A Molecular Stratification of Chilean Gastric Cancer Patients with Potential Clinical Applicability

 , , , , , , , ,
, , , , , , , ,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Results
2.1. A Molecular Subtype Classification for GC Based on CISH/IHC
2.2. NGS Analysis by Subtype
2.3. Differences between GC Subtypes and Patient Prognosis
2.4. Response to Chemotherapy in GC Subtypes
2.5. A Low-Cost Stratification System for GC Patients
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Hematoxylin and Eosin and Immunohistochemistry (IHC) Assays
4.3. IHC for HER2 and PDL1
4.4. Epstein–Barr Virus (EBV) and Mismatch Repair (MMR) Status Determination
4.5. E-Cadherin and p53 Status Determination
4.6. Genomic Analysis
4.7. Outcomes and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2018, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Gullo, I.; Carneiro, F.; Oliveira, C.; Almeida, G.M. Heterogeneity in gastric cancer: From pure morphology to molecular classifications. Pathobiology 2017, 85, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Kato, M.; Asaka, M. Geographic differences in gastric cancer incidence can be explained by differences between helicobacter pylori strains. Intern. Med. 2008, 47, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Etemadi, A.; Safiri, S.; Sepanlou, S.G.; Ikuta, K.; Bisignano, C.; Shakeri, R.; Amani, M.; Fitzmaurice, C.; Nixon, M.; Abbasi, N.; et al. The global, regional, and national burden of stomach cancer in 195 countries, 1990-2017: A systematic analysis for the global burden of disease study 2017. Lancet Gastroenterol. 2019, 5, 42–54. [Google Scholar] [CrossRef]
- Hu, B.; El Hajj, N.; Sittler, S.; Lammert, N.; Barnes, R.; Meloni-Ehrig, A. Gastric cancer: Classification, histology and application of molecular pathology. J. Gastrointest. Oncol. 2012, 3, 251–261. [Google Scholar]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Ma, J.; Shen, H.; Kapesa, L.; Zeng, S. Lauren classification and individualized chemotherapy in gastric cancer. Oncol. Lett. 2016, 11, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Haboubi, N. Pathology and genetics: Tumours of the digestive system. Surg. Oncol. 2000, 9, 144–145. [Google Scholar] [CrossRef]
- Sano, T.; Kodera, Y. Japanese gastric cancer association Japanese classification of gastric carcinoma, 3rd English edition. Gastric Cancer 2011, 14, 101–112. [Google Scholar] [CrossRef]
- Tan, I.B.; Ivanova, T.; Lim, K.H.; Ong, C.W.; Deng, N.; Lee, J.; Tan, S.H.; Wu, J.; Lee, M.H.; Ooi, C.H.; et al. Intrinsic subtypes of gastric cancer, based on gene expression pattern, predict survival and respond differently to chemotherapy. Gastroenterology 2011, 141, 476–485. [Google Scholar] [CrossRef]
