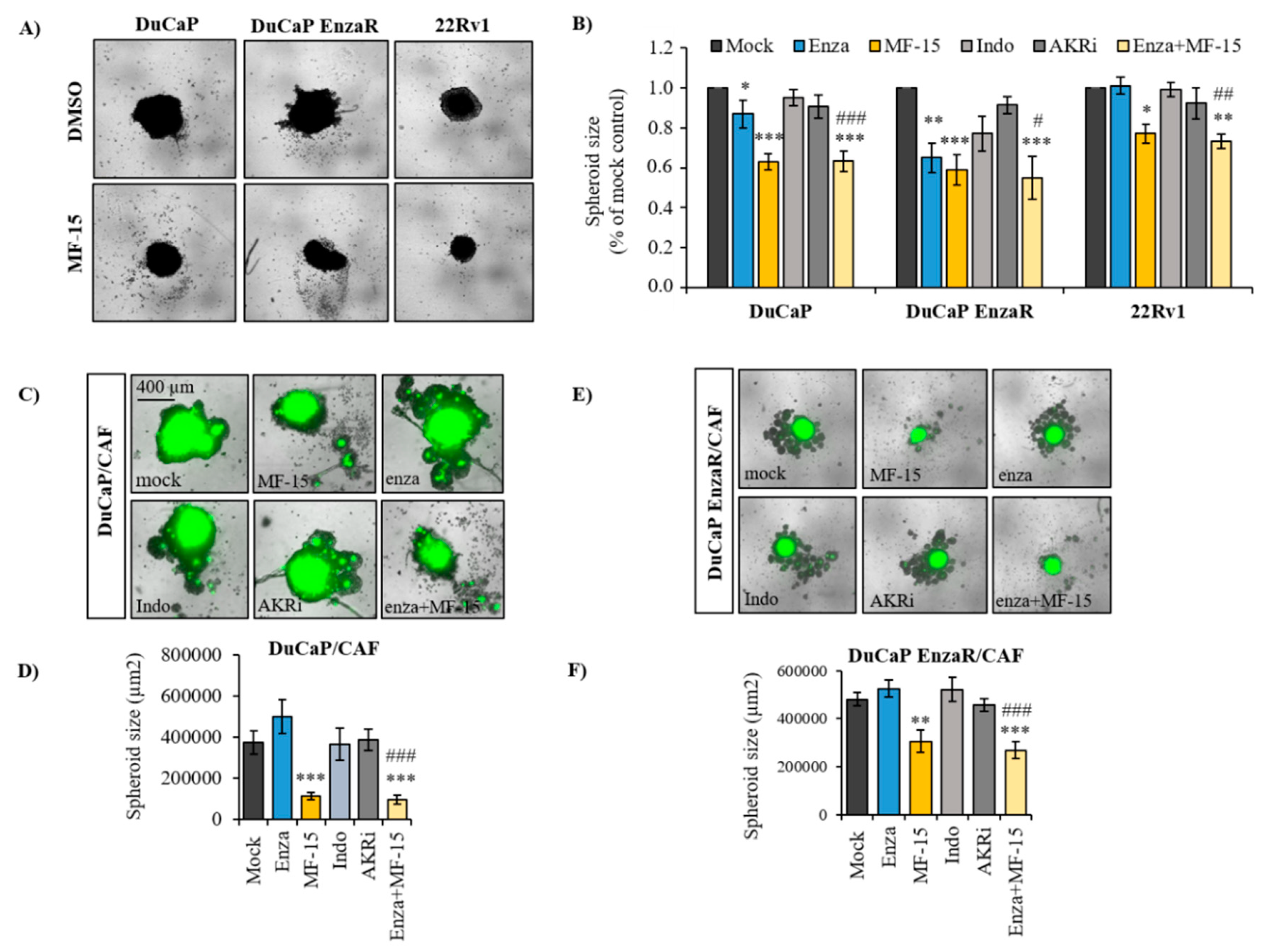
2.3. MF-15 Inhibits 3D Spheroid Growth of Enzalutamide Resistant Du/CAF Co-Cultures
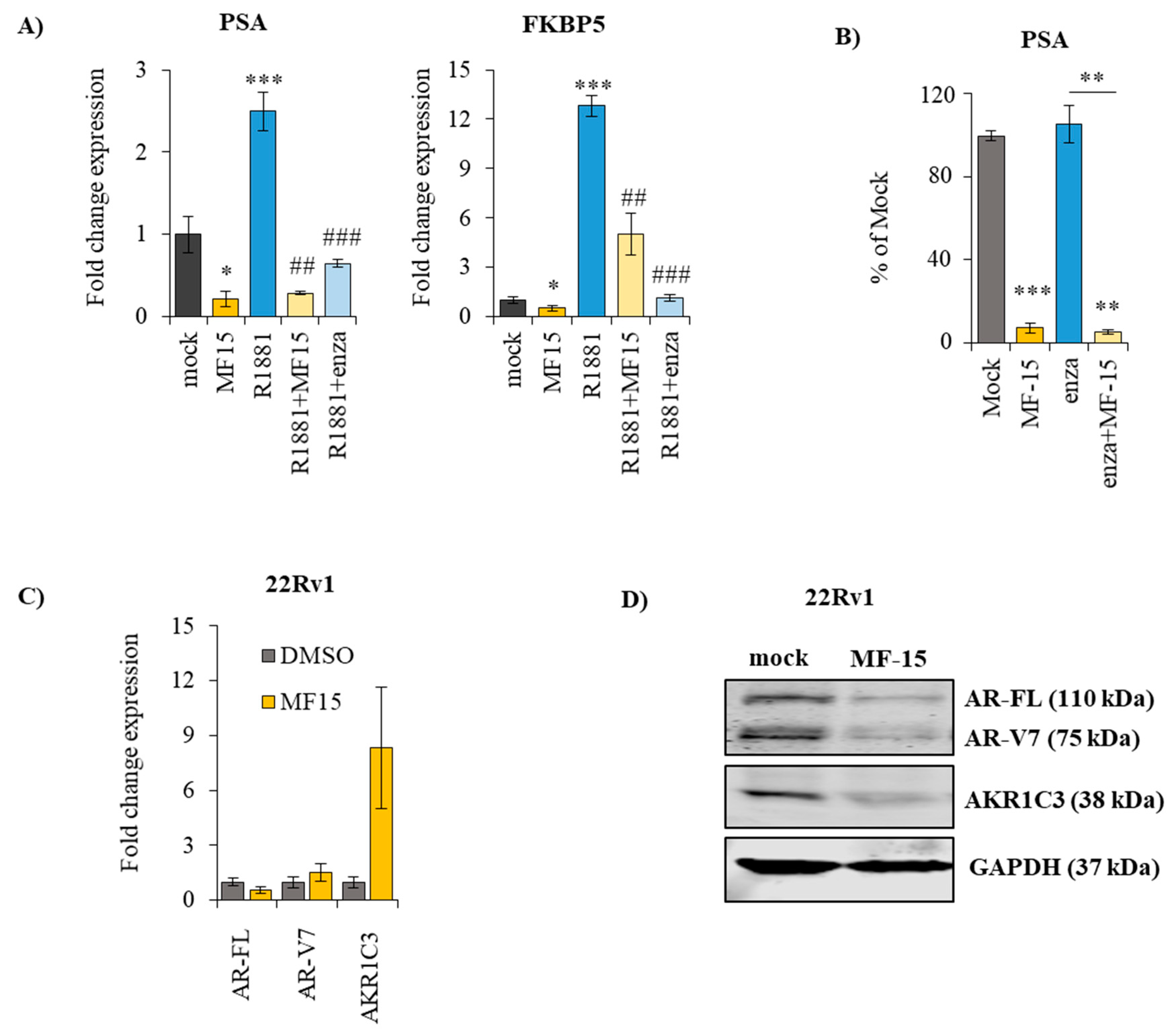
2.4. MF-15 Inhibits Expression of AR Regulated Genes through Downregulation of AR Protein
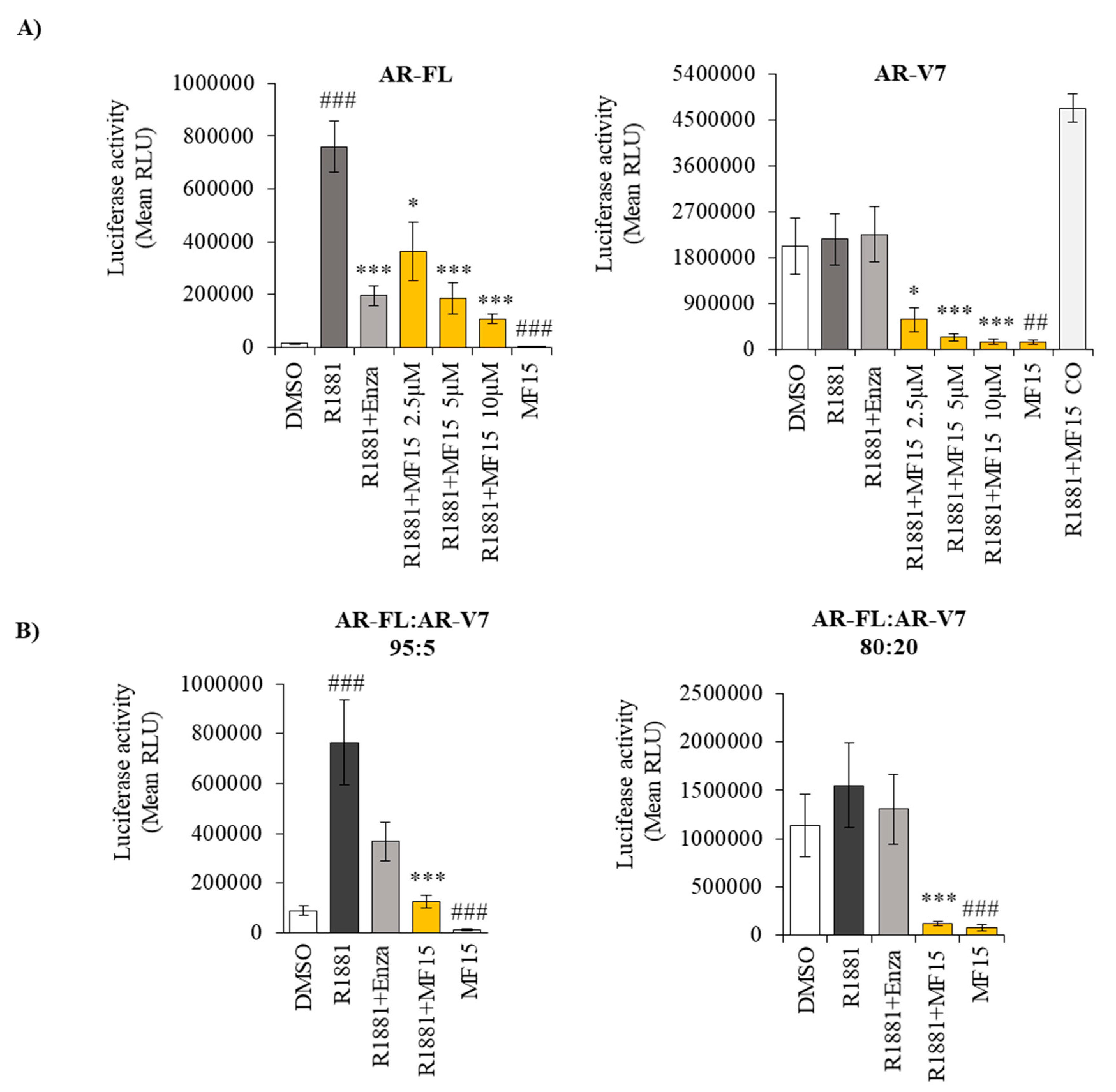
2.5. MF-15 Inhibits AR-FL and AR-V7 Reporter Gene Activity
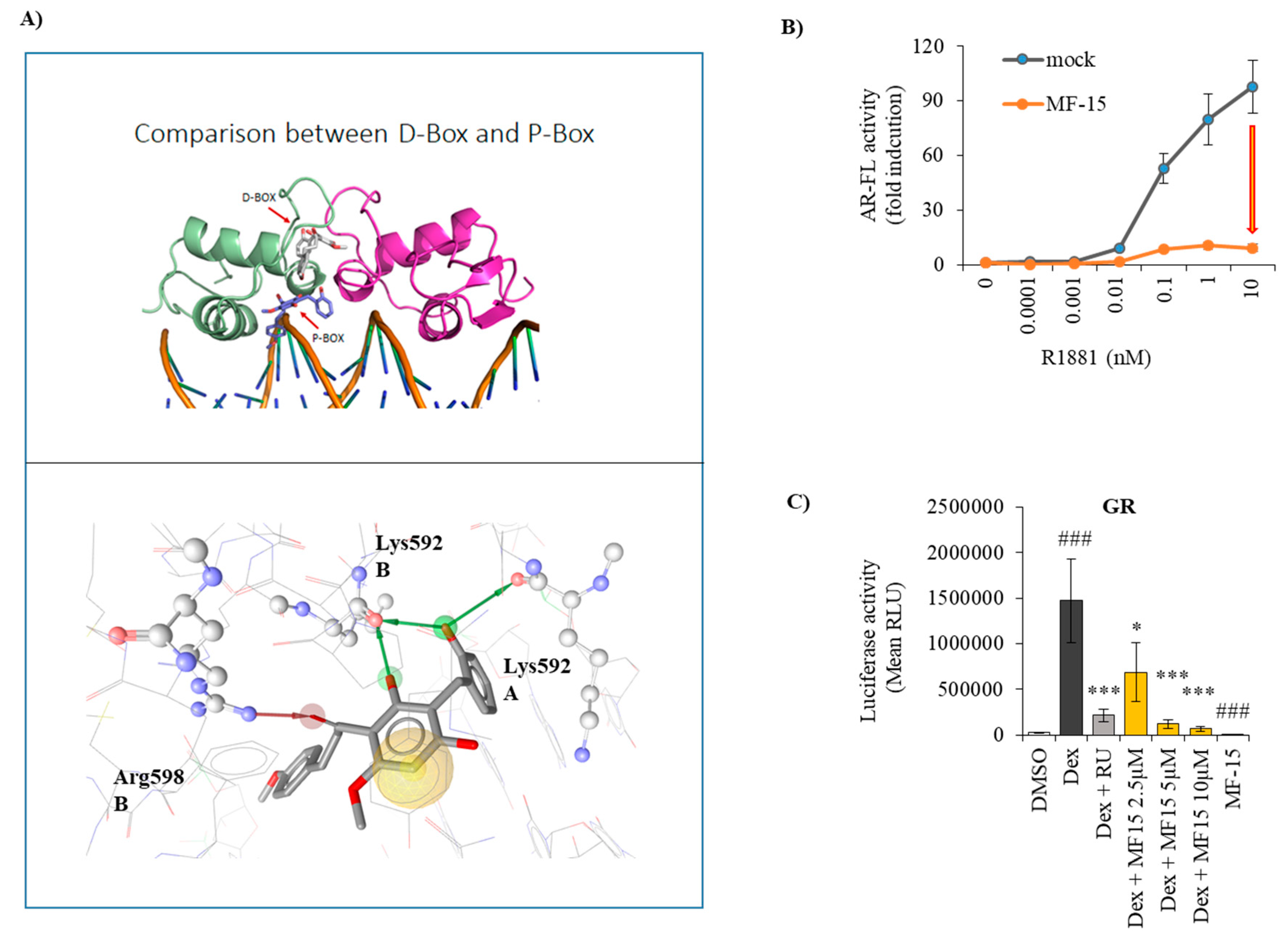
2.6. MF-15 Is a Non-Competitive AR Inhibitor with Probable Binding to the AR-DBD
3. Discussion
4. Materials and Methods
4.1. Isolation of MF-11, MF-14 from Melodorum Fruticosum, and Preparation of MF-15
4.2. Docking Parameters for the AR
4.3. Cell Lines
4.4. Reagents
4.5. Cell Viability
4.6. Spheroid Culture
4.7. Real Time Quantitative RT-PCR (qPCR)
4.8. Western Blotting
4.9. Luciferase Reporter Gene Activity Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Penning, T.M. Aldo-Keto Reductase (AKR) 1C3 inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Lorente, D.; Mateo, J.; Zafeiriou, Z.; Smith, A.D.; Sandhu, S.K.; Ferraldeschi, R.; De Bono, J.S. Switching and withdrawing hormonal agents for castration-resistant prostate cancer. Nat. Rev. Urol. 2015, 12, 37–47. [Google Scholar] [CrossRef]
- Snow, O.; Lallous, N.; Ester, M.; Cherkasov, A. Deep Learning Modelling of Androgen Receptor Responses to Prostate Cancer Therapies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of Intratumoral Androgens in Metastatic Prostate Cancer: A Mechanism for Castration-Resistant Tumor Growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef]
- Potter, G.A.; Barrie, S.E.; Jarman, M.; Rowlands, M.G. Novel Steroidal Inhibitors of Human Cytochrome P45017.alpha.-Hydroxylase-C17,20-lyase): Potential Agents for the Treatment of Prostatic Cancer. J. Med. Chem. 1995, 38, 2463–2471. [Google Scholar] [CrossRef]
