Desmoid-Type Fibromatosis

 , ,
, , 
Abstract
1. Introduction
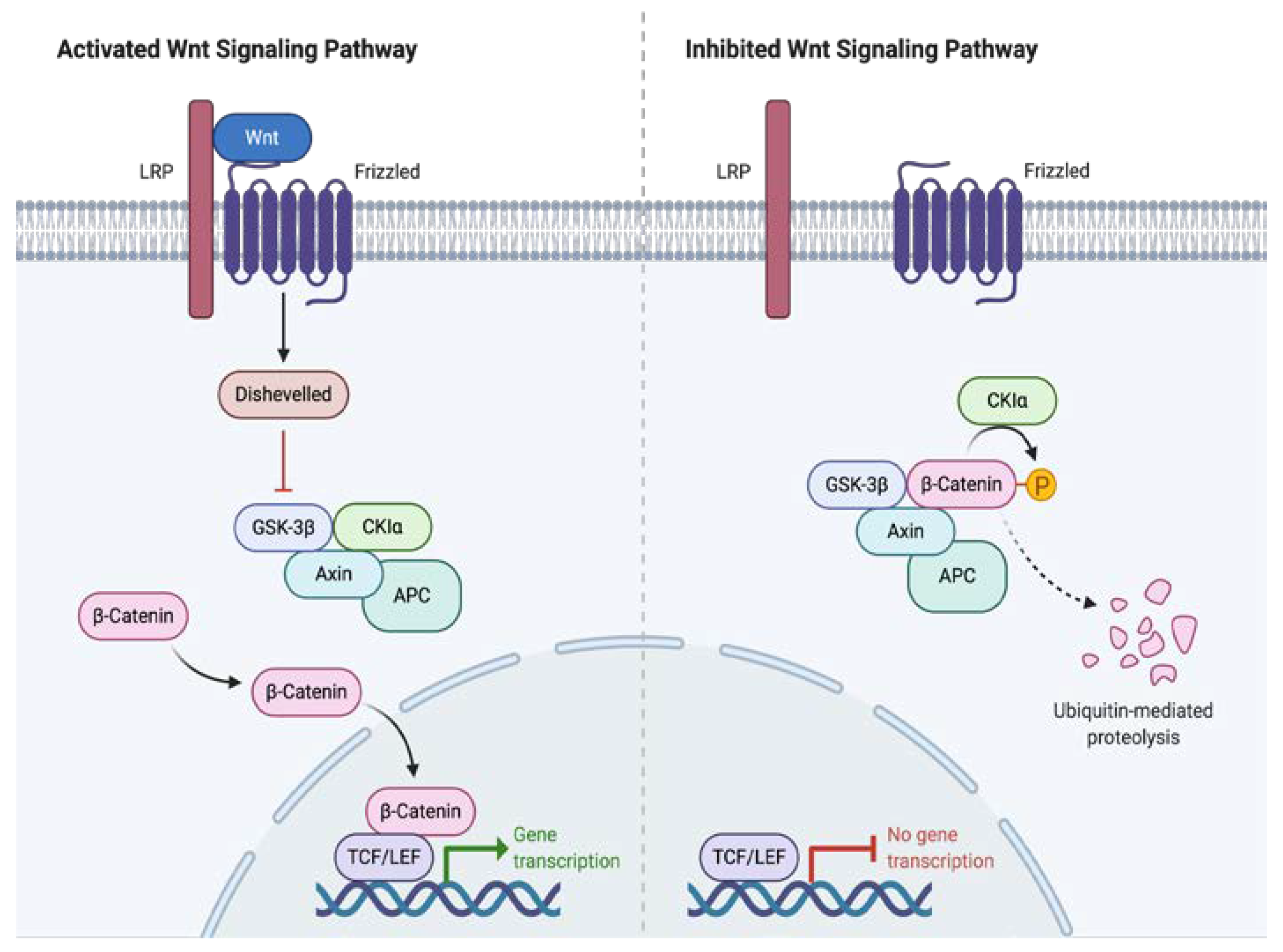
2. Tumor Biology and Signaling Pathways Involved in the Oncogenesis of Desmoid Tumors
3. Histopathology
4. Clinical Features
5. Image Studies
6. Diagnosis
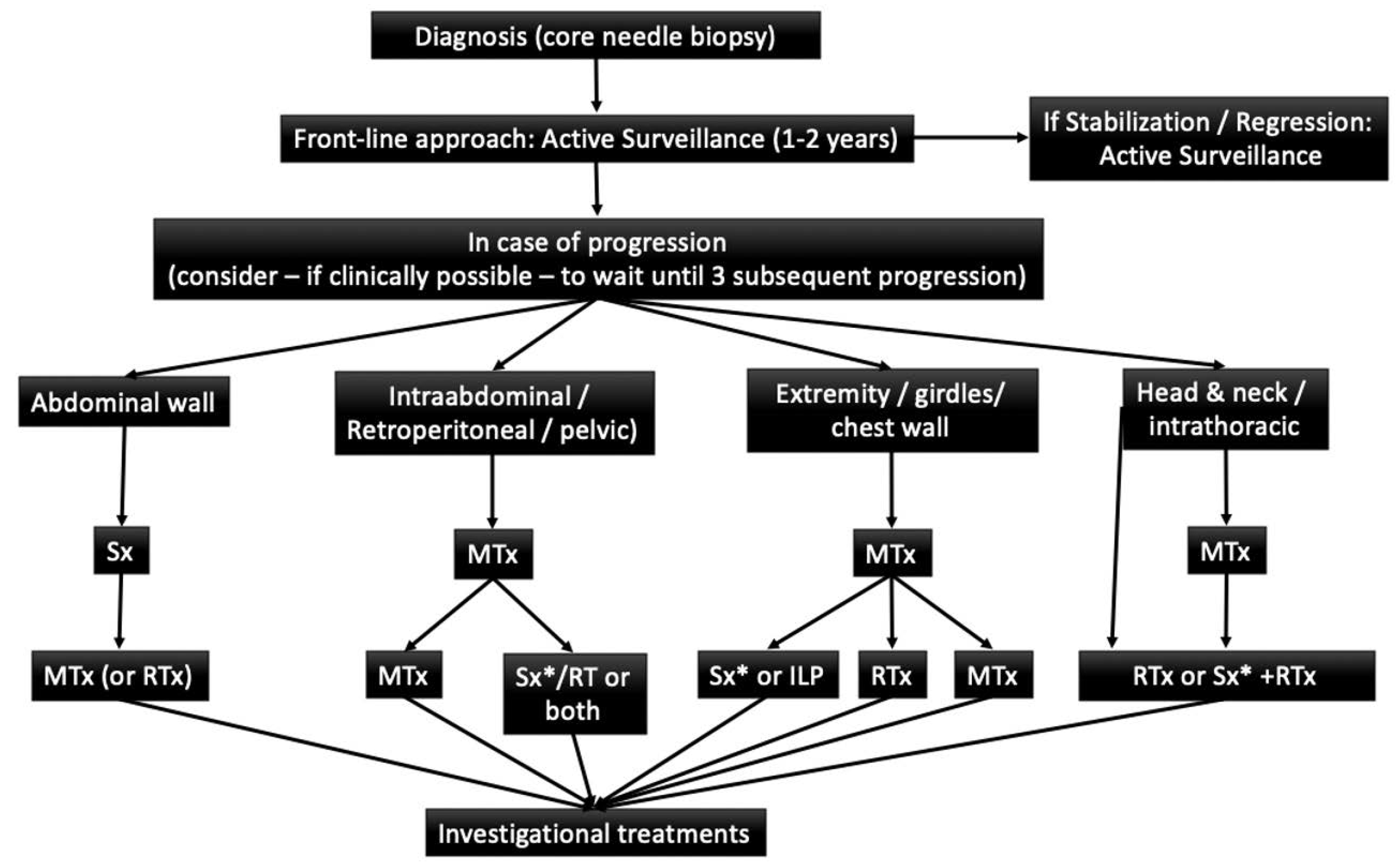
7. Treatment
8. Active Surveillance
9. Surgical Treatment
10. Systemic Therapy
11. Radiotherapy
12. Follow up
13. Quality of Life
14. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Penel, N.; Coindre, J.M.; Bonvalot, S.; Italiano, A.; Neuville, A.; Le Cesne, A.; Terrier, P.; Ray-Coquard, I.; Ranchere-Vince, D.; Robin, Y.M.; et al. Management of desmoid tumours: A nationwide survey of labelled reference centre networks in France. Europe. Eur. J. Cancer 2016, 58–90, e6. [Google Scholar] [CrossRef] [PubMed]
- Kasper, B.; Stroebel, P.; Hohenberger, P. Desmoid tumors–clinical features and treatment options for advanced disease. Oncologist 2011, 16, 682. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.D.M.; Bridge, J.A.; Hogendoorn, P.; Mertens, F. WHO Classification of Tumours of Soft Tissue and Bone (IARC WHO Classification of Tumours), 4th ed.; IARC: Lyon, France, 2013. [Google Scholar]
- Martinez Trufero, J.; Pajares Bernad, I.; Torres Ramon, I.; Hernando, C.J.; Pazo Cid, R. Desmoid Type Fibromatosis: Who, When, and How to Treat. Curr. Treat. Options Oncol. 2017, 18, 29. [Google Scholar] [CrossRef]
- Fiore, M.; MacNeill, A.; Gronchi, A.; Colombo, C. Desmoid-Type Fibromatosis: Evolving Treatment Standards. Surg. Oncol. Clin. N. Am. 2016, 25, 803–826. [Google Scholar] [CrossRef]
- Gurbuz, A.K.; Giardiello, F.M.; Petersen, G.M.; Krush, A.J.; Offerhaus, G.J.; Booker, S.V.; Kerr, M.C.; Hamilton, S.R. Desmoid tumours in familial adenomatous polyposis. Gut 1994, 35, 377–381. [Google Scholar] [CrossRef]
- Koskenvuo, L.; Ristimaki, A.; Lepisto, A. Comparison of sporadic and FAP-associated desmoid-type fibromatoses. J. Surg. Oncol. 2017, 116, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, M.H.; Casparie, M.; Mathus-Vliegen, L.M.; Dekkers, O.M.; Hogendoorn, P.C.; Vasen, H.F. A nation-wide study comparing sporadic and familial adenomatous polyposis-related desmoid-type fibromatoses. Int. J. Cancer 2011, 129, 256–261. [Google Scholar] [CrossRef]
- Angers, S. RTMoon, Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef] [PubMed]
- 10. Salas, S.; Chibon, F.; Noguchi, T.; Terrier, P.; Ranchere-Vince, D.; Lagarde, P.; Benard, J.; Forget, S.; Blanchard, C.; Dômont, J.; et al. Molecular characterization by array comparative genomic hybridization and DNA sequencing of 194 desmoid tumors. Genes Chromosomes Cancer 2010, 49, 560–568. [Google Scholar]