- Lei, Z.; Tan, I.B.; Das, K.; Deng, N.; Zouridis, H.; Pattison, S.; Chua, C.; Feng, Z.; Guan, Y.K.; Ooi, C.H.; et al. Identification of molecular subtypes of gastric cancer with different responses to pi3-kinase inhibitors and 5-fluorouracil. Gastroenterology 2013, 145, 554–565. [Google Scholar] [CrossRef]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Cai, M.; Shuai, X.; Gao, J.; Wang, G.; Tao, K. First-line systemic therapy for advanced gastric cancer: A systematic review and network meta-analysis. Ther. Adv. Med. Oncol. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Setia, N.; Agoston, A.T.; Han, H.S.; Mullen, J.T.; Duda, D.G.; Clark, J.W.; Deshpande, V.; Mino-Kenudson, M.; Srivastava, A.; Lennerz, J.K.; et al. A protein and mRNA expression-based classification of gastric cancer. Mod. Pathol. 2016, 29, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Lee, S.J.; Kim, Y.; Kim, A.; Shin, N.; Choi, K.U.; Lee, C.-H.; Huh, G.Y.; Kim, K.-M.; Setia, N.; et al. High-throughput protein and mrna expression–based classification of gastric cancers can identify clinically distinct subtypes, concordant with recent molecular classifications. Am. J. Surg. Pathol. 2017, 41, 106–115. [Google Scholar] [CrossRef]
- Koh, J.; Lee, K.; Nam, S.K.; Na Seo, A.; Kim, J.; Park, D.J.; Kim, H.; Kim, W.H.; Lee, H.S.; Kim, J.W. Development and validation of an easy-to-implement, practical algorithm for the identification of molecular subtypes of gastric cancer: Prognostic and therapeutic implications. Oncologist 2019, 24, 1321–1330. [Google Scholar] [CrossRef]
- Corvalan, A.H.; Koriyama, C.; Akiba, S.; Eizuru, Y.; Backhouse, C.; Palma, M.; Argandona, J.; Tokunaga, M. Epstein-Barr virus in gastric carcinoma is associated with location in the cardia and with a diffuse histology: A study in one area of Chile. Int. J. Cancer 2001, 94, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Cordova-Delgado, M.; Pinto, M.P.; Retamal, I.N.; Muñoz-Medel, M.; Bravo, M.L.; Fernández, M.F.; Cisternas, B.; Mondaca, S.; Sanchez, C.; Galindo, H.; et al. High proportion of potential candidates for immunotherapy in a chilean cohort of gastric cancer patients: Results of the FORCE1 study. Cancers 2019, 11, 1275. [Google Scholar] [CrossRef]
- Giampieri, R.; Maccaroni, E.; Mandolesi, A.; Del Prete, M.; Andrikou, K.; Faloppi, L.; Bittoni, A.; Bianconi, M.; Scarpelli, M.; Bracci, R.; et al. Mismatch repair deficiency may affect clinical outcome through immune response activation in metastatic gastric cancer patients receiving first-line chemotherapy. J. Gastric Cancer 2016, 20, 156–163. [Google Scholar] [CrossRef]