- SSnow, O.; Lallous, N.; Singh, K.; Lack, N.; Rennie, P.; Cherkasov, A. Androgen receptor plasticity and its implications for prostate cancer therapy. Cancer Treat Rev. 2019, 81, 101871. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, N.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; De Wit, R.; Mülders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, D.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2018, 378, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ban, F.; Dalal, K.; Leblanc, E.; Frewin, K.; Ma, D.; Adomat, H.; Rennie, P.S.; Cherkasov, A. Discovery of Small-Molecule Inhibitors Selectively Targeting the DNA-Binding Domain of the Human Androgen Receptor. J. Med. Chem. 2014, 57, 6458–6467. [Google Scholar]
- Liu, C.; Lou, W.; Zhu, Y.; Yang, J.C.; Nadiminty, N.; Gaikwad, N.W.; Evans, C.P.; Gao, A.C. Intracrine Androgens and AKR1C3 Activation Confer Resistance to Enzalutamide in Prostate Cancer. Cancer Res. 2015, 75, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men with Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2156. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013, 3, 1030–1043. [Google Scholar] [CrossRef]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef]
- Knudsen, K.E.; Penning, T. Partners in crime: Deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol. Metab. 2010, 21, 315–324. [Google Scholar] [CrossRef]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased Expression of Genes Converting Adrenal Androgens to Testosterone in Androgen-Independent Prostate Cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef]