- Li, J.; Wang, C.Y. TBL1-TBLR1 and beta-catenin recruit each other to Wnt target-gene promoter for transcription activation and oncogenesis. Nat. Cell Biol. 2008, 10, 160–169. [Google Scholar] [CrossRef]
- Crago, A.M.; Chmielecki, J.; Rosenberg, M.; O’Connor, R.; Byrne, C.; Wilder, F.G.; Thorn, K.; Agius, P.; Kuk, D.; Socci, N.D.; et al. Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid-type fibromatosis by whole-exome sequencing and genomic analysis. Genes Chromosomes Cancer 2015, 54, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.J.; Tuvin, D.; Hajibashi, S.; Habeeb, S.; Bolshakov, S.; Mayordomo-Aranda, E.; Warneke, C.L.; Lopez-Terrada, D.; Pollock, R.E.; Lev, D. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am. J. Pathol. 2008, 173, 1518–1527. [Google Scholar] [CrossRef]
- Hamada, S.; Futamura, N.; Ikuta, K.; Urakawa, H.; Kozawa, E.; Ishiguro, N.; Nishida, Y. CTNNB1 S45F mutation predicts poor efficacy of meloxicam treatment for desmoid tumors: A pilot study. PLoS ONE 2014, 9, 6391. [Google Scholar] [CrossRef] [PubMed]
- Church, J.; Xhaja, X.; LaGuardia, L.; O’Malley, M.; Burke, C.; Kalady, M. Desmoids and Genotype in Fam. Adenomatous Polyposis. Dis. Colon Rectum 2015, 58, 444–448. [Google Scholar] [CrossRef]
- Escobar, C.; Munker, R.; Thomas, J.O.; Li, B.D.; Burton, G.V. Update on desmoid tumors. Ann. Oncol. 2012, 23, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Sobin, L.H.; Shekitka, K.M.; Federspiel, B.H.; Helwig, E.B. Intra-abdominal fibromatosis. A pathologic analysis of 130 tumors with comparison of clinical subgroups. Am. J. Surg. Pathol. 1990, 14, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Kotiligam, D.; Lazar, A.J.F.; Pollock, R.; Lev, D. Desmoid tumor: A disease opportune for molecular insights. Histol. Histopathol. 2008, 23, 117–126. [Google Scholar] [PubMed]
- Owens, C.L.; Sharma, R.; Ali, S.Z. Deep fibromatosis (desmoid tumor): Cytopathologic characteristics, clinicoradiologic features, and immunohistochemical findings on fine-needle aspiration. Cancer 2007, 111, 166–172. [Google Scholar] [CrossRef]
- Devata, S.; Chugh, R. Desmoid tumors: A comprehensive review of the evolving biology, unpredictable behavior, and myriad of management options. Hematol. Oncol. Clin. N. Am. 2013, 27, 989–1005. [Google Scholar] [CrossRef]
- Chew, C.; Reid, R.; O’Dwyer, P.J. Evaluation of the Long Term Outcome of Patients with Extremity. Desmoids. Eur. J. Surg. Oncol. 2004, 30, 428–432. [Google Scholar] [CrossRef]
- Mantello, M.T.; Haller, J.O.; Marquis, J.R. Sonography of abdominal desmoid tumors in adolescents. J. Ultrasound Med. 1989, 8, 467–470. [Google Scholar] [CrossRef]
