The LIM Protein Ajuba Augments Tumor Metastasis in Colon Cancer

 ,
,  ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Ajuba Expression Is Increased in Human Colon Cancer Cell Lines and Metastatic Tumor Tissue
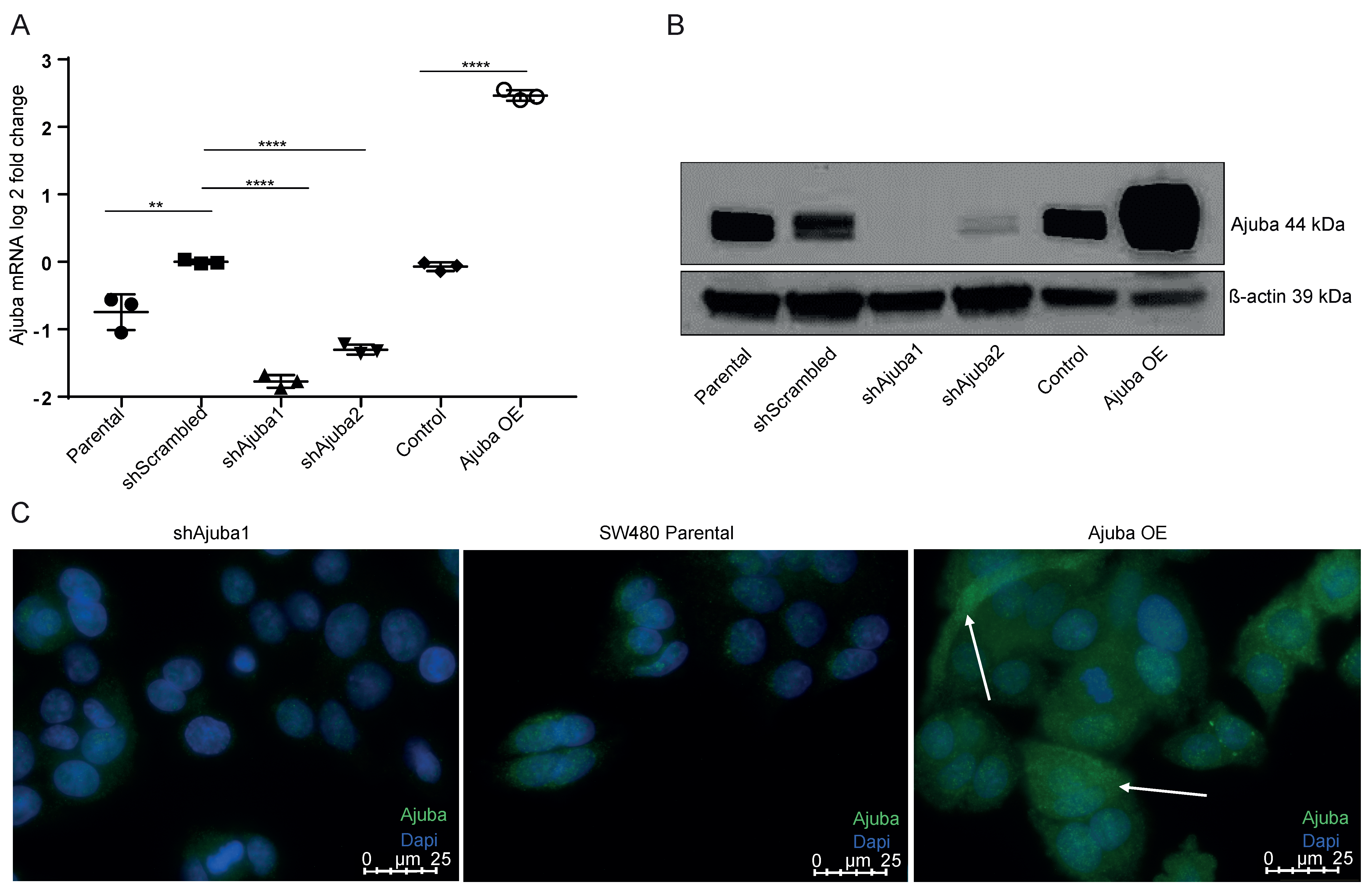
2.2. Ajuba Is Efficiently Knocked down and Overexpressed Using Lentiviral Transduction
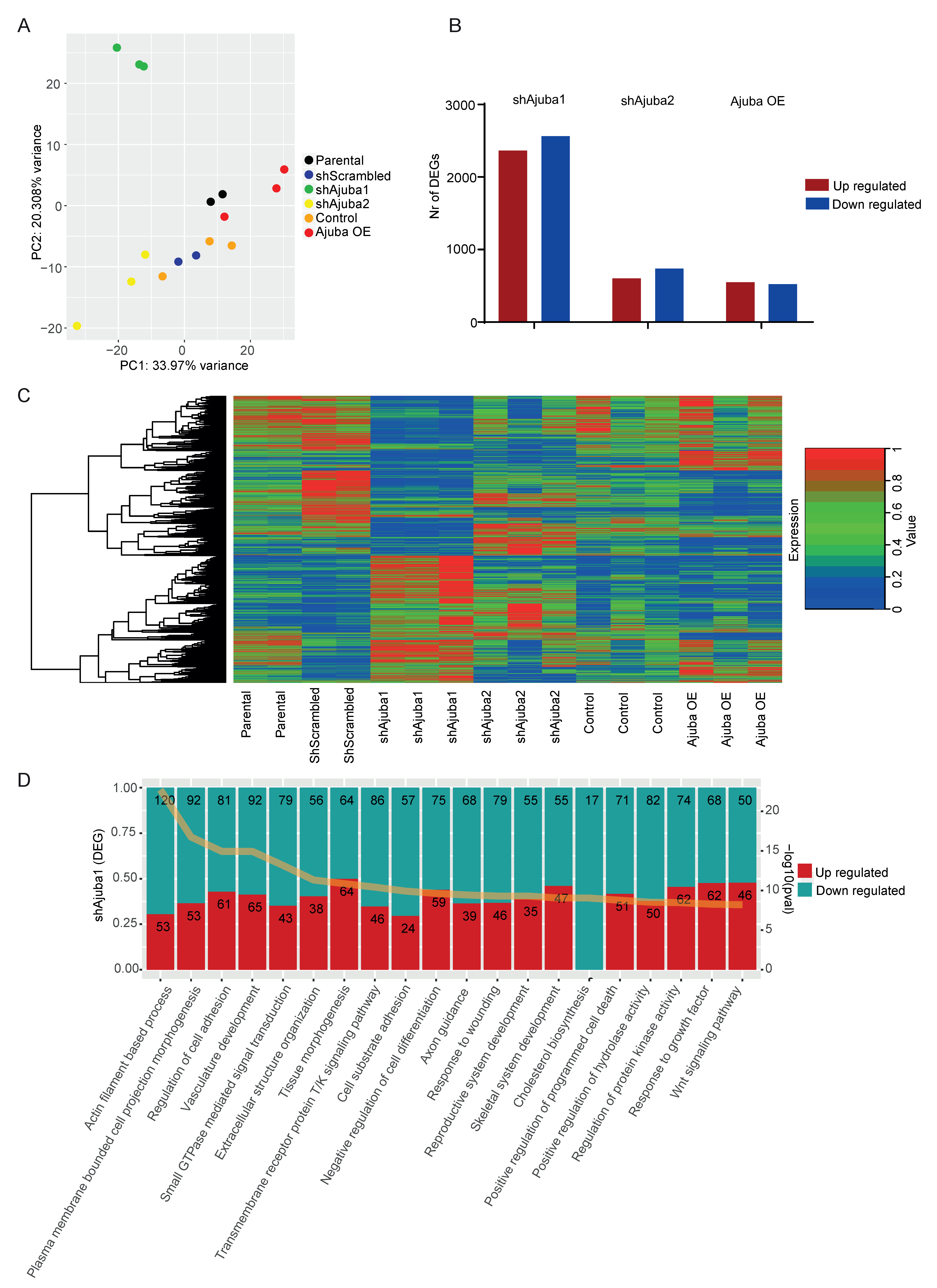
2.3. RNA-Seq Analysis of SW480 with Ajuba KD and OE
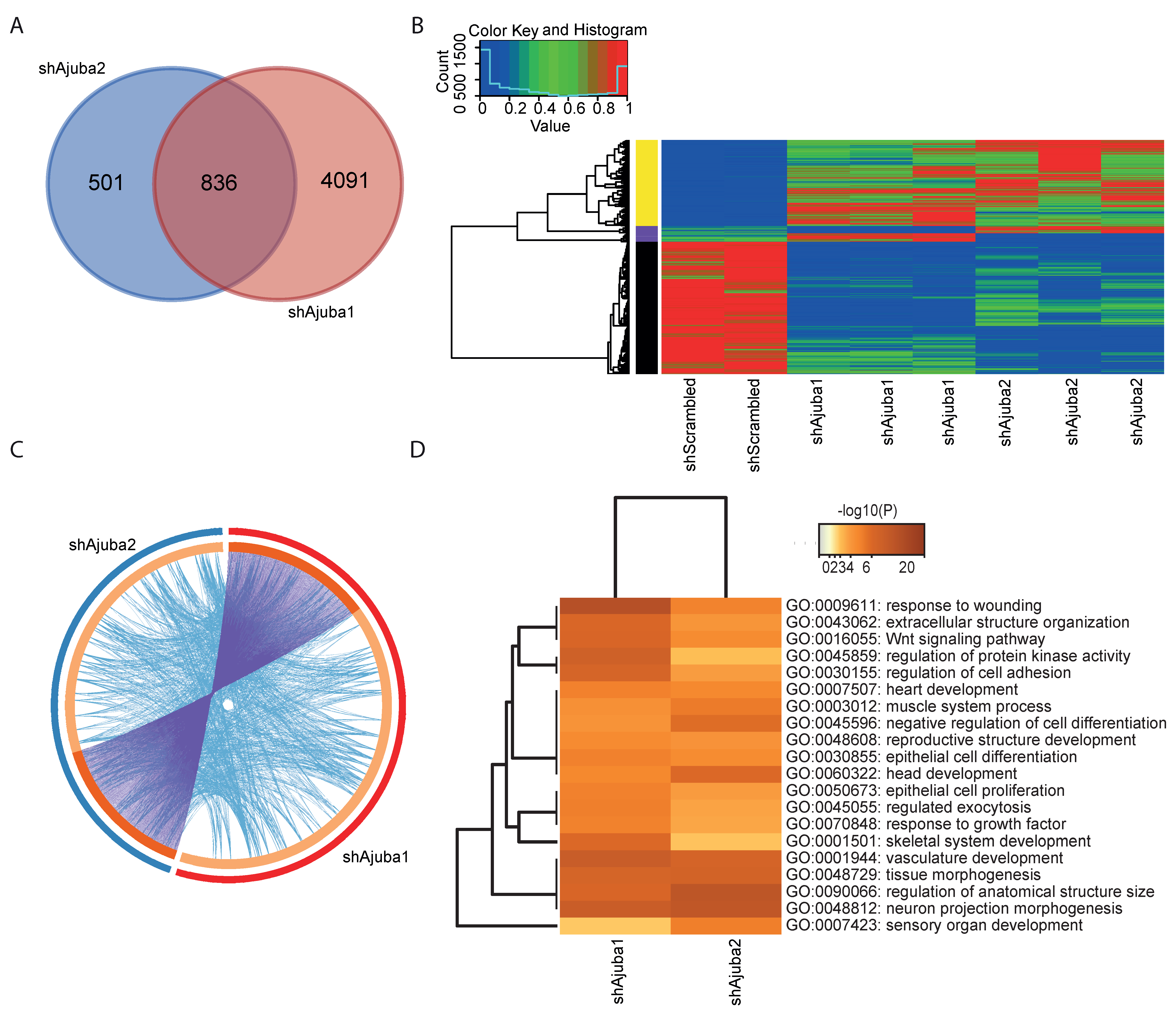
2.4. Pathway Enrichment Analysis Using Common DEG from the Ajuba KD Cell Lines Showed Ajuba Involvement in Cell Adhesion, Cell Differentiation and Proliferation
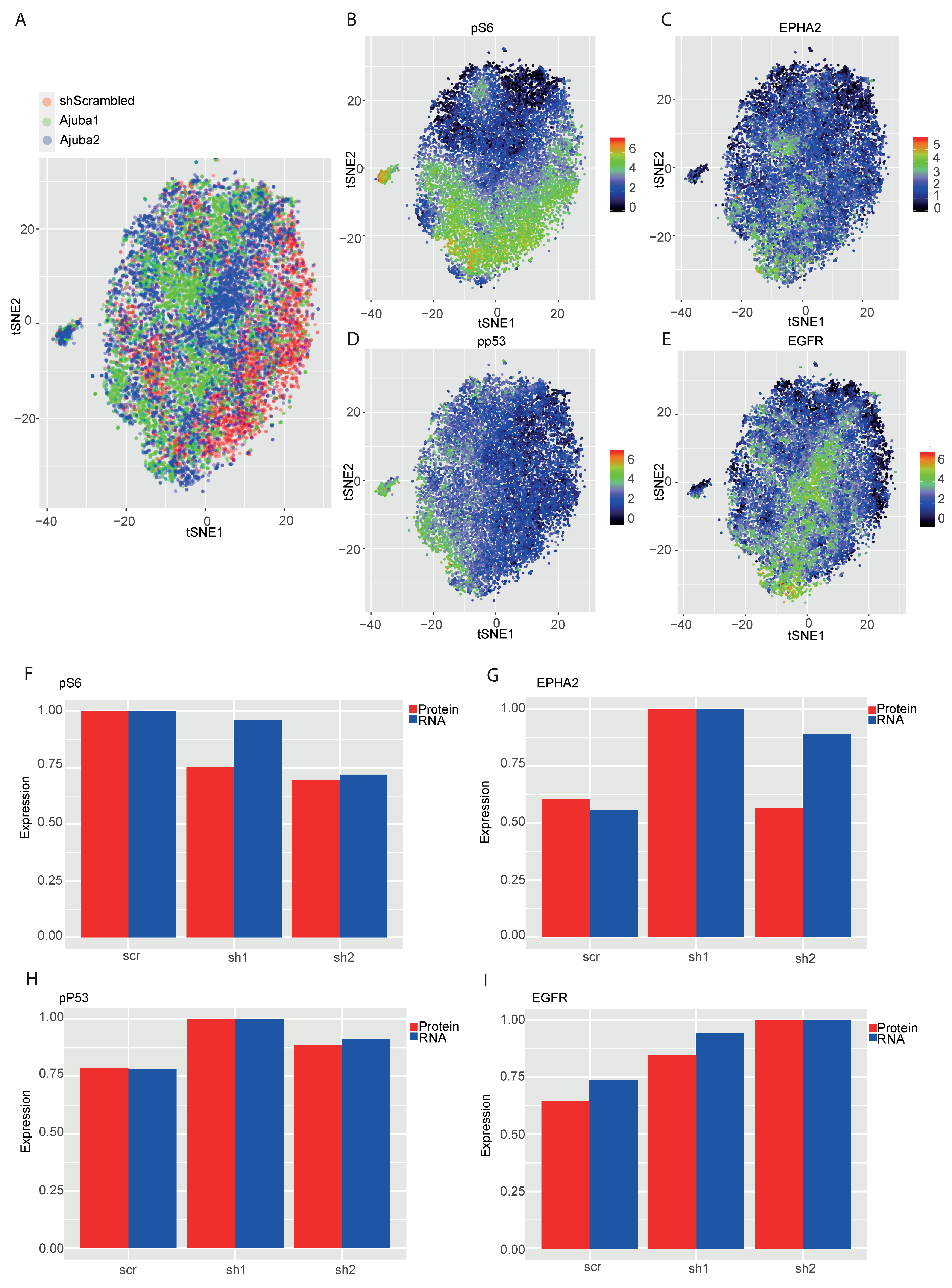
2.5. Mass Cytometry Analysis of Ajuba KD Reveals Differential Expression of Cancer Associated Proteins
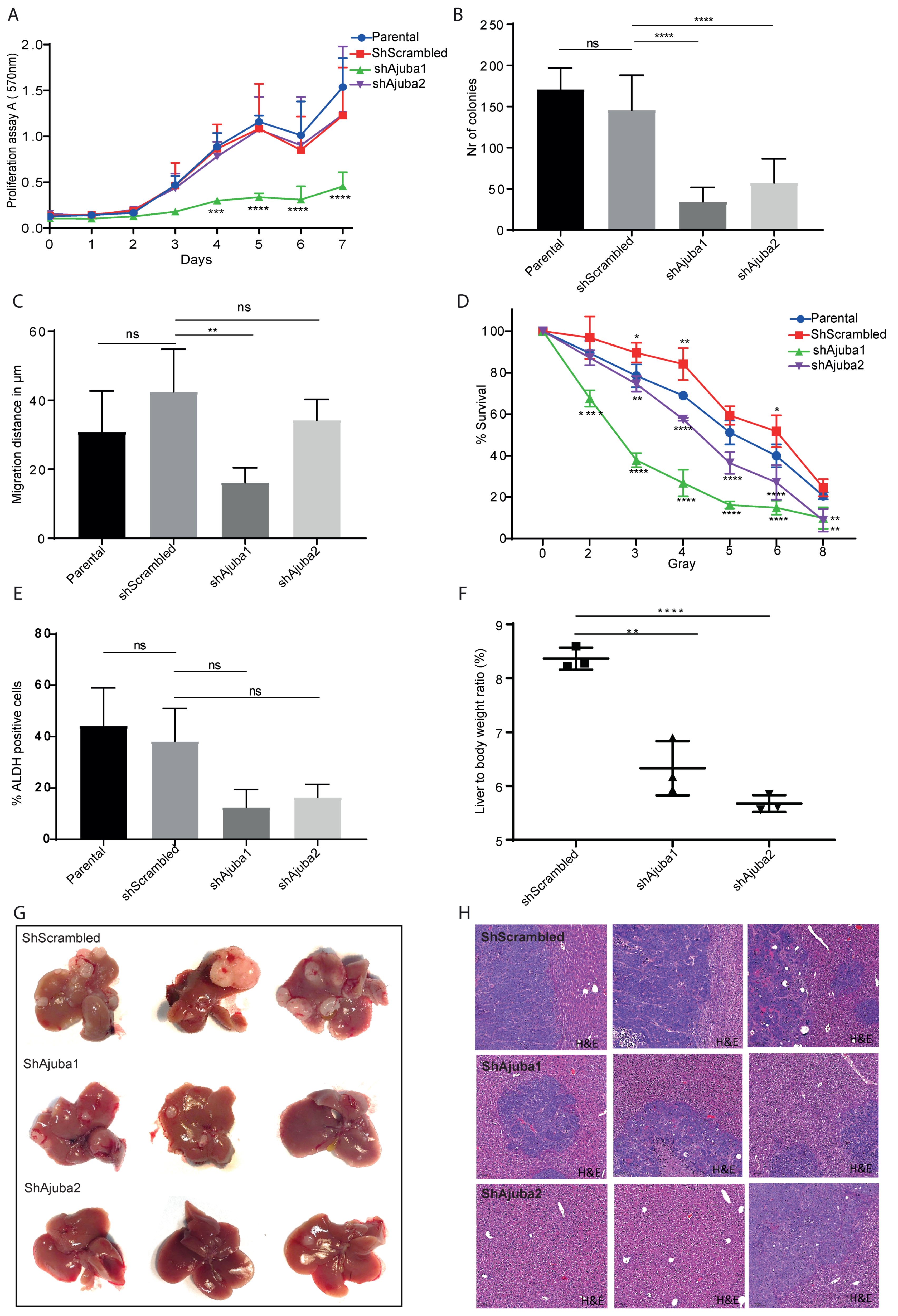
2.6. Ajuba KD Reduces Cell Migration, Colony Formation and Metastatic Tumor Burden in Rag2−/−Gamma(c) Mice
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Clinical Samples
4.3. Public Data Acquisition
4.4. Differential Expression
4.5. Survival Analysis
4.6. Western Blot
4.7. Quantitative Real-Time Reverse Transcription PCR
4.8. Lentiviral Transduction
- Clone shAjuba1: targeting the coding region NM-032876.4-1385s1c1
- Clone shAjuba2: targeting the non-coding region 2URT NM_032876.4-2786s1c1
- Clone shScrambled: pLKO.1-puro Non-Mammalian shRNA control plasmid
- To overexpress Ajuba, SW480 cells were transduced with lentivirus containing an Ajuba overexpressing construct
- Clone Ajuba OE Harvard PlasmID:Phage_CMV_C_FLAG_HA_IRES_PURO
4.9. Immunofluorescence
4.10. RNA-Sequencing
4.11. Alignment
4.12. Data Visualization
4.13. Venn Diagram
4.14. Pathway Enrichment Analysis
4.15. Mass Cytometry by Time-of-Flight (CyTOF)
4.16. Mass Cytometry Analysis
4.17. Comparing RNA-Seq with Mass Cytometry Results
4.18. Cell Proliferation Assay
4.19. Colony Formation Assay
4.20. Migration Assay
4.21. Irradiation
4.22. ALDH Assay
