Harnessing NK Cell Checkpoint-Modulating Immunotherapies


Abstract
1. Introduction
2. NK Cells as a Potential Therapeutic Strategy
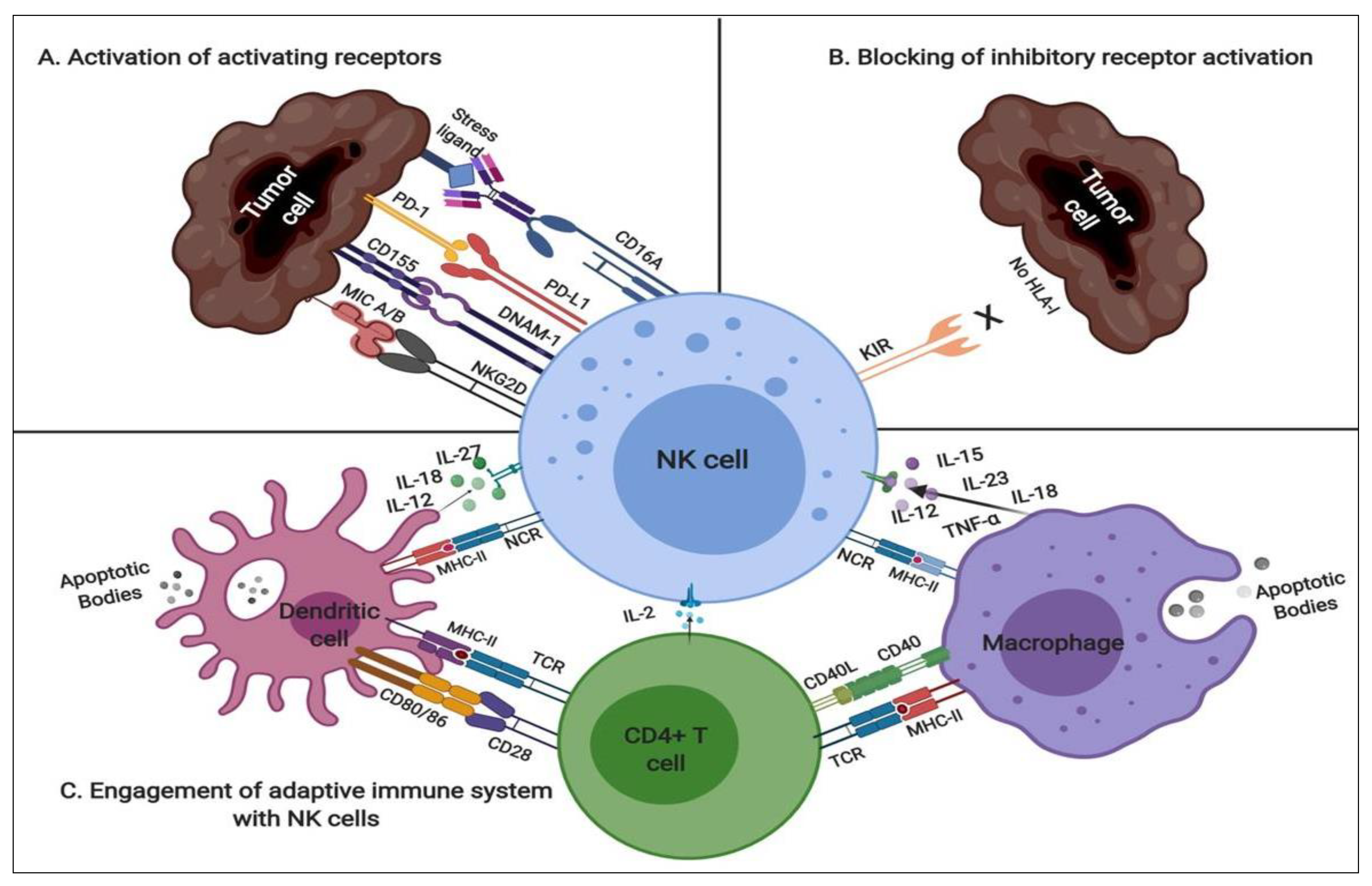
2.1. NK Modulatory Mechanisms
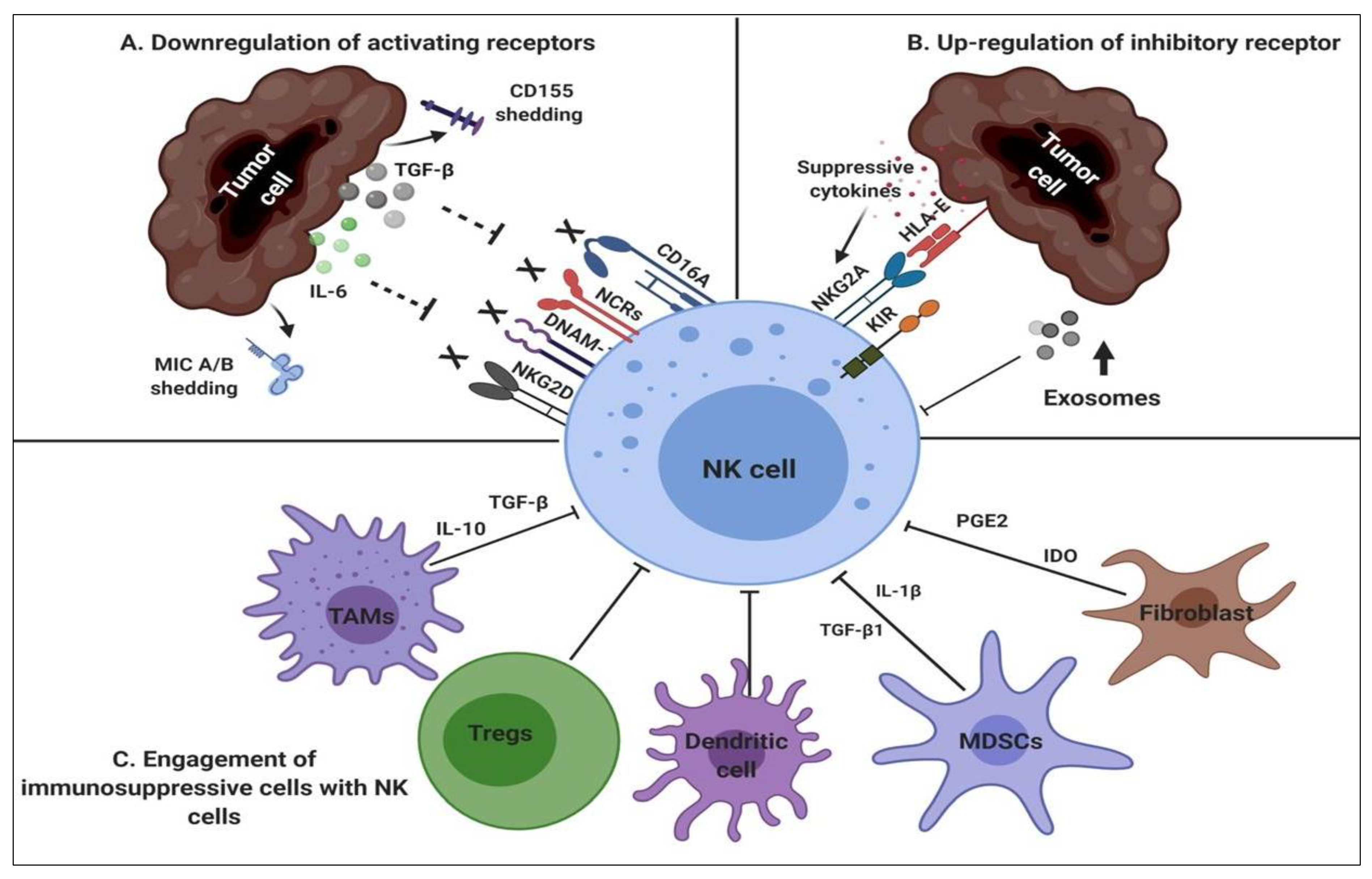
2.2. NK-Suppressive Mechanisms
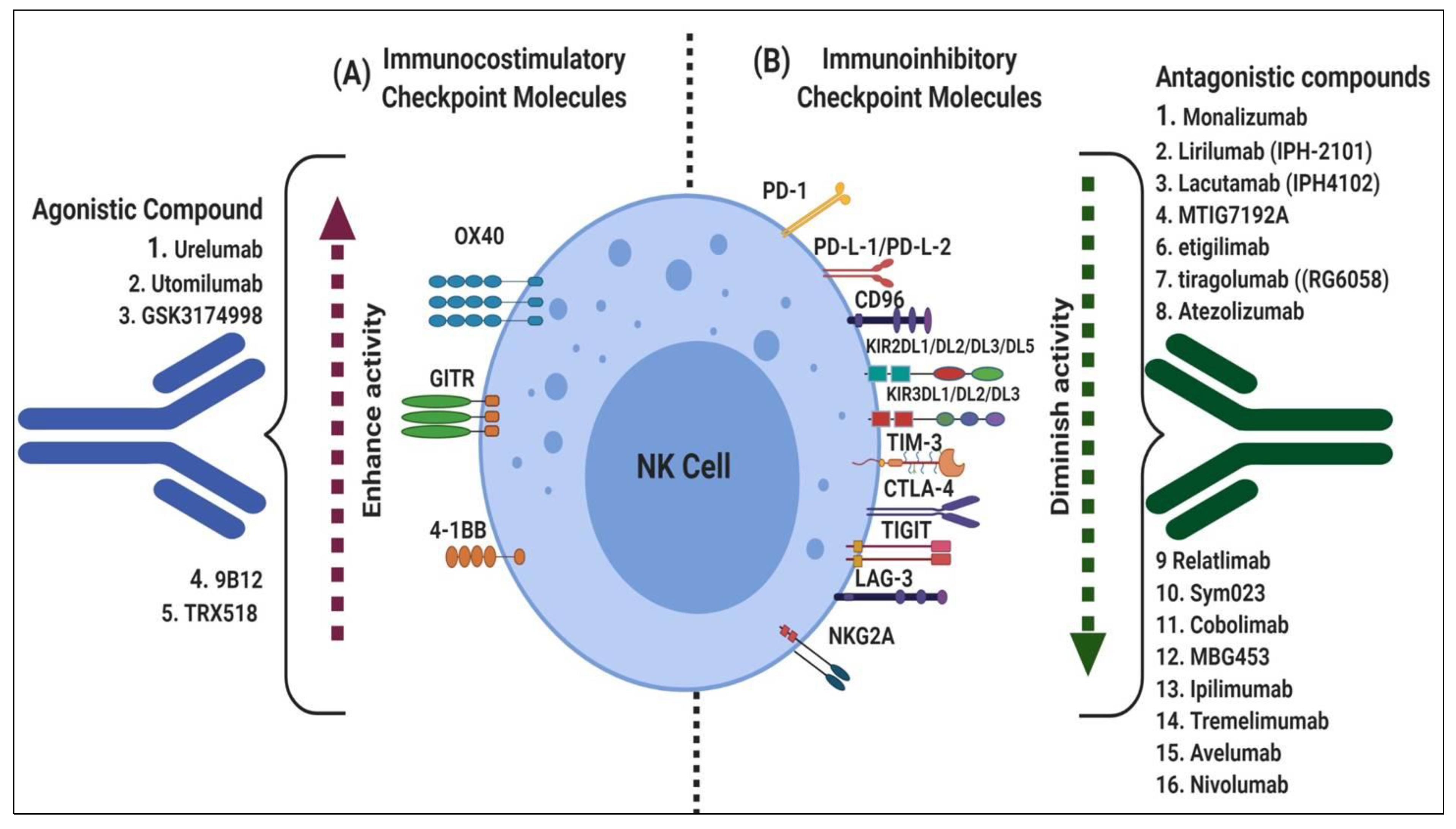
3. Fundamental Mechanisms of Immune Checkpoints
3.1. Immune Checkpoint Costimulators
3.2. Immune Checkpoint Inhibitors
4. Genetic Modification of NK Cells
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matsunaga, T.; Rahman, A. What brought the adaptive immune system to vertebrates? The jaw hypothesis and the seahorse. Immunol. Rev. 1998, 166, 177–186. [Google Scholar] [CrossRef]
- Oldham, R.K. Natural killer cells: Artifact to reality: An odyssey in biology. Cancer Metastasis Rev. 1983, 2, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Colucci, F.; Caligiuri, M.A.; Di Santo, J.P. What does it take to make a natural killer? Nat. Rev. Immunol. 2003, 3, 413–425. [Google Scholar] [CrossRef]
- Veluchamy, J.P.; Kok, N.; van der Vliet, H.J.; Verheul, H.M.W.; de Gruijl, T.D.; Spanholtz, J. The Rise of Allogeneic Natural Killer Cells as a Platform for Cancer Immunotherapy: Recent Innovations and Future Developments. Front. Immunol. 2017, 8, 631. [Google Scholar] [CrossRef]
- Wu, J.; Lanier, L.L. Natural killer cells and cancer. Adv. Cancer Res. 2003, 90, 127–156. [Google Scholar]
- Raulet, D.H.; Guerra, N. Oncogenic stress sensed by the immune system: Role of natural killer cell receptors. Nat. Rev. Immunol. 2009, 9, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Farnault, L.; Sanchez, C.; Baier, C.; Le Treut, T.; Costello, R.T. Hematological malignancies escape from NK cell innate immune surveillance: Mechanisms and therapeutic implications. Clin. Dev. Immunol. 2012, 2012, 421702. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. Follow the leader: NK cell receptors for classical and nonclassical MHC class I. Cell 1998, 92, 705–707. [Google Scholar] [CrossRef]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Lanier, L.L. Activating and inhibitory NK cell receptors. Adv. Exp. Med. Biol. 1998, 452, 13–18. [Google Scholar] [PubMed]
- Vivier, E.; Nunes, J.A.; Vely, F. Natural killer cell signaling pathways. Science 2004, 306, 1517–1519. [Google Scholar] [CrossRef] [PubMed]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef]