- Huang, C.C.; Ko, S.F.; Yeh, M.C.; Ng, S.H.; Huang, H.Y.; Lee, C.C.; Lee, T.Y. Aggressive fibromatosis of the chest wall: Sonographic appearance of the fascial tail and staghorn patterns. J. Ultrasound Med. 2009, 28, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Murphey, M.D.; Ruble, C.M.; Tyszko, S.M.; Zbojniewicz, A.M.; Potter, B.K.; Miettinen, M. From the archives of the AFIP: Musculoskeletal fibromatoses: Radiologic-pathologic correlation. Radiographics 2009, 29, 2143–2173. [Google Scholar] [CrossRef] [PubMed]
- Dinauer, P.A.; Brixey, C.J.; Moncur, J.T.; Fanburg-Smith, J.C.; Murphey, M.D. Pathologic and MR imaging features of benign fibrous soft-tissue tumors in adults. Radiographics 2007, 27, 173–187. [Google Scholar] [CrossRef]
- Cassidy, M.R.; Lefkowitz, R.A.; Long, N.; Qin, L.X.; Kirane, A.; Sbaity, E.; Hameed, M.; Coit, D.G.; Brennan, M.F.; Singer, S.; et al. Association of MRI T2 Signal Intensity with Desmoid Tumor Progression During Active Observation: A Retrospective Cohort Study. Ann. Surg. 2018. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Nair, N.; Banavali, S. Uptake characteristics of fluorodeoxyglucose (FDG) in deep fibromatosis and abdominal desmoids: Potential clinical role of FDG-PET in the management. Br. J. Radiol. 2007, 80, 750–756. [Google Scholar] [CrossRef]
- Kasper, B.; Baumgarten, C.; Garcia, J.; Bonvalot, S.; Haas, R.; Haller, F.; Hohenberger, P.; Penel, N.; Messiou, C.; van der Graaf, W.T.; et al. An update on the management of sporadic desmoid-type fibromatosis. Ann. Oncol. 2017, 28, 2399–2408. [Google Scholar] [CrossRef]
- Desmoid Tumor Working Group. The management of desmoid tumours: A joint global consensus-based guideline approach for adult and paediatric patients. Eur J. Cancer 2020, 127, 96–107. [Google Scholar] [CrossRef]
- Improta, L.; Tzanis, D.; Bouhadiba, T.; Abdelhafidh, K.; Bonvalot, S. Desmoid tumours in the surveillance era: What are the remaining indications for surgery? Eur. J. Surg. Oncol. 2020, 46, 1310–1314. [Google Scholar] [CrossRef]
- Al-Jazrawe, M.; Au, M.; Alman, B. Optimal therapy for desmoid tumors: Current options and challenges for the future. Expert Rev. Anticancer Ther. 2015, 15, 1443–1458. [Google Scholar] [CrossRef]
- Bonvalot, S.; Eldweny, H.; Haddad, V.; Rimareix, F.; Missenard, G.; Oberlin, O.; Vanel, D.; Terrier, P.; Blay, J.Y.; Le Cesne, A.; et al. Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur. J. Surg. Oncol. 2008, 34, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Salas, S.; Dufresne, A.; Bui, B.; Blay, J.Y.; Terrier, P.; Ranchere-Vince, D.; Bonvalot, S.; Stoeckle, E.; Guillou, L.; Le Cesne, A.; et al. Prognostic factors influencing progression-free survival determined from a series of sporadic desmoid tumors: A wait-and-see policy according to tumor presentation. J. Clin. Oncol. 2011, 29, 3553–3558. [Google Scholar] [CrossRef] [PubMed]