4.23. In Vivo Metastases
4.24. Graphs and Statistical Analysis
4.25. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goyal, R.K.; Lin, P.; Kanungo, J.; Payne, A.S.; Muslin, A.J.; Longmore, G.D. Ajuba, a novel LIM protein, interacts with Grb2, augments mitogen-activated protein kinase activity in fibroblasts, and promotes meiotic maturation of Xenopus oocytes in a Grb2- and Ras-dependent manner. Mol. Cell. Biol. 1999, 19, 4379–4389. [Google Scholar] [CrossRef]
- Srichai, M.B.; Konieczkowski, M.; Padiyar, A.; Konieczkowski, D.J.; Mukherjee, A.; Hayden, P.S.; Kamat, S.; El-Meanawy, M.A.; Khan, S.; Mundel, P.; et al. A WT1 co-regulator controls podocyte phenotype by shuttling between adhesion structures and nucleus. J. Biol. Chem. 2004, 279, 14398–14408. [Google Scholar] [CrossRef]
- Sharp, T.V.; Munoz, F.; Bourboulia, D.; Presneau, N.; Darai, E.; Wang, H.W.; Cannon, M.; Butcher, D.N.; Nicholson, A.G.; Klein, G.; et al. LIM domains-containing protein 1 (LIMD1), a tumor suppressor encoded at chromosome 3p21.3, binds pRB and represses E2F-driven transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 16531–16536. [Google Scholar] [CrossRef] [PubMed]
- Kadrmas, J.L.; Beckerle, M.C. The LIM domain: From the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol. 2004, 5, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Das Thakur, M.; Feng, Y.; Jagannathan, R.; Seppa, M.J.; Skeath, J.B.; Longmore, G.D. Ajuba LIM proteins are negative regulators of the Hippo signaling pathway. Curr. Biol. 2010, 20, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Ma, F.; Tian, R.; Zhou, Y.; Lan, W.; Song, Q.; Cheng, X. AJUBA increases the cisplatin resistance through hippo pathway in cervical cancer. Gene 2018, 644, 148–154. [Google Scholar] [CrossRef]
- Shi, X.; Chen, Z.; Hu, X.; Luo, M.; Sun, Z.; Li, J.; Shi, S.; Feng, X.; Zhou, C.; Li, Z.; et al. AJUBA promotes the migration and invasion of esophageal squamous cell carcinoma cells through upregulation of MMP10 and MMP13 expression. Oncotarget 2016, 7, 36407–36418. [Google Scholar] [CrossRef]