- Steckelbroeck, S.; Jin, Y.; Gopishetty, S.; Oyesanmi, B.; Penning, T.M. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity implications for steroid hormone metabolism and action. J. Biol. Chem. 2004, 279, 10784–10795. [Google Scholar] [CrossRef] [PubMed]
- Verma, K.; Gupta, N.; Zang, T.; Wangtrakluldee, P.; Srivastava, S.K.; Penning, T.M.; Trippier, P.C.; Wangtrakuldee, P. AKR1C3 Inhibitor KV-37 Exhibits Antineoplastic Effects and Potentiates Enzalutamide in Combination Therapy in Prostate Adenocarcinoma Cells. Mol. Cancer Ther. 2018, 17, 1833–1845. [Google Scholar] [CrossRef] [PubMed]
- Neuwirt, H.; Bouchal, J.; Kharaishvili, G.; Ploner, C.; Jöhrer, K.; Pitterl, F.; Weber, A.; Klocker, H.; Eder, I.E. Cancer-associated fibroblasts promote prostate tumor growth and progression through upregulation of cholesterol and steroid biosynthesis. Cell Commun. Signal. 2020, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Armstrong, C.M.; Lou, W.; Lombard, A.; Evans, C.P.; Gao, A.C. Inhibition of AKR1C3 Activation Overcomes Resistance to Abiraterone in Advanced Prostate Cancer. Mol Cancer Ther. 2017, 16, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Byrns, M.C.; Jin, Y.; Penning, T.M. Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): Overview and structural insights. J. Steroid Biochem. Mol. Biol. 2011, 125, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Adeniji, A.O.; Chen, M.; Penning, T.M. AKR1C3 as a target in castrate resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2013, 137, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.J.; Hayden, R.E.; Simpson, P.; Birtwistle, J.; Mayer, K.; Ride, J.P.; Bunce, C.M. AKR1C isoforms represent a novel cellular target for jasmonates alongside their mitochondrial-mediated effects. Cancer Res. 2009, 69, 4769–4775. [Google Scholar] [CrossRef]