- Grignol, V.P.; Pollock, R.; Howard, J.H.H. Management of Desmoids. Surg. Clin. N. Am. 2016, 96, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- de Bénazé, G.D.; Vigan, M.; Corradini, N.; Minard-Colin, V.; Marie-Cardine, A.; Verite, C.; Defachelles, A.S.; Thebaud, E.; Castex, M.P.; Sirvent, N.; et al. Functional analysis of young patients with desmoid-type fibromatosis: Initial Surveillance Does not Jeopardize Long Term Quality of life. Eur. J. Surg. Oncol. 2020, 46, 1294–1300. [Google Scholar] [CrossRef]
- van Houdt, W.J.; Husson, O.; Patel, A.; Jones, R.L.; Smith, M.J.; Miah, A.B.; Messiou, C.; Moskovic, E.; Al-Muderis, O.; Benson, C.; et al. Outcome of primary desoid tumors at all anatomic locations initially managed with active surveillance. Ann. Surg. Oncol. 2019, 26, 4699–4706. [Google Scholar] [CrossRef]
- Penel, N.; Chibon, F.; Salas, S. Adult desmoid tumors: Biology, management and ongoing trials. Curr. Opin. Oncol. 2017, 29, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Stojadinovic, A.; Leung, D.H.; Hoos, A.; Jaques, D.P.; Lewis, J.J.; Brennan, M.F. Analysis of the prognostic significance of microscopic margins in 2084 localized primary adult soft tissue sarcomas. Ann. Surg. 2002, 235, 424–434. [Google Scholar] [CrossRef]
- Gronchi, A.; Casali, P.G.; Mariani, L.; Lo Vullo, S.; Colecchia, M.; Lozza, L.; Bertulli, R.; Fiore, M.; Olmi, P.; Santinami, M.; et al. Quality of surgery and outcome in extra-abdominal aggressive fibromatosis: A series of patients surgically treated at a single institution. J. Clin. Oncol. 2003, 21, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.J.; Boland, P.J.; Leung, D.H.; Woodruff, J.M.; Brennan, M.F. The enigma of desmoid tumors. Ann. Surg. 1999, 229, 866–872. [Google Scholar] [CrossRef]
- Posner, M.C.; Shiu, M.H.; Newsome, J.L.; Hajdu, S.I.; Gaynor, J.J.; Brennan, M.F. The desmoid tumor. Not a benign disease. Arch. Surg. 1989, 124, 191–196. [Google Scholar] [CrossRef]
- Ballo, M.T.; Zagars, G.K.; Pollack, A.; Pisters, P.W.; Pollock, R.A. Desmoid tumor: Prognostic factors and outcome after surgery, radiation therapy, or combined surgery and radiation therapy. J. Clin. Oncol. 1999, 17, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Merchant, N.B.; Lewis, J.J.; Woodruff, J.M.; Leung, D.H.; Brennan, M.F. Extremity and trunk desmoid tumors: A multifactorial analysis of outcome. Cancer 1999, 86, 2045–2052. [Google Scholar] [CrossRef]
