Correlation of MET and PD-L1 Expression in Malignant Melanoma



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
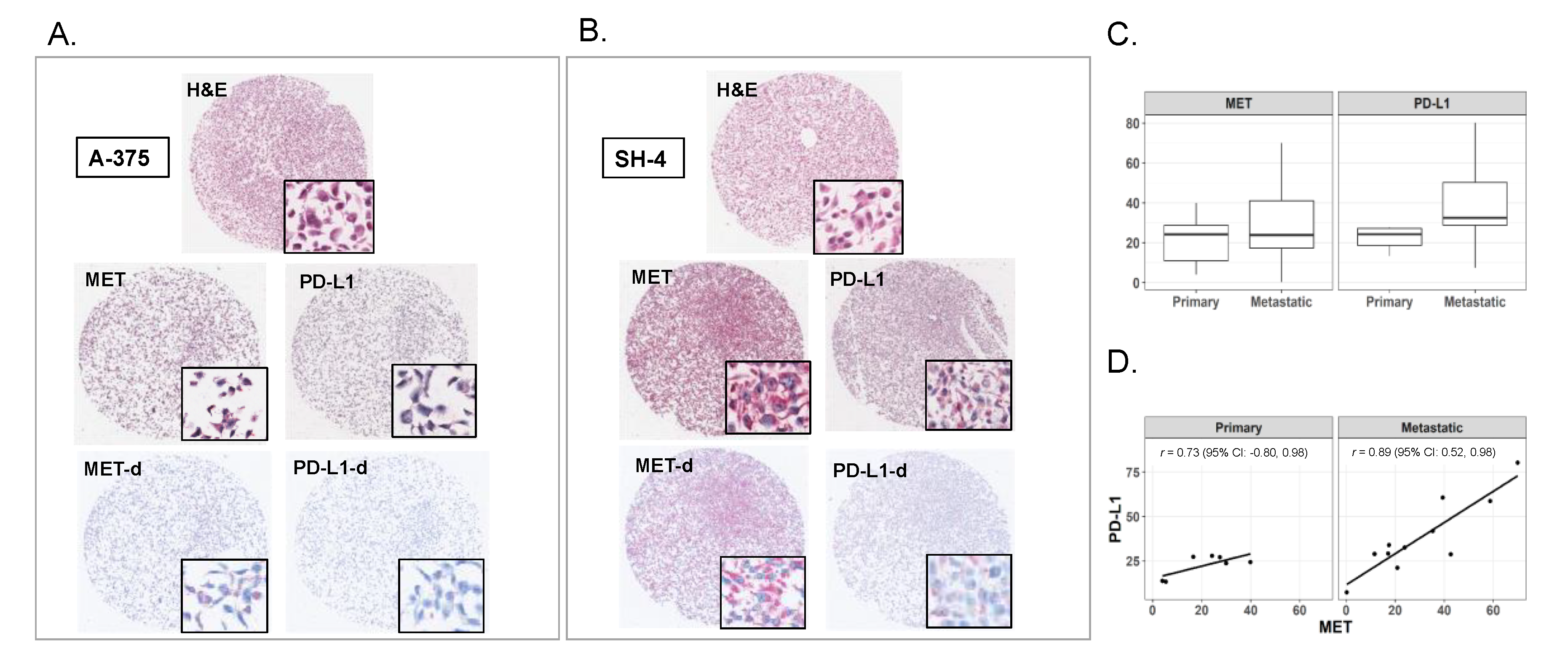
2.1. Expression of MET and PD-L1 in Melanoma Cell Lines
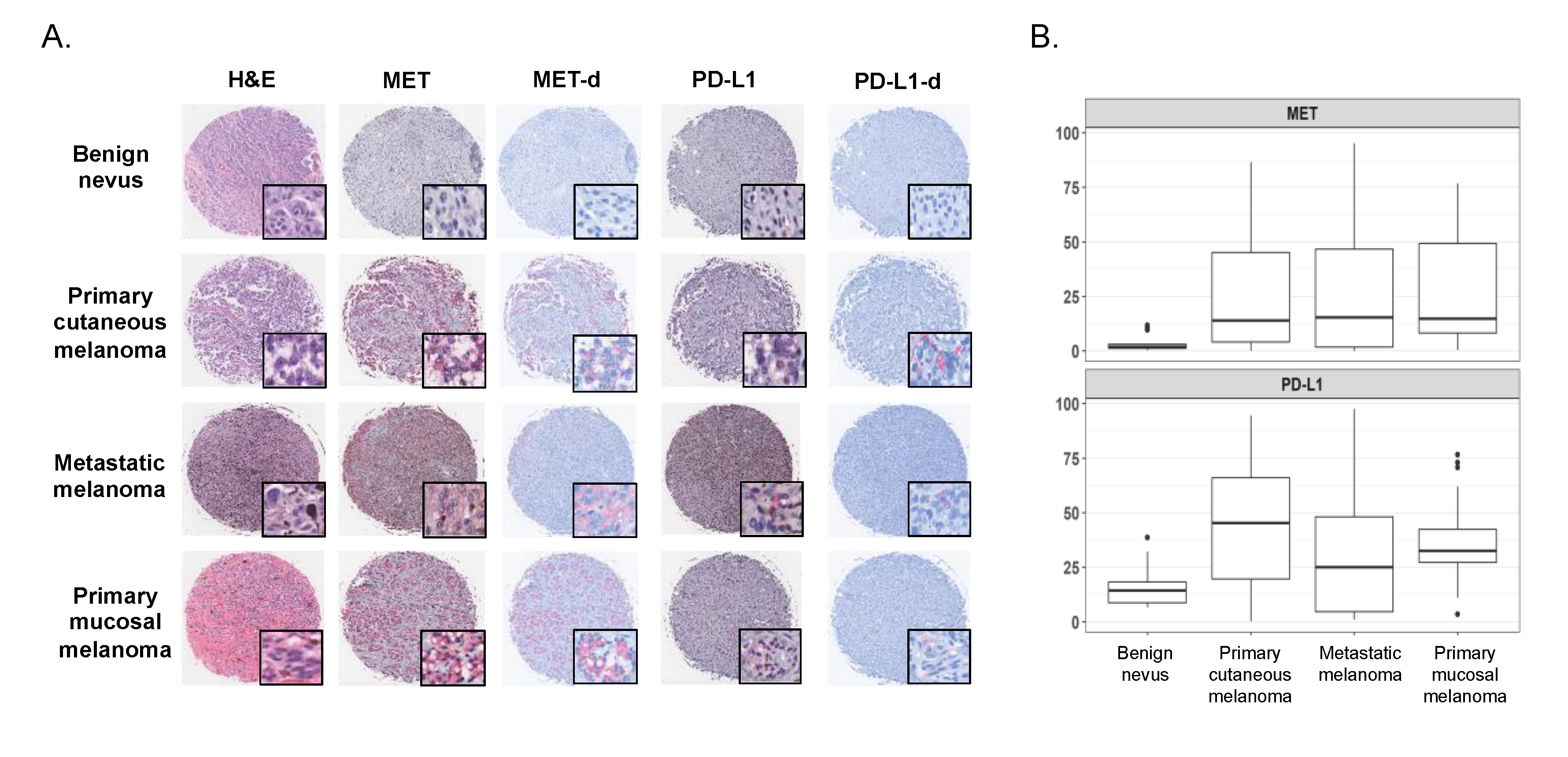
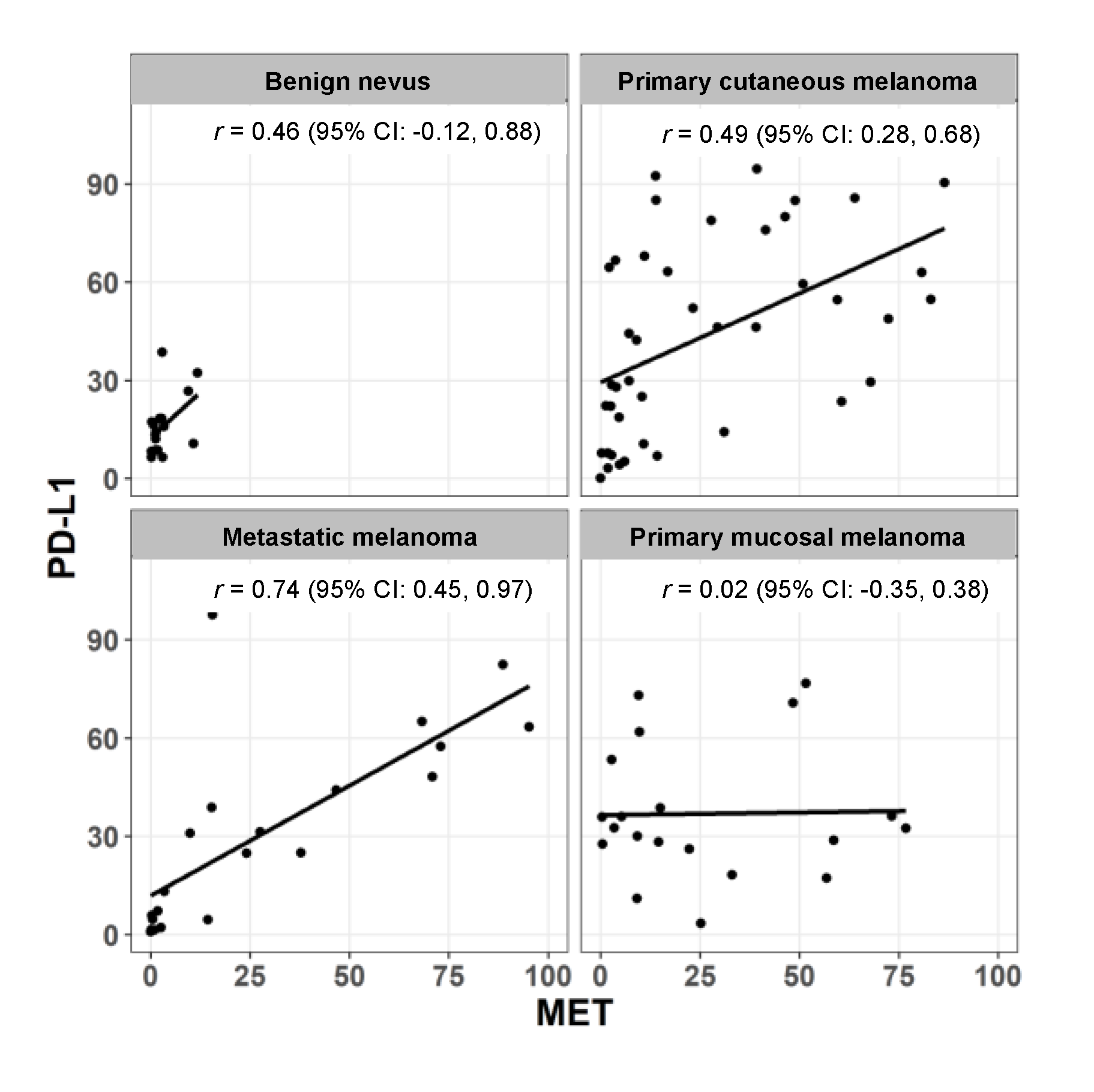
2.2. Expression of MET and PD-L1 in a Human Melanoma Tissue Microarray
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Microarray
4.2. Tissue Microarray and Patient Characteristics
4.3. Immunohistochemistry (IHC)
4.4. Image Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Umitsu, M.; De Silva, D.M.; Roy, A.; Bottaro, D.P. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 2017, 108, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-Genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Hubel, J.; Hieronymus, T. HGF/Met-Signaling Contributes to Immune Regulation by Modulating Tolerogenic and Motogenic Properties of Dendritic Cells. Biomedicines 2015, 3, 138–148. [Google Scholar] [CrossRef]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Roger Thompson, A.A.; Wauters, E.; Wamsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349–353. [Google Scholar] [CrossRef]
- Gibbons Johnson, R.M.; Dong, H. Functional Expression of Programmed Death-Ligand 1 (B7-H1) by Immune Cells and Tumor Cells. Front. Immunol. 2017, 8, 961. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Teng, F.; Meng, X.; Kong, L.; Yu, J. Progress and challenges of predictive biomarkers of anti PD-1/PD-L1 immunotherapy: A systematic review. Cancer Lett. 2018, 414, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniach, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Mackensen, A. Contribution of the PD-L1/PD-1 pathway to T-Cell exhaustion: An update on implications for chronic infections and tumor evasion. Cancer Immunol. Immunother. 