- Liang, X.H.; Zhang, G.X.; Zeng, Y.B.; Yang, H.F.; Li, W.H.; Liu, Q.L.; Tang, Y.L.; He, W.G.; Huang, Y.N.; Zhang, L.; et al. LIM protein JUB promotes epithelial-mesenchymal transition in colorectal cancer. Cancer Sci. 2014, 105, 660–666. [Google Scholar] [CrossRef]
- Song, K.; Su, W.; Liu, Y.; Zhang, J.; Liang, Q.; Li, N.; Guan, Q.; He, J.; Bai, X.; Zhao, W.; et al. Identification of genes with universally upregulated or downregulated expressions in colorectal cancer. J. Gastroenterol. Hepatol. 2019, 34, 880–889. [Google Scholar] [CrossRef]
- Tanaka, I.; Osada, H.; Fujii, M.; Fukatsu, A.; Hida, T.; Horio, Y.; Kondo, Y.; Sato, A.; Hasegawa, Y.; Tsujimura, T.; et al. LIM-domain protein AJUBA suppresses malignant mesothelioma cell proliferation via Hippo signaling cascade. Oncogene 2015, 34, 73–83. [Google Scholar] [CrossRef]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kaneda, A.; Tsuji, S.; Isagawa, T.; Yamamoto, S.; Fujita, T.; Yamanaka, R.; Tanaka, Y.; Nukiwa, T.; Marquez, V.E.; et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci. Rep. 2013, 3, 1911. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Ding, X.; Kong, L.; Zhou, X.; Zhang, Z.; Ju, H.; Ding, S. NFATC2 is a novel therapeutic target for colorectal cancer stem cells. Onco Targets Ther. 2018, 11, 6911–6924. [Google Scholar] [CrossRef] [PubMed]
- Howlander, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review; posted to the SEER web site; National Cancer Institute: Bethesda, MD, USA, 2019.
- (ATCC) TATCC. SW480 [SW-480] (ATCC® CCL-228TM). Available online: https://www.lgcstandards-atcc.org/Products/All/CCL-228.aspx?geo_country=ch#generalinformation (accessed on 7 May 2020).
- Brodie, T.M.; Tosevski, V.; Medová, M. OMIP-045: Characterizing human head and neck tumors and cancer cell lines with mass cytometry. Cytometry A 2018, 93, 406–410. [Google Scholar] [CrossRef]