- Huang, K.; Fu, H.; Shi, Y.Q.; Zhou, Y.; Du, C.Y. Prognostic factors for extra-abdominal and abdominal wall desmoids: A 20-year experience at a single institution. J. Surg. Oncol. 2009, 100, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.T.; DeLaney, T.F.; Kobayashi, W.K.; Szymonifka, J.; Yeap, B.Y.; Chen, Y.L.; Rosenberg, A.E.; Harmon, D.C.; Choy, E.; Yoon, S.S.; et al. Desmoid tumor: Analysis of prognostic factors and outcomes in a surgical series. Ann. Surg. Oncol. 2012, 19, 4028–4035. [Google Scholar] [CrossRef] [PubMed]
- Crago, A.M.; Denton, B.; Salas, S.; Dufresne, A.; Mezhir, J.J.; Hameed, M.; Gonen, M.; Singer, S.; Brennan, M.F. A prognostic nomogram for prediction of recurrence in desmoid fibromatosis. Ann. Surg. 2013, 258, 347–353. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Jiang, H.; Wang, Y.; Li, Z.; Lu, H. Effective treatment of aggressive fibromatosis with celecoxib guided by genetic testing. Cancer Biol. Ther. 2017, 18, 757–760. [Google Scholar] [CrossRef]
- Janinis, J.; Patriki, M.; Vini, L.; Aravantinos, G.; Whelan, J.S. The pharmacological treatment of aggressive fibromatosis: A systematic review. Ann. Oncol. 2003, 14, 181–190. [Google Scholar] [CrossRef]
- Garbay, D.; Le Cesne, A.; Penel, N.; Chevreau, C.; Marec-Berard, P.; Blay, J.Y.; Debled, M.; Isambert, N.; Thyss, A.; Bompas, E.; et al. Chemotherapy in patients with desmoid tumors: A study from the French Sarcoma Group (FSG). Ann. Oncol. 2012, 23, 182–186. [Google Scholar] [CrossRef]
- Skubitz, K.M.; Manivel, J.C.; Clohisy, D.R.; Frolich, J.W. Response of imatinib-resistant extra-abdominal aggressive fibromatosis to sunitinib: Case report and review of the literature on response to tyrosine kinase inhibitors. Cancer Chemother Pharmacol. 2009, 64, 635–640. [Google Scholar] [CrossRef]
- Chugh, R.; Wathen, J.K.; Patel, S.R.; Maki, R.G.; Meyers, P.A.; Schuetze, S.M.; Priebat, D.A.; Thomas, D.G.; Jacobson, J.A.; Samuels, B.L.; et al. Efficacy of imatinib in aggressive fibromatosis: Results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin. Cancer Res. 2010, 16, 4884–4891. [Google Scholar] [CrossRef]
- Toulmonde, M.; Pulido, M.; Ray-Coquard, I.; Andre, T.; Isambert, N.; Chevreau, C.; Penel, N.; Bompas, E.; Saada, E.; Bertucci, F.; et al. Pazopanib or methotrexate–vinblastine combination chemotherapy in adult patients with progressive desmoid tumours (DESMOPAZ): A non-comparative, randomised, open-label, multicentre, phase 2 study. Lancet 2019, 20, 1263–1272. [Google Scholar] [CrossRef]