- Pietra, G.; Manzini, C.; Rivara, S.; Vitale, M.; Cantoni, C.; Petretto, A.; Balsamo, M.; Conte, R.; Benelli, R.; Minghelli, S.; et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res. 2012, 72, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Ljunggren, H.G.; Karre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Deng, W.; Gowen, B.G.; Zhang, L.; Wang, L.; Lau, S.; Iannello, A.; Xu, J.; Rovis, T.L.; Xiong, N.; Raulet, D.H. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science 2015, 348, 136–139. [Google Scholar] [CrossRef]
- Semeraro, M.; Rusakiewicz, S.; Minard-Colin, V.; Delahaye, N.F.; Enot, D.; Vely, F.; Marabelle, A.; Papoular, B.; Piperoglou, C.; Ponzoni, M.; et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci. Transl. Med. 2015, 7, 283ra55. [Google Scholar] [CrossRef]
- Garni-Wagner, B.A.; Purohit, A.; Mathew, P.A.; Bennett, M.; Kumar, V. A novel function-associated molecule related to non-MHC-restricted cytotoxicity mediated by activated natural killer cells and T cells. J. Immunol. 1993, 151, 60–70. [Google Scholar]
- Makkouk, A.; Chester, C.; Kohrt, H.E. Rationale for anti-CD137 cancer immunotherapy. Eur. J. Cancer 2016, 54, 112–119. [Google Scholar] [CrossRef]
- Seidel, U.J.; Schlegel, P.; Lang, P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front. Immunol. 2013, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Sojka, D.K.; Bruniquel, D.; Schwartz, R.H.; Singh, N.J. IL-2 secretion by CD4+ T cells in vivo is rapid, transient, and influenced by TCR-specific competition. J. Immunol. 2004, 172, 6136–6143. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.S.; Ritz, J.; Frank, D.A. IL-2 induces STAT4 activation in primary NK cells and NK cell lines, but not in T cells. J. Immunol. 1999, 162, 299–304. [Google Scholar] [PubMed]
- Niehrs, A.; Altfeld, M. Regulation of NK-Cell Function by HLA Class II. Front. Cell. Infect. Microbiol. 2020, 10, 55. [Google Scholar] [CrossRef]
- Goyvaerts, C.; Breckpot, K. Pros and Cons of Antigen-Presenting Cell Targeted Tumor Vaccines. J. Immunol. Res. 2015, 2015, 785634. [Google Scholar] [CrossRef]
- Ziblat, A.; Nunez, S.Y.; Raffo Iraolagoitia, X.L.; Spallanzani, R.G.; Torres, N.I.; Sierra, J.M.; Secchiari, F.; Domaica, C.I.; Fuertes, M.B.; Zwirner, N.W. Interleukin (IL)-23 Stimulates IFN-gamma Secretion by CD56(bright) Natural Killer Cells and Enhances IL-18-Driven Dendritic Cells Activation. Front. Immunol. 2017, 8, 1959. [Google Scholar] [CrossRef] [PubMed]