- Haraguchi, K.; Ohsugi, M.; Abe, Y.; Semba, K.; Akiyama, T.; Yamamoto, T. Ajuba negatively regulates the Wnt signaling pathway by promoting GSK-3beta-mediated phosphorylation of beta-catenin. Oncogene 2008, 27, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, R.; Schimizzi, G.V.; Zhang, K.; Loza, A.J.; Yabuta, N.; Nojima, H.; Longmore, G.D. AJUBA LIM Proteins Limit Hippo Activity in Proliferating Cells by Sequestering the Hippo Core Kinase Complex in the Cytosol. Mol. Cell. Biol. 2016, 36, 2526–2542. [Google Scholar] [CrossRef]
- Lange, A.W.; Sridharan, A.; Xu, Y.; Stripp, B.R.; Perl, A.K.; Whitsett, J.A. Hippo/Yap signaling controls epithelial progenitor cell proliferation and differentiation in the embryonic and adult lung. J. Mol. Cell Biol. 2015, 7, 35–47. [Google Scholar] [CrossRef]
- Marie, H.; Pratt, S.J.; Betson, M.; Epple, H.; Kittler, J.T.; Meek, L.; Moss, S.J.; Troyanovsky, S.; Attwell, D.; Longmore, G.D.; et al. The LIM protein Ajuba is recruited to cadherin-dependent cell junctions through an association with alpha-catenin. J. Biol. Chem. 2003, 278, 1220–1228. [Google Scholar] [CrossRef]
- Yang, D.; Hou, T.; Li, L.; Chu, Y.; Zhou, F.; Xu, Y.; Hou, X.; Song, H.; Zhu, K.; Hou, Z.; et al. Smad1 promotes colorectal cancer cell migration through Ajuba transactivation. Oncotarget 2017, 8, 110415–110425. [Google Scholar] [CrossRef]
- Hou, Z.; Peng, H.; Ayyanathan, K.; Yan, K.P.; Langer, E.M.; Longmore, G.D.; Rauscher, F.J. The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol. Cell. Biol. 2008, 28, 3198–3207. [Google Scholar] [CrossRef]
- Kisseleva, M.; Feng, Y.; Ward, M.; Song, C.; Anderson, R.A.; Longmore, G.D. The LIM protein Ajuba regulates phosphatidylinositol 4,5-bisphosphate levels in migrating cells through an interaction with and activation of PIPKI alpha. Mol. Cell. Biol. 2005, 25, 3956–3966. [Google Scholar] [CrossRef]