- Škarydová, L.; Živná, L.; Xiong, G.; Maser, E.; Wsol, V. AKR1C3 as a potential target for the inhibitory effect of dietary flavonoids. Chem. Biol. Interact. 2009, 178, 138–144. [Google Scholar] [CrossRef]
- Liedtke, A.J.; Adeniji, A.O.; Chen, M.; Byrns, M.C.; Jin, Y.; Christianson, D.W.; Marnett, L.J.; Penning, T.M. Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (Type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J. Med. Chem. 2013, 56, 2429–2446. [Google Scholar]
- Féau, C.; Arnold, L.A.; Kosinski, A.; Zhu, F.; Connelly, M.; Guy, R.K. Novel flufenamic acid analogues as inhibitors of androgen receptor mediated transcription. ACS Chem. Biol. 2009, 4, 834–843. [Google Scholar] [CrossRef][Green Version]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Jiao, L.; Xu, C.; Yu, Y.; Zhang, Z.; Chang, Z.; Deng, Z.; Sun, Y. Neural protein gamma-synuclein interacting with androgen receptor promotes human prostate cancer progression. BMC Cancer 2012, 12, 593. [Google Scholar] [CrossRef] [PubMed]
- Wangtrakuldee, P.; Adeniji, A.O.; Zang, T.; Duan, L.; Khatri, B.; Twenter, B.M.; Estrada, M.A.; Higgins, T.F.; Winkler, J.D.; Penning, T.M. A 3-(4-nitronaphthen-1-yl) amino-benzoate analog as a bifunctional AKR1C3 inhibitor and AR antagonist: Head to head comparison with other advanced AKR1C3 targeted therapeutics. J. Steroid Biochem. Mol. Biol. 2019, 192, 105283. [Google Scholar] [CrossRef]
- Sadar, M.D. Discovery of drugs that directly target the intrinsically disordered region of the androgen receptor. Expert Opin. Drug Discov. 2020, 15, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Bank RPD. RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 14 April 2020).