- Ziblat, A.; Domaica, C.I.; Spallanzani, R.G.; Iraolagoitia, X.L.; Rossi, L.E.; Avila, D.E.; Torres, N.I.; Fuertes, M.B.; Zwirner, N.W. IL-27 stimulates human NK-cell effector functions and primes NK cells for IL-18 responsiveness. Eur. J. Immunol. 2015, 45, 192–202. [Google Scholar] [CrossRef]
- Zwirner, N.W.; Ziblat, A. Regulation of NK Cell Activation and Effector Functions by the IL-12 Family of Cytokines: The Case of IL-27. Front. Immunol. 2017, 8, 25. [Google Scholar] [CrossRef]
- Bluman, E.M.; Bartynski, K.J.; Avalos, B.R.; Caligiuri, M.A. Human natural killer cells produce abundant macrophage inflammatory protein-1 alpha in response to monocyte-derived cytokines. J. Clin. Invest. 1996, 97, 2722–2727. [Google Scholar] [CrossRef]
- Chan, C.J.; Smyth, M.J.; Martinet, L. Molecular mechanisms of natural killer cell activation in response to cellular stress. Cell Death Differ. 2014, 21, 5–14. [Google Scholar] [CrossRef]
- Vales-Gomez, M.; Chisholm, S.E.; Cassady-Cain, R.L.; Roda-Navarro, P.; Reyburn, H.T. Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. Cancer Res. 2008, 68, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.R.; Hurt, E.M.; Farrar, W.L. The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer 2005, 41, 2502–2512. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Chytil, A.; Washington, K.; Romero-Gallo, J.; Gorska, A.E.; Wirth, P.S.; Gautam, S.; Moses, H.L.; Grady, W.M. Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res. 2004, 64, 4687–4692. [Google Scholar] [CrossRef]
- Romagne, F.; Andre, P.; Spee, P.; Zahn, S.; Anfossi, N.; Gauthier, L.; Capanni, M.; Ruggeri, L.; Benson, D.M., Jr.; Blaser, B.W.; et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood 2009, 114, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.D.; Higgins, L.M.; Steinle, A.; Cosman, D.; Haugk, K.; Plymate, S.R. Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J. Clin. Invest. 2004, 114, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.; Ferrone, S. Tumor escape mechanisms: Potential role of soluble HLA antigens and NK cells activating ligands. Tissue Antigens 2008, 72, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Correa, B.; Gayoso, I.; Bergua, J.M.; Casado, J.G.; Morgado, S.; Solana, R.; Tarazona, R. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol. Cell Biol. 2012, 90, 109–115. [Google Scholar] [CrossRef]