- Chan, W.W.L.; Lam, K.; Lee, V.H.F.; Davidson, M.; So, T.; Li, J.; Chau, I.; Kwong, D. Gastric cancer-from aetiology to management: Differences between the east and the west. Clin. Oncol. 2019, 31, 570–577. [Google Scholar] [CrossRef]
- Crew, K.D.; I Neugut, A. Epidemiology of gastric cancer. World J. Gastroenterol. 2006, 12, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, A.; Yoshida, S.; Saijo, N. Disparities in gastric cancer chemotherapy between the east and west. J. Clin. Oncol. 2006, 24, 2188–2196. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.A.; Sano, T. The difference in gastric cancer between Japan, USA and Europe: What are the facts? What are the suggestions? Crit. Rev. Oncol. Hematol. 2001, 40, 77–94. [Google Scholar] [CrossRef]
- Kubota, Y.; Kawazoe, A.; Sasaki, A.; Mishima, S.; Sawada, K.; Nakamura, Y.; Kotani, D.; Kuboki, Y.; Taniguchi, H.; Kojima, T.; et al. The impact of molecular subtype on efficacy of chemotherapy and checkpoint inhibition in advanced gastric cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Huang, S.C.; Ng, K.F.; Yeh, T.S.; Cheng, C.T.; Lin, J.S.; Liu, Y.J.; Chuang, H.C.; Chen, T.C. Subtraction of Epstein–Barr virus and microsatellite instability genotypes from the Lauren histotypes: Combined molecular and histologic subtyping with clinicopathological and prognostic significance validated in a cohort of 1,248 cases. Int. J. Cancer 2019, 145, 3218–3230. [Google Scholar] [CrossRef] [PubMed]
- Corallo, S.; Fuca, G.; Morano, F.; Salati, M.; Spallanzani, A.; Gloghini, A.; Volpi, C.C.; Trupia, D.V.; Lobefaro, R.; Guarini, V.; et al. Clinical behavior and treatment response of epstein-barr virus–positive metastatic gastric cancer: Implications for the development of future trials. Oncologist 2020. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.F.K.P.; Pereira, M.A.; Amorim, L.C.; Mello, E.S.; Faraj, S.F.; Ribeiro, U.; Hoff, P.M.G.; Cecconello, I.; Castria, T.B. Gastric cancer molecular classification and adjuvant therapy: Is there a different benefit according to the subtype? J. Surg. Oncol. 2019, 121, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Bang, Y.J.; Fuchs, C.S.; Ohtsu, A.; Kher, U.; Lam, B.; Koshiji, M.; Van Cutsem, E. KEYNOTE-062: Phase III study of pembrolizumab (MK-3475) alone or in combination with chemotherapy versus chemotherapy alone as first-line therapy for advanced gastric or gastroesophageal junction (GEJ) adenocarcinoma. J. Clin. Oncol. 2016, 34, TPS185. [Google Scholar] [CrossRef]