- Gounder, M.M.; Mahoney, M.R.; Van Tine, B.A.; Ravi, V.; Attia, S.; Deshpande, H.A.; Gupta, A.A.; Milhem, M.M.; Conry, R.M.; Movva, S.; et al. Sorafenib for Advanced and Refractory Desmoid Tumors. J. Med. 2018, 379, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Skubitz, K.M. Biology and Treatment of Aggressive Fibromatosis or Desmoid Tumor. Mayo Clin. Proc. 2017, 92, 947–964. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, K.; Church, J.M.; Jagelman, D.G.; Fazio, V.W.; McGannon, E.; George, C.R.; Schroeder, T.; Lavery, I.; Oakley, J. Noncytotoxic drug therapy for intraabdominal desmoid tumor in patients with familial adenomatous polyposis. Dis. Colon. Rectum 1992, 35, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Fiore, M.; Colombo, C.; Radaelli, S.; Prestianni, P.; Sanfilippo, R.; Morosi, C.; Stacchiotti, S.; Casali, P.G.; Gronchi, A. Activity of toremifene in sporadic desmoid-type fibromatosis. J. Clin. Oncol. 2011, 29, 10033. [Google Scholar] [CrossRef]
- Azzarelli, A.; Gronchi, A.; Bertulli, R.; Tesoro Tess, J.D.; Baratti, D.; Pennacchioli, E.; Dileo, P.; Rasponi, A.; Ferrari, A.; Pilotti, S.; et al. Low-dose chemotherapy with methotrexate and vinblastine for patients with advanced aggressive fibromatosis. Cancer 2001, 92, 1259–1264. [Google Scholar] [CrossRef]
- Constantinidou, A.; Jones, R.L.; Scurr, M.; Al-Muderis, O.; Judson, I. Pegylated liposomal doxorubicin, an effective, well tolerated treatment for refractory aggressive fibromatosis. Eur. J. Cancer 2009, 45, 2930–2934. [Google Scholar] [CrossRef]
- Patel, S.; Evans, H.; Benjamin, R. Combination chemotherapy in adult desmoid tumors. Cancer 1993, 72, 3244–3247. [Google Scholar] [CrossRef]
- Kasper, B.; Gruenwald, V.; Reichardt, P.; Bauer, S.; Hohenberger, P.; Haller, F. Correlation of CTNNB1 mutation status with progression arrest rate in RECIST progressive desmoid-type fibromatosis treated with imatinib: Translational research results from a phase 2 study of the German Interdisciplinary Sarcoma Group (GISG-01). Ann. Surg. Oncol. 2016, 23, 1924–1927. [Google Scholar] [CrossRef]
- Penel, N.; Le Cesne, A.; Bui, B.N.; Perol, D.; Brain, E.G.; Ray-Coquard, I.; Guillemet, C.; Chevreau, C.; Cupissol, D.; Chabaud, S.; et al. Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): An FNCLCC/French Sarcoma Group phase II trial with a long-term follow-up. Ann. Oncol. 2011, 22, 452–457. [Google Scholar] [CrossRef]