- Kearney, C.J.; Ramsbottom, K.M.; Voskoboinik, I.; Darcy, P.K.; Oliaro, J. Loss of DNAM-1 ligand expression by acute myeloid leukemia cells renders them resistant to NK cell killing. OncoImmunology 2016, 5, e1196308. [Google Scholar] [CrossRef]
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760. [Google Scholar] [CrossRef]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-beta. Front. Immunol. 2019, 10, 2689. [Google Scholar] [CrossRef]
- Groth, A.; Kloss, S.; von Strandmann, E.P.; Koehl, U.; Koch, J. Mechanisms of tumor and viral immune escape from natural killer cell-mediated surveillance. J. Innate Immun. 2011, 3, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Elkabets, M.; Ribeiro, V.S.; Dinarello, C.A.; Ostrand-Rosenberg, S.; Di Santo, J.P.; Apte, R.N.; Vosshenrich, C.A. IL-1beta regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur. J. Immunol. 2010, 40, 3347–3357. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Nunez, S.Y.; Ziblat, A.; Secchiari, F.; Torres, N.I.; Sierra, J.M.; Raffo Iraolagoitia, X.L.; Araya, R.E.; Domaica, C.I.; Fuertes, M.B.; Zwirner, N.W. Human M2 Macrophages Limit NK Cell Effector Functions through Secretion of TGF-beta and Engagement of CD85j. J. Immunol. 2018, 200, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Sitrin, J.; Ring, A.; Garcia, K.C.; Benoist, C.; Mathis, D. Regulatory T cells control NK cells in an insulitic lesion by depriving them of IL-2. J. Exp. Med. 2013, 210, 1153–1165. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Sarhan, D.; Palma, M.; Mao, Y.; Adamson, L.; Kiessling, R.; Mellstedt, H.; Osterborg, A.; Lundqvist, A. Dendritic cell regulation of NK-cell responses involves lymphotoxin-alpha, IL-12, and TGF-beta. Eur. J. Immunol. 2015, 45, 1783–1793. [Google Scholar] [CrossRef]
- Zappasodi, R.; Merghoub, T.; Wolchok, J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 2018, 33, 581–598. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Korman, A.J.; Peggs, K.S.; Allison, J.P. Checkpoint blockade in cancer immunotherapy. Adv. Immunol. 