- Dalal, K.; Ban, F.; Li, H.; Morin, H.; Roshan-Moniri, M.; Tam, K.J.; Shepherd, A.; Sharma, A.; Peacock, J.; Carlson, M.L.; et al. Selectively targeting the DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J. Biol. Chem. 2015, 290, 25850. [Google Scholar] [CrossRef]
- Li, Y.; Xie, N.; Gleave, M.E.; Rennie, P.S.; Dong, X. AR-v7 protein expression is regulated by protein kinase and phosphatase. Oncotarget 2015, 6, 33743–33754. [Google Scholar] [CrossRef]
- Hoefer, J.; Akbor, M.; Handle, F.; Ofer, P.; Puhr, M.; Parson, W.; Culig, Z.; Klocker, H.; Heidegger, I. Critical role of androgen receptor level in prostate cancer cell resistance to new generation antiandrogen enzalutamide. Oncotarget 2016, 7, 59781–59794. [Google Scholar] [CrossRef] [PubMed]
- Mayr, F.; Moeller, G.; Garscha, U.; Fischer, J.; Castano, P.R.; Inderbinen, S.G.; Temml, V.; Waltenberger, B.; Schwaiger, S.; Hartmann, R.W.; et al. Finding New Molecular Targets of Familiar Natural Products Using In Silico Target Prediction. bioRxiv 2020. [Google Scholar] [CrossRef]
- Schuster, D.; Kowalik, D.; Kirchmair, J.; Laggner, C.; Markt, P.; Aebischer-Gumy, C.; Ströhle, F.; Möller, G.; Wolber, G.; Wilckens, T.; et al. Identification of chemically diverse, novel inhibitors of 17β-hydroxysteroid dehydrogenase type 3 and 5 by pharmacophore-based virtual screening. J. Steroid Biochem. Mol. Biol. 2011, 125, 148–161. [Google Scholar] [CrossRef]
- Eder, T.; Weber, A.; Neuwirt, H.; Gruenbacher, G.; Ploner, C.; Klocker, H.; Sampson, N.; Eder, I.E. Cancer-Associated Fibroblasts Modify the Response of Prostate Cancer Cells to Androgen and Anti-Androgens in Three-Dimensional Spheroid Culture. Int. J. Mol. Sci. 2016, 17, 1458. [Google Scholar] [CrossRef]