- Ayyanathan, K.; Peng, H.; Hou, Z.; Fredericks, W.J.; Goyal, R.K.; Langer, E.M.; Longmore, G.D.; Rauscher, F.J. The Ajuba LIM domain protein is a corepressor for SNAG domain mediated repression and participates in nucleocytoplasmic Shuttling. Cancer Res. 2007, 67, 9097–9106. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Unternaehrer, J.J. Epithelial-mesenchymal Transition and Cancer Stem Cells: At the Crossroads of Differentiation and Dedifferentiation. Dev. Dyn. 2019, 248, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Torres, M.; Allan, A.L. Aldehyde dehydrogenase as a marker and functional mediator of metastasis in solid tumors. Clin. Exp. Metastasis 2016, 33, 97–113. [Google Scholar] [CrossRef]
- Kalan, S.; Matveyenko, A.; Loayza, D. LIM Protein Ajuba Participates in the Repression of the ATR-Mediated DNA Damage Response. Front. Genet. 2013, 4, 95. [Google Scholar] [CrossRef]
- Fowler, S.; Maguin, P.; Kalan, S.; Loayza, D. LIM Protein Ajuba associates with the RPA complex through direct cell cycle-dependent interaction with the RPA70 subunit. Sci. Rep. 2018, 8, 9536. [Google Scholar] [CrossRef]
- Hutchinson, J.A.; Shanware, N.P.; Chang, H.; Tibbetts, R.S. Regulation of ribosomal protein S6 phosphorylation by casein kinase 1 and protein phosphatase 1. J. Biol. Chem. 2011, 286, 8688–8696. [Google Scholar] [CrossRef] [PubMed]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef]
- Walker-Daniels, J.; Coffman, K.; Azimi, M.; Rhim, J.S.; Bostwick, D.G.; Snyder, P.; Kerns, B.J.; Waters, D.J.; Kinch, M.S. Overexpression of the EphA2 tyrosine kinase in prostate cancer. Prostate 1999, 41, 275–280. [Google Scholar] [CrossRef]
- Martini, G.; Cardone, C.; Vitiello, P.P.; Belli, V.; Napolitano, S.; Troiani, T.; Ciardiello, D.; Della Corte, C.M.; Morgillo, F.; Matrone, N.; et al. EPHA2 Is a Predictive Biomarker of Resistance and a Potential Therapeutic Target for Improving Antiepidermal Growth Factor Receptor Therapy in Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 845–855. [Google Scholar] [CrossRef]