2006, 90, 297–339. [Google Scholar]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Nuebling, T.; Schumacher, C.E.; Hofmann, M.; Hagelstein, I.; Schmiedel, B.J.; Maurer, S.; Federmann, B.; Rothfelder, K.; Roerden, M.; Dorfel, D.; et al. The Immune Checkpoint Modulator OX40 and Its Ligand OX40L in NK-Cell Immunosurveillance and Acute Myeloid Leukemia. Cancer Immunol. Res. 2018, 6, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Baltz, K.M.; Krusch, M.; Bringmann, A.; Brossart, P.; Mayer, F.; Kloss, M.; Baessler, T.; Kumbier, I.; Peterfi, A.; Kupka, S.; et al. Cancer immunoediting by GITR (glucocorticoid-induced TNF-related protein) ligand in humans: NK cell/tumor cell interactions. FASEB J. 2007, 21, 2442–2454. [Google Scholar] [CrossRef] [PubMed]
- Navabi, S.; Doroudchi, M.; Tashnizi, A.H.; Habibagahi, M. Natural Killer Cell Functional Activity After 4-1BB Costimulation. Inflammation 2015, 38, 1181–1190. [Google Scholar] [CrossRef]
- Vidard, L.; Dureuil, C.; Baudhuin, J.; Vescovi, L.; Durand, L.; Sierra, V.; Parmantier, E. CD137 (4-1BB) Engagement Fine-Tunes Synergistic IL-15- and IL-21-Driven NK Cell Proliferation. J. Immunol. 2019, 203, 676–685. [Google Scholar] [CrossRef]
- Hurtado, J.C.; Kim, S.H.; Pollok, K.E.; Lee, Z.H.; Kwon, B.S. Potential role of 4-1BB in T cell activation. Comparison with the costimulatory molecule CD28. J. Immunol. 1995, 155, 3360–3367. [Google Scholar]
- Shao, Z.; Schwarz, H. CD137 ligand, a member of the tumor necrosis factor family, regulates immune responses via reverse signal transduction. J. Leukoc. Biol. 2011, 89, 21–29. [Google Scholar] [CrossRef]
- Croft, M. The role of TNF superfamily members in T-cell function and diseases. Nat. Rev. Immunol. 2009, 9, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Shuford, W.W.; Klussman, K.; Tritchler, D.D.; Loo, D.T.; Chalupny, J.; Siadak, A.W.; Brown, T.J.; Emswiler, J.; Raecho, H.; Larsen, C.P.; et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J. Exp. Med. 1997, 186, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Bukczynski, J.; Watts, T.H. 4-1BB ligand-mediated costimulation of human T cells induces CD4 and CD8 T cell expansion, cytokine production, and the development of cytolytic effector function. J. Immunol. 2002, 168, 4897–4906. [Google Scholar] [CrossRef]
- Vinay, D.S.; Kwon, B.S. Differential expression and costimulatory effect of 4-1BB (CD137) and CD28 molecules on cytokine-induced murine CD8(+) Tc1 and Tc2 cells. Cell. Immunol. 1999, 192, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Kwon, B.S. Role of 4-1BB in immune responses. Semin. Immunol. 1998, 10, 481–489. [Google Scholar] [CrossRef]