- Schalken, J.A.; Fitzpatrick, J.M. Enzalutamide: Targeting the androgen signalling pathway in metastatic castration-resistant prostate cancer. BJU Int. 2016, 117, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-Q.; Gu, X.; Gao, X.-S.; Li, Y.; Yu, H.; Xiong, W.; Yu, H.; Wang, W.; Li, Y.; Teng, Y.; et al. Overexpression of AKR1C3 significantly enhances human prostate cancer cells resistance to radiation. Oncotarget 2016, 7, 48050–48058. [Google Scholar] [CrossRef]
- Wako, K.; Kawasaki, T.; Yamana, K.; Suzuki, K.; Jiang, S.; Umezu, H.; Nishiyama, T.; Takahashi, K.; Hamakubo, T.; Kodama, T.; et al. Expression of androgen receptor through androgen-converting enzymes is associated with biological aggressiveness in prostate cancer. J. Clin. Pathol. 2008, 61, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhao, L.; Zhang, H.; Liu, X.; Zhao, L.; Zhao, X.; Li, Y.; Li, J. AKR1C3 overexpression may serve as a promising biomarker for prostate cancer progression. Diagn. Pathol. 2014, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Hamid, A.R.A.H.; Pfeiffer, M.J.; Verhaegh, G.W.; Schaafsma, E.; Brandt, A.; Sweep, F.; Sedelaar, J.P.M.; Schalken, J.A. Aldo-keto Reductase Family 1 Member C3 (AKR1C3) Is a Biomarker and Therapeutic Target for Castration-Resistant Prostate Cancer. Mol. Med. 2012, 18, 1449–1455. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Armstrong, A.J.; Dehm, S.M.; Luo, J. Androgen receptor variant-driven prostate cancer: Clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis. 2016, 19, 231–241. [Google Scholar] [CrossRef]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, N.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, U.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef]