- Zelinski, D.P.; Zantek, N.D.; Stewart, J.C.; Irizarry, A.R.; Kinch, M.S. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001, 61, 2301–2306. [Google Scholar]
- Zhou, Y.; Sakurai, H. Emerging and Diverse Functions of the EphA2 Noncanonical Pathway in Cancer Progression. Biol. Pharm. Bull. 2017, 40, 1616–1624. [Google Scholar] [CrossRef]
- Dohn, M.; Jiang, J.; Chen, X. Receptor tyrosine kinase EphA2 is regulated by p53-family proteins and induces apoptosis. Oncogene 2001, 20, 6503–6515. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Krasinskas, A.M. EGFR Signaling in Colorectal Carcinoma. Patholog. Res. Int. 2011, 2011, 932932. [Google Scholar] [CrossRef] [PubMed]
- Spano, J.P.; Fagard, R.; Soria, J.C.; Rixe, O.; Khayat, D.; Milano, G. Epidermal growth factor receptor signaling in colorectal cancer: Preclinical data and therapeutic perspectives. Ann. Oncol. 2005, 16, 189–194. [Google Scholar] [CrossRef]
- Jia, H.; Song, L.; Cong, Q.; Wang, J.; Xu, H.; Chu, Y.; Li, Q.; Zhang, Y.; Zou, X.; Zhang, C.; et al. The LIM protein AJUBA promotes colorectal cancer cell survival through suppression of JAK1/STAT1/IFIT2 network. Oncogene 2017, 36, 2655–2666. [Google Scholar] [CrossRef]
- Liu, M.; Jiang, K.; Lin, G.; Liu, P.; Yan, Y.; Ye, T.; Yao, G.; Barr, M.P.; Liang, D.; Wang, Y.; et al. Ajuba inhibits hepatocellular carcinoma cell growth via targeting of β-catenin and YAP signaling and is regulated by E3 ligase Hakai through neddylation. J. Exp. Clin. Cancer Res. 2018, 37, 165. [Google Scholar] [CrossRef]
- Nola, S.; Daigaku, R.; Smolarczyk, K.; Carstens, M.; Martin-Martin, B.; Longmore, G.; Bailly, M.; Braga, V.M. Ajuba is required for Rac activation and maintenance of E-cadherin adhesion. J. Cell Biol. 2011, 195, 855–871. [Google Scholar] [CrossRef]
- Abe, Y.; Ohsugi, M.; Haraguchi, K.; Fujimoto, J.; Yamamoto, T. LATS2-Ajuba complex regulates gamma-tubulin recruitment to centrosomes and spindle organization during mitosis. FEBS Lett. 2006, 580, 782–788. [Google Scholar] [CrossRef]