2007, 56, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Balan, M.; Mier y Teran, E.; Waaga-Gasser, A.M.; Gasser, M.; Choueiri, T.K.; Freeman, G.; Pal, S. Novel roles of c-Met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J. Biol. Chem. 2015, 290, 8110–8120. [Google Scholar] [CrossRef]
- Martin, V.; Chiriaco, C.; Modica, C.; Acquadro, A.; Cortese, M.; Galimi, F.; Perera, T.; Gammaitoni, L.; Aglietta, M.; Comoglio, P.M.; et al. Met inhibition revokes IFNgamma-Induction of PD-1 ligands in MET-amplified tumours. Br. J. Cancer 2019, 120, 527–536. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin.Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meeting 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Zhou, A.Y.; Johnson, D.B. Combinatorial Therapies in Melanoma: MAPK Inhibitors and Beyond. Am. J. Clin. Dermatol. 2018, 19, 181–193. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, K.Y.; Giubellino, A. The Role of MET in Melanoma and Melanocytic Lesions. Am. J. Pathol. 2019, 189, 2138–2148. [Google Scholar] [CrossRef]
- Oba, J.; Kim, S.-H.; Wang, W.-L.; Macedo, M.P.; Carapeto, F.; McKean, M.A.; Van Arnam, J.; Eterovic, A.K.; Sen, S.; Kale, C.R.; et al. Targeting the HGF/MET Axis Counters Primary Resistance to KIT Inhibition in KIT-Mutant Melanoma. JCO Precis. Oncol. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Caenepeel, S.; Cooke, K.; Wadsworth, S.; Huang, G.; Robert, L.; Homet Moreno, B.; Parisi, G.; Cajulis, E.; Kendall, R.; Beltran, P.; et al. MAPK pathway inhibition induces MET and GAB1 levels, priming BRAF mutant melanoma for rescue by hepatocyte growth factor. Oncotarget 2017, 8, 17795–17809. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Molnarfi, N.; Benkhoucha, M.; Funakoshi, H.; Nakamura, T.; Lalive, P.H. Hepatocyte growth factor: A regulator of inflammation and autoimmunity. Autoimmun. Rev. 2015, 14, 293–303. [Google Scholar] [CrossRef]