- Sarwar, M.; Semenas, J.; Miftakhova, R.; Simoulis, A.; Robinson, B.; Wingren, A.G.; Mongan, N.P.; Heery, D.; Johnson, H.; Abrahamsson, P.-A.; et al. Targeted suppression of AR-V7 using PIP5K1α inhibitor overcomes enzalutamide resistance in prostate cancer cells. Oncotarget 2016, 7, 63065–63081. [Google Scholar] [CrossRef]
- Cao, B.; Qi, Y.; Zhang, G.; Xu, D.; Zhan, Y.; Álvarez, X.; Guo, Z.; Fu, X.; Plymate, S.R.; Sartor, O.; et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget 2014, 5, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Qiu, Y. Role of androgen receptor splice variants in prostate cancer metastasis. Asian J. Urol. 2016, 3, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar] [CrossRef]
- Bernemann, C.; Schnoeller, T.; Luedeke, M.; Steinestel, K.; Boegemann, M.; Schrader, A.J.; Steinestel, J. Expression of AR-V7 in Circulating Tumour Cells Does Not Preclude Response to Next Generation Androgen Deprivation Therapy in Patients with Castration Resistant Prostate Cancer. Eur. Urol. 2017, 71, 1–3. [Google Scholar] [CrossRef]
- Myung, J.-K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef]
- Endo, S.; Matsunaga, T.; Kanamori, A.; Otsuji, Y.; Nagai, H.; Sundaram, K.; El-Kabbani, O.; Toyooka, N.; Ohta, S.; Hara, A. Selective Inhibition of Human Type-5 17β-Hydroxysteroid Dehydrogenase (AKR1C3) by Baccharin, a Component of Brazilian Propolis. J. Nat. Prod. 2012, 75, 716–721. [Google Scholar] [CrossRef]
- Yin, Y.D.; Fu, M.; Brooke, D.G.; Heinrich, D.M.; Denny, W.A.; Jamieson, S.M.F. The Activity of SN33638, an Inhibitor of AKR1C3, on Testosterone and 17β-Estradiol Production and Function in Castration-Resistant Prostate Cancer and ER-Positive Breast Cancer. Front. Oncol. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2014.00159/full (accessed on 10 July 2020).