- Langer, E.M.; Feng, Y.; Zhaoyuan, H.; Rauscher, F.J.; Kroll, K.L.; Longmore, G.D. Ajuba LIM proteins are snail/slug corepressors required for neural crest development in Xenopus. Dev. Cell. 2008, 14, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Montoya-Durango, D.E.; Velu, C.S.; Kazanjian, A.; Rojas, M.E.; Jay, C.M.; Longmore, G.D.; Grimes, H.L. Ajuba functions as a histone deacetylase-dependent co-repressor for autoregulation of the growth factor-independent-1 transcription factor. J. Biol. Chem. 2008, 283, 32056–32065. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, J.; Pratt, S.J.; Marie, H.; Longmore, G.D. Ajuba, a cytosolic LIM protein, shuttles into the nucleus and affects embryonal cell proliferation and fate decisions. Mol. Biol. Cell. 2000, 11, 3299–3313. [Google Scholar] [CrossRef] [PubMed]
- Dawid, I.B.; Breen, J.J.; Toyama, R. LIM domains: Multiple roles as adapters and functional modifiers in protein interactions. Trends Genet. 1998, 14, 156–162. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Therneau, T.M. Modeling Survival Data: Extending the Cox Model; Grambsch, P.M., Ed.; Springer: New York, NY, USA, 2000. [Google Scholar]
- Way, M. ggkm: Kaplan-Meier Survival Curves with Numbers at Risk. R package version 1.0. 2016. Available online: https://rdrr.io/github/michaelway/ggkm/ (accessed on 24 September 2019).
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic. Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Warnes, G.R. Gplots: Various R Programming Tools for Plotting Data. 2019. Available online: https://CRAN.R-project.org/package=gplots (accessed on 24 September 2019).
- Diagrams CadcV. Available online: http://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed on 25 September 2019).