- Melero, I.; Johnston, J.V.; Shufford, W.W.; Mittler, R.S.; Chen, L. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell. Immunol. 1998, 190, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, R.A.; Tamada, K.; Strome, S.E.; Chen, L. Signaling through NK cell-associated CD137 promotes both helper function for CD8+ cytolytic T cells and responsiveness to IL-2 but not cytolytic activity. J. Immunol. 2002, 169, 4230–4236. [Google Scholar] [CrossRef]
- Wong, H.Y.; Schwarz, H. CD137 / CD137 ligand signalling regulates the immune balance: A potential target for novel immunotherapy of autoimmune diseases. J. Autoimmun. 2020. [Google Scholar] [CrossRef]
- Kang, Y.J.; Kim, S.O.; Shimada, S.; Otsuka, M.; Seit-Nebi, A.; Kwon, B.S.; Watts, T.H.; Han, J. Cell surface 4-1BBL mediates sequential signaling pathways 'downstream' of TLR and is required for sustained TNF production in macrophages. Nat. Immunol. 2007, 8, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Dharmadhikari, B.; Wu, M.; Abdullah, N.S.; Rajendran, S.; Ishak, N.D.; Nickles, E.; Harfuddin, Z.; Schwarz, H. CD137 and CD137L signals are main drivers of type 1, cell-mediated immune responses. OncoImmunology 2016, 5, e1113367. [Google Scholar] [CrossRef]
- Wilcox, R.A.; Chapoval, A.I.; Gorski, K.S.; Otsuji, M.; Shin, T.; Flies, D.B.; Tamada, K.; Mittler, R.S.; Tsuchiya, H.; Pardoll, D.M.; et al. Cutting edge: Expression of functional CD137 receptor by dendritic cells. J. Immunol. 2002, 168, 4262–4267. [Google Scholar] [CrossRef] [PubMed]
- Freeman, Z.T.; Nirschl, T.R.; Hovelson, D.H.; Johnston, R.J.; Engelhardt, J.J.; Selby, M.J.; Kochel, C.M.; Lan, R.Y.; Zhai, J.; Ghasemzadeh, A.; et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J. Clin. Invest. 2020, 130, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Houot, R.; Goldstein, M.J.; Kohrt, H.E.; Myklebust, J.H.; Alizadeh, A.A.; Lin, J.T.; Irish, J.M.; Torchia, J.A.; Kolstad, A.; Chen, L.; et al. Therapeutic effect of CD137 immunomodulation in lymphoma and its enhancement by Treg depletion. Blood 2009, 114, 3431–3438. [Google Scholar] [CrossRef]
- Chester, C.; Ambulkar, S.; Kohrt, H.E. 4-1BB agonism: Adding the accelerator to cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 1243–1248. [Google Scholar] [CrossRef]