- Mehta, R.; Almhanna, K. Predictive markers in gastric cancer immunotherapy treatment-are we there yet? Transl. Gastroenterol. Hepatol. 2019, 4, 4. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Krook, M.A.; Lenyo, A.; Wilberding, M.; Barker, H.; Dantuono, M.; Bailey, K.M.; Chen, H.Z.; Reeser, J.W.; Wing, M.R.; Miya, J.; et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-fusion cholangiocarcinoma. Mol. Cancer Ther. 2020, 19, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Kim, J.-H.; Kim, M.; Kim, S.-Y. Genomic and epigenomic heterogeneity in molecular subtypes of gastric cancer. World J. Gastroenterol. 2016, 22, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, M.; Jiang, Z.; Wang, X. ARID1A mutations are associated with increased immune activity in gastrointestinal cancer. Cells 2019, 8, 678. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, L.C.; Saito, Y.; Wang, T.; Matsuda, Y.; Oosting, J.; Silva, A.N.S.; Slaney, H.L.; Melotte, V.; Hutchins, G.; Tan, P.; et al. KRAS status is related to histological phenotype in gastric cancer: Results from a large multicentre study. Gastric Cancer 2019, 22, 1193–1203. [Google Scholar] [CrossRef]
- Yoon, C.; Till, J.E.; Cho, S.J.; Chang, K.K.; Lin, J.X.; Huang, C.M.; Ryeom, S.; Lin, J.X. KRAS activation in gastric adenocarcinoma stimulates epithelial-to-mesenchymal transition to cancer stem-like cells and promotes metastasis. Mol. Cancer Res. 2019, 17, 1945–1957. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Knelson, E.H.; Sequist, L.V. A bright future for KRAS inhibitors. Nat. Cancer 2020, 1, 25–27. [Google Scholar] [CrossRef]
- Bernal, C.; Vargas, M.; Ossandon, F.; Santibanez, E.; Urrutia, J.; Luengo, V.; Zavala, L.F.; Backhouse, C.; Palma, M.; Argandona, J.; et al. DNA methylation profile in diffuse type gastric cancer: Evidence for hypermethylation of the BRCA1 promoter region in early-onset gastric carcinogenesis. Biol. Res. 2009, 41, 303–315. [Google Scholar] [CrossRef]
- Carrasco-Avino, G.; Diaz, J.; Valbuena, J.R.; Araya, G.; Torres, J.; Duarte, I.; Aravena, E.; Mena, F.; Barrientos, C.; Corvalan, A.H.; et al. Overexpression of p73 as a tissue marker for high-risk gastritis. Clin. Cancer Res. 2010, 16, 3253–3259. [Google Scholar] [CrossRef]
- Ho, S.W.T.; Tan, P. Dissection of gastric cancer heterogeneity for precision oncology. Cancer Sci. 2019, 110, 3405–3414. [Google Scholar] [CrossRef]