- Benkhoucha, M.; Molnarfi, N.; Kaya, G.; Belnoue, E.; Bjarnadóttir, K.; Dietrich, P.-Y.; Walker, P.R.; Martinvalet, D.; Derouazi, M.; Lalive, P.H. Identification of a novel population of highly cytotoxic c-Met-Expressing CD8(+) T lymphocytes. EMBO Rep. 2017, 18, 1545–1558. [Google Scholar] [CrossRef] [PubMed]
- Sagi, Z.; Hieronymus, T. The Impact of the Epithelial-Mesenchymal Transition Regulator Hepatocyte Growth Factor Receptor/Met on Skin Immunity by Modulating Langerhans Cell Migration. Front. Immunol. 2018, 9, 517. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Loughrey, M.B.; Bankhead, P.; Coleman, H.G.; Hagan, R.S.; Craig, S.; McCorry, A.; Gray, R.T.; McQuaid, S.; Dunne, P.D.; Hamilton, P.W.; et al. Validation of the systematic scoring of immunohistochemically stained tumour tissue microarrays using QuPath digital image analysis. Histopathology 2018, 73, 327–338. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Fisher, D.E. Treatment of Advanced Melanoma in 2020 and Beyond. J. Investig. Dermatol. Available online: https://pubmed.ncbi.nlm.nih.gov/32268150/ (accessed on 8 July 2020).
- Lim, S.Y.; Menzies, A.M.; Rizos, H. Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer 2017, 123, 2118–2129. [Google Scholar] [CrossRef]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-Genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef]
- Larkin, J.; Charion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, D.C.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Grob, J.J.; Stoyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Henderson, L.; Xiao, S.-Y.; Patel, P.; Yauch, R.L.; Hegde, P.; Zha, J.; Pandita, A.; Peterson, A.; Salgia, R. Durable complete response of metastatic gastric cancer with Anti-Met therapy followed by resistance at recurrence. Cancer Discov. 