- Souza-Fonseca-Guimaraes, F.; Blake, S.J.; Makkouk, A.; Chester, C.; Kohrt, H.E.; Smyth, M.J. Anti-CD137 enhances anti-CD20 therapy of systemic B-cell lymphoma with altered immune homeostasis but negligible toxicity. OncoImmunology 2016, 5, e1192740. [Google Scholar] [CrossRef][Green Version]
- Kohrt, H.E.; Houot, R.; Goldstein, M.J.; Weiskopf, K.; Alizadeh, A.A.; Brody, J.; Muller, A.; Pachynski, R.; Czerwinski, D.; Coutre, S.; et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood 2019, 134, 658. [Google Scholar]
- Kohrt, H.E.; Houot, R.; Weiskopf, K.; Goldstein, M.J.; Scheeren, F.; Czerwinski, D.; Colevas, A.D.; Weng, W.K.; Clarke, M.F.; Carlson, R.W.; et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J. Clin. Invest. 2012, 122, 1066–1075. [Google Scholar] [CrossRef]
- Kohrt, H.E.; Colevas, A.D.; Houot, R.; Weiskopf, K.; Goldstein, M.J.; Lund, P.; Mueller, A.; Sagiv-Barfi, I.; Marabelle, A.; Lira, R.; et al. Targeting CD137 enhances the efficacy of cetuximab. J. Clin. Invest. 2014, 124, 2668–2682. [Google Scholar] [CrossRef]
- Yonezawa, A.; Dutt, S.; Chester, C.; Kim, J.; Kohrt, H.E. Boosting Cancer Immunotherapy with Anti-CD137 Antibody Therapy. Clin. Cancer Res. 2015, 21, 3113–3120. [Google Scholar] [CrossRef]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Sznol, M.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Di Gravio, D.; Huang, B.; Gambhire, D.; Chen, Y.; et al. Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5349–5357. [Google Scholar] [CrossRef]
- Calderhead, D.M.; Buhlmann, J.E.; van den Eertwegh, A.J.; Claassen, E.; Noelle, R.J.; Fell, H.P. Cloning of mouse Ox40: A T cell activation marker that may mediate T-B cell interactions. J. Immunol. 1993, 151, 5261–5271. [Google Scholar]
- Willoughby, J.; Griffiths, J.; Tews, I.; Cragg, M.S. OX40: Structure and function—What questions remain? Mol. Immunol. 2017, 83, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Sun, X.; Li, W.; Liu, K.; Tian, D.; Dong, Y.; Sun, X.; Xu, H.; Zhang, D. Critical role of OX40 in the expansion and survival of CD4 T-cell-derived double-negative T cells. Cell Death Dis. 2018, 9, 616. [Google Scholar] [CrossRef] [PubMed]