- Uchôa Guimarães, C.T.U.; Martins, N.N.F.; Oliveira, K.C.D.S.; Almeida, C.M.; Pinheiro, T.M.; Gigek, C.O.; Cavallero, S.R.D.A.; Assumpcao, P.P.; Smith, M.A.C.; Burbano, R.R.; et al. Liquid biopsy provides new insights into gastric cancer. Oncotarget 2018, 9, 15144–15156. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Corso, G.; Carvalho, J.; Marrelli, D.; Vindigni, C.; Carvalho, B.; Seruca, R.; Roviello, F.; Oliveira, C. Somatic mutations and deletions of the e-cadherin gene predict poor survival of patients with gastric cancer. J. Clin. Oncol. 2013, 31, 868–875. [Google Scholar] [CrossRef]
- Rüschoff, J.; Hanna, W.; Bilous, M.; Hofmann, M.; Osamura, R.Y.; Penault-Llorca, F.; van de Vijver, M.; Viale, G. HER2 testing in gastric cancer: A practical approach. Mod. Pathol. 2012, 25, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.; Yates, C.; Shepard, C.R. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin. Exp. Metastasis 2008, 25, 621–628. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Owen, G.I.; Pinto, M.P.; Retamal, I.N.; Fernádez, M.F.; Cisternas, B.; Mondaca, S.; Sánchez, C.; Galindo, H.; Nervi, B.; Ibañez, C.; et al. Chilean gastric cancer task force. Medicine (Baltimore) 2018, 97, e0419. [Google Scholar] [CrossRef]




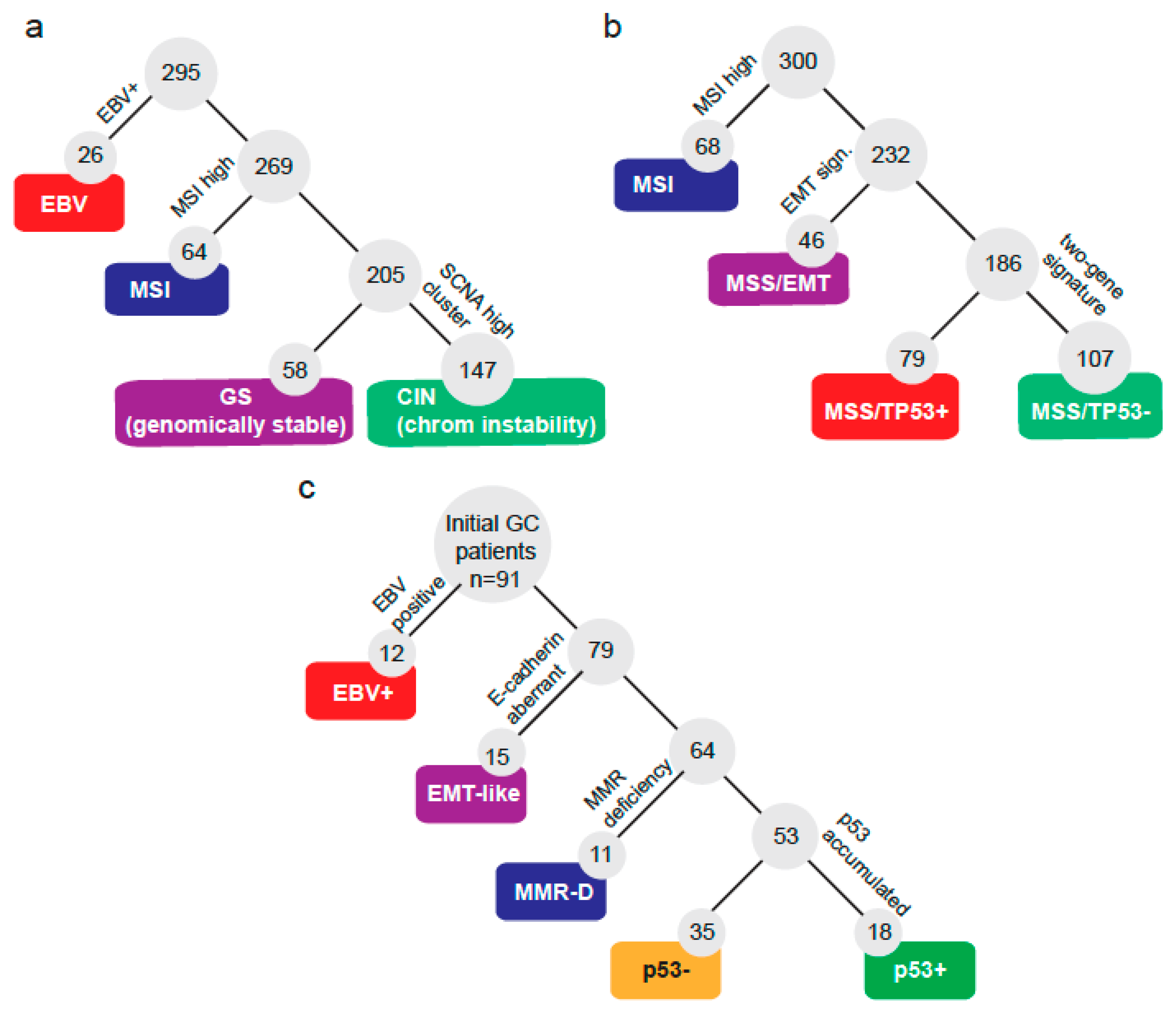{kind=link}
{kind=link}
{kind=link}
| Characteristic | Units (IQR or %) |
|---|---|
| Median OS; months | 30 (16–61) |
| Males | 60 (66) |
| Females | 31 (34) |
| Median age; years | 63 (55–73) |
| Stage | |
| I/II | 33 (36) |
| III | 46 (51) |
| IV | 12 (13) |
| Lauren histotype | |
| Intestinal | 27 (37) |
| Diffuse | 31 (42) |
| Mixed | 15 (21) |
| WHO classification | |
| Adenocarcinoma | 77 (88) |
| Undifferentiated carcinoma | 8 (9) |
| Adeno-squamous carcinoma | 3 (3) |
| Primary tumor location | |
| Proximal | 26 (30) |
| Medial | 30 (34) |
| Distal | 27 (31) |
| Multiple | 4 (5) |
| Signet ring cells | |
| None | 58 (64) |
| <50% | 27 (30) |
| ≥50% | 6 (6) |
| HER2+ status | 12 (13) |
| PDL1+ status | 26 (29) |
| EBV+ status | 12 (13) |
| MMR-deficient | 13 (14) |
| p53+ status | 39 (43) |
| E-cadherin loss | 15 (17) |
| Comorbidities | |
| None | 46 (51) |
| 1 | 20 (22) |
| 2 | 18 (20) |
| ≥3 | 7 (8) |
| Characteristic | MMR-D n = 11 | EBV+ n = 12 | p53+ n = 18 | p53– n = 35 | EMT-Like n = 15 | Total n = 91 | p-Value |
|---|---|---|---|---|---|---|---|
| Median OS; Months (range) | 64 (17–98) | 38 (20.5–57) | 28 (13–40) | 39.5 (18–62) | 16 (12–29) | 30 (16–61) | 0.01 |
| Males; n (%) | 6 (55) | 11 (92) | 11 (61) | 22 (63) | 10 (67) | 60 (66) | 0.31 |
| Median age; years (IQR 1) | 66 (64–70) | 57 (52–69) | 63.5 (56–71) | 64.5 (55–76) | 57 (50–75) | 63.5 (55–73) | 0.22 |
| Stage; n (%) | |||||||