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Gherardini, P.F. Premessa: R Package for Pre-Progessing of Flow and Mass Cytometry. 2019. Available online: https://github.com/ParkerICI/premessa (accessed on 30 September 2019).
- Haaland, E.B. FlowCore: Basic Structures for Flow Cytometry Data. 2019. Available online: https://www.bioconductor.org/packages/release/bioc/html/flowCore.html (accessed on 30 September 2019).
- Van Der Maaten, L. Accelerating t-SNE using Tree-Based Algorithms. J. Mach. Learn. 2014, 15, 3221–3245. [Google Scholar]
- Soares, K.C.; Foley, K.; Olino, K.; Leubner, A.; Mayo, S.C.; Jain, A.; Jaffee, E.; Schulick, R.D.; Yoshimura, K.; Edil, B.; et al. A preclinical murine model of hepatic metastases. J. Vis. Exp. 2014, 51677. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2:Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dommann, N.; Sánchez-Taltavull, D.; Eggs, L.; Birrer, F.; Brodie, T.; Salm, L.; Baier, F.A.; Medová, M.; Humbert, M.; Tschan, M.P.; et al. The LIM Protein Ajuba Augments Tumor Metastasis in Colon Cancer. Cancers 2020, 12, 1913. https://doi.org/10.3390/cancers12071913
Dommann N, Sánchez-Taltavull D, Eggs L, Birrer F, Brodie T, Salm L, Baier FA, Medová M, Humbert M, Tschan MP, et al. The LIM Protein Ajuba Augments Tumor Metastasis in Colon Cancer. Cancers. 2020; 12(7):1913. https://doi.org/10.3390/cancers12071913
Chicago/Turabian StyleDommann, Noëlle, Daniel Sánchez-Taltavull, Linda Eggs, Fabienne Birrer, Tess Brodie, Lilian Salm, Felix Alexander Baier, Michaela Medová, Magali Humbert, Mario P. Tschan, and et al. 2020. "The LIM Protein Ajuba Augments Tumor Metastasis in Colon Cancer" Cancers 12, no. 7: 1913. https://doi.org/10.3390/cancers12071913
APA StyleDommann, N., Sánchez-Taltavull, D., Eggs, L., Birrer, F., Brodie, T., Salm, L., Baier, F. A., Medová, M., Humbert, M., Tschan, M. P., Beldi, G., Candinas, D., & Stroka, D. (2020). The LIM Protein Ajuba Augments Tumor Metastasis in Colon Cancer. Cancers, 12(7), 1913. https://doi.org/10.3390/cancers12071913