- Jo, J.C.; Hong, Y.S.; Kim, K.P.; Lee, J.L.; Lee, J.; Park, Y.S.; Kim, S.Y.; Ryu, J.S.; Lee, J.S.; Kim, T.W. A prospective multicenter phase II study of sunitinib in patients with advanced aggressive fibromatosis. Invest. New Drugs 2014, 32, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.M.; Lefkowitz, R.A.; Keohan, M.L.; D’Adamo, D.R.; Hameed, M.; Antonescu, C.R.; Singer, S.; Stout, K.; Ahn, L.; Maki, R.G.; et al. Activity of Sorafenib against desmoid tumor/deep fibromatosis. Clin. Cancer Res. 2011, 17, 4082–4090. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A. A Pilot Study Evaluating the Use of mTor Inhibitor Sirolimus in Children and Young Adults with Desmoid-Type Fibromatosis. Available online: https://clinicaltrials.gov/ct2/show/NCT01265030 (accessed on 9 July 2020).
- Gronchi, A.; Colombo, C.; Le Péchoux, C.; Dei Tos, A.P.; Le Cesne, A.; Marrari, A.; Penel, N.; Grignani, G.; Blay, J.Y.; Casali, P.G.; et al. Sporadic desmoid-type fibromatosis: A stepwise approach to a non-metastasising neoplasm--A position paper from the Italian and the French Sarcoma Group. Ann. Oncol. 2014, 25, 578–583. [Google Scholar] [CrossRef]
- Janssen, M.L.; van Broekhoven, D.L.; Cates, J.M.; Salas, S.; Bonvalot, S.; Grünhagen, D.J.; Verhoef, C. Meta-analysis of the influence of surgical margin and adjuvant radiotherapy on local recurrence after resection of sporadic desmoid-type fibromatosis. Br. J. Surg. 2017, 104, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Guadagnolo, B.A.; Zagars, G.K.; Ballo, MT. Long-term outcomes for desmoid tumors treated with radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 441–447. [Google Scholar] [CrossRef]
- Fiore, M.; Rimareix, F.; Mariani, L.; Domont, J.; Collini, P.; Le Péchoux, C.; Casali, P.G.; Le Cesne, A.; Gronchi, A.; Bonvalot, S. Desmoid-type fibromatosis: A front-line conservative approach to select patients for surgical treatment. Ann. Surg. Oncol. 2009, 16, 2587–2593. [Google Scholar] [CrossRef]
- Husson, O.; Younger, E.; Dunlop, A.; Dean, L.; Strauss, D.C.; Benson, C.; Hayes, A.J.; Miah, A.; van Houdt, W.; Zaidi, S.; et al. Desmoid fibromatosis through the patients’ eyes: Time to change the focus and organisation of care? Support Care Cancer 2019, 27, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Timbergen, M.J.; van de Poll-Franse, L.V.; Grünhagen, D.J.; van der Graaf, W.T.; Sleijfer, S.; Verhoef, C.; Husson, O. Identification and assessment of health-related quality of life issues in patients with sporadic desmoid-type fibromatosis: A literature review and focus group study. Qual. Life Res. 2018, 27, 3097–3111. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.M.; Maddux, L.; Paty, J.; Atkinson, T.M. Prospective development of a patient reported outcomes (PRO) tool in desmoid tumors: A novel clinical trial endpoint. J. Clin. Oncol. 2017, 35, 11022. [Google Scholar]
- Foundation, T.D.T.R. DTRF Patient Registry. 2017. Available online: https://dtrf.org/patient-registry/ (accessed on 9 July 2020).