- Sekine, Y.; Nakayama, H.; Miyazawa, Y.; Kato, H.; Furuya, Y.; Arai, S.; Koike, H.; Matsui, H.; Shibata, Y.; Ito, K.; et al. Simvastatin in combination with meclofenamic acid inhibits the proliferation and migration of human prostate cancer PC-3 cells via an AKR1C3 mechanism. Oncol. Lett. 2018, 15, 3167–3172. [Google Scholar] [CrossRef]
- Engels, N.S.H. Discovery of Novel Anti-inflammatory Natural Products Isolated from Plants Used in Traditional Vietnamese Medicine. Ph.D. Dissertation, Universität Innsbruck, Innsbruck, Austria, March 2019. [Google Scholar]
- Finetti, F.; Terzuoli, E.; Giachetti, A.; Santi, R.; Villari, D.; Hanaka, H.; Radmark, O.; Ziche, M.; Donnini, S. mPGES-1 in prostate cancer controls stemness and amplifies epidermal growth factor receptor-driven oncogenicity. Endocr. Relat. Cancer 2015, 22, 665–678. [Google Scholar] [CrossRef]
- Shaffer, P.L.; Jivan, A.; Dollins, D.E.; Claessens, F.; Gewirth, D.T. Structural basis of androgen receptor binding to selective androgen response elements. Proc. Natl. Acad. Sci. USA 2004, 101, 4758–4763. [Google Scholar] [CrossRef]
- Madar, S.; Brosh, R.; Buganim, Y.; Ezra, O.; Goldstein, I.; Solomon, H.; Kogan, I.; Goldfinger, N.; Klocker, H.; Rotter, V. Modulated expression of WFDC1 during carcinogenesis and cellular senescence. Carcinogenesis 2009, 30, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Hille, C.; Gorges, T.M.; Riethdorf, S.; Mazel, M.; Steuber, T.; Von Amsberg, G.; König, F.; Peine, S.; Alix-Panabières, C.; Pantel, K. Detection of Androgen Receptor Variant 7 (ARV7) mRNA Levels in EpCAM-Enriched CTC Fractions for Monitoring Response to Androgen Targeting Therapies in Prostate Cancer. Cells 2019, 8, 1067. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6770695/ (accessed on 10 July 2020).
- Prawat, U.; Chairerk, O.; Phupornprasert, U.; Salae, A.-W.; Tuntiwachwuttikul, P. Two New C-benzylated Dihydrochalcone Derivatives from the Leaves of Melodorum siamensis. Planta Med. 2013, 79, 83–86. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kafka, M.; Mayr, F.; Temml, V.; Möller, G.; Adamski, J.; Höfer, J.; Schwaiger, S.; Heidegger, I.; Matuszczak, B.; Schuster, D.; et al. Dual Inhibitory Action of a Novel AKR1C3 Inhibitor on Both Full-Length AR and the Variant AR-V7 in Enzalutamide Resistant Metastatic Castration Resistant Prostate Cancer. Cancers 2020, 12, 2092. https://doi.org/10.3390/cancers12082092
Kafka M, Mayr F, Temml V, Möller G, Adamski J, Höfer J, Schwaiger S, Heidegger I, Matuszczak B, Schuster D, et al. Dual Inhibitory Action of a Novel AKR1C3 Inhibitor on Both Full-Length AR and the Variant AR-V7 in Enzalutamide Resistant Metastatic Castration Resistant Prostate Cancer. Cancers. 2020; 12(8):2092. https://doi.org/10.3390/cancers12082092
Chicago/Turabian StyleKafka, Mona, Fabian Mayr, Veronika Temml, Gabriele Möller, Jerzy Adamski, Julia Höfer, Stefan Schwaiger, Isabel Heidegger, Barbara Matuszczak, Daniela Schuster, and et al. 2020. "Dual Inhibitory Action of a Novel AKR1C3 Inhibitor on Both Full-Length AR and the Variant AR-V7 in Enzalutamide Resistant Metastatic Castration Resistant Prostate Cancer" Cancers 12, no. 8: 2092. https://doi.org/10.3390/cancers12082092
APA StyleKafka, M., Mayr, F., Temml, V., Möller, G., Adamski, J., Höfer, J., Schwaiger, S., Heidegger, I., Matuszczak, B., Schuster, D., Klocker, H., Bektic, J., Stuppner, H., & Eder, I. E. (2020). Dual Inhibitory Action of a Novel AKR1C3 Inhibitor on Both Full-Length AR and the Variant AR-V7 in Enzalutamide Resistant Metastatic Castration Resistant Prostate Cancer. Cancers, 12(8), 2092. https://doi.org/10.3390/cancers12082092