2011, 1, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, H.; Yu, W.; Zha, J.; Pandita, A.; Penuel, E.; Rangell, L.; Raja, R.; Mohan, S.; Patel, R.; Desai, R.; et al. Biomarker analyses from a placebo-controlled phase II study evaluating erlotinib±onartuzumab in advanced non-small cell lung cancer: MET expression levels are predictive of patient benefit. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 4488–4498. [Google Scholar] [CrossRef]
- Bouattour, M.; Raymond, E.; Qin, S.; Cheng, A.-L.; Stammberger, U.; Locatelli, G.; Faivre, S. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatol. Baltim. Md 2018, 67, 1132–1149. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Plimack, E.; Arkenau, H.-T.; Jonasch, E.; Heng, D.Y.C.; Powless, T.; Frigault, M.M.; Clark, E.A.; Handzel, A.A.; Gardner, H.; et al. Biomarker-Based Phase II Trial of Savolitinib in Patients With Advanced Papillary Renal Cell Cancer. J. Clin. Oncol. Off. J. Am.Soc. Clin. Oncol. 2017, 35, 2993–3001. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, K.Y.; Desar, S.; Pengo, T.; Shanley, R.; Giubellino, A. Correlation of MET and PD-L1 Expression in Malignant Melanoma. Cancers 2020, 12, 1847. https://doi.org/10.3390/cancers12071847
Song KY, Desar S, Pengo T, Shanley R, Giubellino A. Correlation of MET and PD-L1 Expression in Malignant Melanoma. Cancers. 2020; 12(7):1847. https://doi.org/10.3390/cancers12071847
Chicago/Turabian StyleSong, Kyu Young, Sabina Desar, Thomas Pengo, Ryan Shanley, and Alessio Giubellino. 2020. "Correlation of MET and PD-L1 Expression in Malignant Melanoma" Cancers 12, no. 7: 1847. https://doi.org/10.3390/cancers12071847
APA StyleSong, K. Y., Desar, S., Pengo, T., Shanley, R., & Giubellino, A. (2020). Correlation of MET and PD-L1 Expression in Malignant Melanoma. Cancers, 12(7), 1847. https://doi.org/10.3390/cancers12071847