| I/II | 7 (64) | 6 (50) | 9 (50) | 10 (29) | 1 (7) | 33 (36) | 0.058 |
| III | 4 (36) | 4 (33) | 7 (39) | 20 (57) | 11 (73) | 46 (51) | |
| IV | 0 | 2 (17) | 2 (11) | 5 (14) | 3 (20) | 12 (13) | |
| Primary tumor; n (%) | |||||||
| Proximal | 1 (9) | 9 (75) | 5 (29) | 8 (25) | 3 (20) | 26 (30) | 0.081 |
| Medial | 3 (27) | 2 (17) | 5 (29) | 14 (44) | 6 (40) | 30 (34) | |
| Distal | 7 (64) | 1 (8) | 6 (35) | 8 (25) | 5 (33) | 27 (31) | |
| Multiple | 0 | 0 | 1 (6) | 2 (6) | 1 (7) | 4 (5) | |
| IHC 2; n (%) | |||||||
| HER2+ | 1 (9) | 0 | 6 (33) | 5 (14) | 0 | 12 (13) | 0.037 |
| PDL1+ | 3 (27) | 11 (92) | 5 (28) | 5 (14) | 2 (13) | 26 (29) | <0.001 |
| Signet ring cells; n (%) | |||||||
| No | 7 (64) | 12 (100) | 14 (78) | 18 (51) | 7 (47) | 58 (64) | 0.017 |
| <50% | 4 (36) | 0 | 3 (17) | 12 (34) | 8 (53) | 27 (30) | |
| ≥50% | 0 | 0 | 1 (6) | 5 (14) | 0 | 6 (7) | |
| Lauren histotype; n (%) | |||||||
| Diffuse | 5 (56) | 2 (29) | 3 (20) | 14 (50) | 7 (50) | 31 (42) | 0.038 |
| Intestinal | 2 (22) | 4 (57) | 10 (67) | 10 (36) | 1 (7) | 29 (40) | |
| Mixed | 2 (22) | 1 (14) | 2 (13) | 4 (14) | 6 (43) | 13 (18) | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, M.P.; Córdova-Delgado, M.; Retamal, I.N.; Muñoz-Medel, M.; Bravo, M.L.; Durán, D.; Villanueva, F.; Sanchez, C.; Acevedo, F.; Mondaca, S.; et al. A Molecular Stratification of Chilean Gastric Cancer Patients with Potential Clinical Applicability. Cancers 2020, 12, 1863. https://doi.org/10.3390/cancers12071863
Pinto MP, Córdova-Delgado M, Retamal IN, Muñoz-Medel M, Bravo ML, Durán D, Villanueva F, Sanchez C, Acevedo F, Mondaca S, et al. A Molecular Stratification of Chilean Gastric Cancer Patients with Potential Clinical Applicability. Cancers. 2020; 12(7):1863. https://doi.org/10.3390/cancers12071863
Chicago/Turabian StylePinto, Mauricio P., Miguel Córdova-Delgado, Ignacio N. Retamal, Matías Muñoz-Medel, M. Loreto Bravo, Doris Durán, Francisco Villanueva, César Sanchez, Francisco Acevedo, Sebastián Mondaca, and et al. 2020. "A Molecular Stratification of Chilean Gastric Cancer Patients with Potential Clinical Applicability" Cancers 12, no. 7: 1863. https://doi.org/10.3390/cancers12071863
APA StylePinto, M. P., Córdova-Delgado, M., Retamal, I. N., Muñoz-Medel, M., Bravo, M. L., Durán, D., Villanueva, F., Sanchez, C., Acevedo, F., Mondaca, S., Koch, E., Ibañez, C., Galindo, H., Madrid, J., Nervi, B., Peña, J., Torres, J., Owen, G. I., Corvalán, A. H., ... Garrido, M. (2020). A Molecular Stratification of Chilean Gastric Cancer Patients with Potential Clinical Applicability. Cancers, 12(7), 1863. https://doi.org/10.3390/cancers12071863