{kind=link}
{kind=link}
| Author/Year | N | Primary/Recurrent | Median Months | 5 Years DFS Margins (+) | 5 Years DFS Margins (−) |
|---|---|---|---|---|---|
| Posner, et al. 1989 [41] | 128 | 78/53 | 88 | 85 | 50 |
| Ballo, et al. 1999 [42] | 189 | 85/104 | 112 | 75 | 50 |
| Merchant, et al. 1999 [43] | 105 | 105/0 | 49 | 70 | 78 |
| Gronchi, et al. 2003 [39] | 203 | 128/75 | 130/153 | 82/65 | 79/47 |
| Huang, et al. 2009 [44] | 151 | 113/38 | 102 | 80 | 80 |
| Mullen, et al. 2012 [45] | 177 | 133/44 | 40 | 82 | 52 |
| Crago, et al. 2013 [46] | 57 | 382/113 | 60 | 69 | 69 |
| Treatment | Type of Study | N | Objective Response Rate | Other Response Rate | Reference |
|---|---|---|---|---|---|
| Sulindac | Retrospective | 14 | 57% | - | [55] |
| Toremifene | Retrospective | 27 | 22% | 6-month PFS: 76% | [56] |
| Metotrexate-Vinblastina | phase II | 27 | 15% | 10-year PFS: 67% | [57] |
| liposomal Doxorubicin | Retrospective | 14 | 33% | - | [58] |
| Doxorubicin + dacarbazine | Retrospective | 12 | 50% | - | [59] |
| Imatinib 800 mg/d | phase II | 51 | 6% | 1-year PFS: 66% | [51] |
| Imatinib 800 mg/d | phase II | 37 | 6% | 6-month PFS: 65% | [60] |
| Imatinib 400 mg/d | phase II | 50 | 12% | 1-year PFS: 67% | [61] |
| Sunitinib | phase II | 19 | 26% | 1-year PFS: 80% | [62] |
| Sorafenib | Retrospective | 26 | 26% | - | [63] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Ortega, D.Y.; Martín-Tellez, K.S.; Cuellar-Hubbe, M.; Martínez-Said, H.; Álvarez-Cano, A.; Brener-Chaoul, M.; Alegría-Baños, J.A.; Martínez-Tlahuel, J.L. Desmoid-Type Fibromatosis. Cancers 2020, 12, 1851. https://doi.org/10.3390/cancers12071851
Garcia-Ortega DY, Martín-Tellez KS, Cuellar-Hubbe M, Martínez-Said H, Álvarez-Cano A, Brener-Chaoul M, Alegría-Baños JA, Martínez-Tlahuel JL. Desmoid-Type Fibromatosis. Cancers. 2020; 12(7):1851. https://doi.org/10.3390/cancers12071851
Chicago/Turabian StyleGarcia-Ortega, Dorian Yarih, Karla Susana Martín-Tellez, Mario Cuellar-Hubbe, Héctor Martínez-Said, Alethia Álvarez-Cano, Moises Brener-Chaoul, Jorge Adán Alegría-Baños, and Jorge Luis Martínez-Tlahuel. 2020. "Desmoid-Type Fibromatosis" Cancers 12, no. 7: 1851. https://doi.org/10.3390/cancers12071851
APA StyleGarcia-Ortega, D. Y., Martín-Tellez, K. S., Cuellar-Hubbe, M., Martínez-Said, H., Álvarez-Cano, A., Brener-Chaoul, M., Alegría-Baños, J. A., & Martínez-Tlahuel, J. L. (2020). Desmoid-Type Fibromatosis. Cancers, 12(7), 1851. https://doi.org/10.3390/cancers12071851