- Stuber, E.; Neurath, M.; Calderhead, D.; Fell, H.P.; Strober, W. Cross-linking of OX40 ligand, a member of the TNF/NGF cytokine family, induces proliferation and differentiation in murine splenic B cells. Immunity 1995, 2, 507–521. [Google Scholar] [CrossRef]
- Ohshima, Y.; Tanaka, Y.; Tozawa, H.; Takahashi, Y.; Maliszewski, C.; Delespesse, G. Expression and function of OX40 ligand on human dendritic cells. J. Immunol. 1997, 159, 3838–3848. [Google Scholar]
- Takasawa, N.; Ishii, N.; Higashimura, N.; Murata, K.; Tanaka, Y.; Nakamura, M.; Sasaki, T.; Sugamura, K. Expression of gp34 (OX40 ligand) and OX40 on human T cell clones. Jpn. J. Cancer Res. 2001, 92, 377–382. [Google Scholar] [CrossRef]
- Gramaglia, I.; Weinberg, A.D.; Lemon, M.; Croft, M. Ox-40 ligand: A potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 1998, 161, 6510–6517. [Google Scholar]
- Weinberg, A.D.; Wegmann, K.W.; Funatake, C.; Whitham, R.H. Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J. Immunol. 1999, 162, 1818–1826. [Google Scholar]
- Murata, K.; Ishii, N.; Takano, H.; Miura, S.; Ndhlovu, L.C.; Nose, M.; Noda, T.; Sugamura, K. Impairment of antigen-presenting cell function in mice lacking expression of OX40 ligand. J. Exp. Med. 2000, 191, 365–374. [Google Scholar] [CrossRef]
- Gramaglia, I.; Jember, A.; Pippig, S.D.; Weinberg, A.D.; Killeen, N.; Croft, M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J. Immunol. 2000, 165, 3043–3050. [Google Scholar] [CrossRef] [PubMed]
- Rogers, P.R.; Song, J.; Gramaglia, I.; Killeen, N.; Croft, M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity 2001, 15, 445–455. [Google Scholar] [CrossRef]
- Zingoni, A.; Sornasse, T.; Cocks, B.G.; Tanaka, Y.; Santoni, A.; Lanier, L.L. Cross-talk between activated human NK cells and CD4+ T cells via OX40-OX40 ligand interactions. J. Immunol. 2004, 173, 3716–3724. [Google Scholar] [CrossRef] [PubMed]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, Y.; Lizee, G.; Qin, H.; Liu, S.; Rabinovich, B.; Kim, G.J.; Wang, Y.H.; Ye, Y.; Sikora, A.G.; et al. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J. Clin. Invest. 2008, 118, 1165–1175. [Google Scholar] [CrossRef]
- Turaj, A.H.; Cox, K.L.; Penfold, C.A.; French, R.R.; Mockridge, C.I.; Willoughby, J.E.; Tutt, A.L.; Griffiths, J.; Johnson, P.W.M.; Glennie, M.J.; et al. Augmentation of CD134 (OX40)-dependent NK anti-tumour activity is dependent on antibody cross-linking. Sci Rep. 2018, 8, 2278. [Google Scholar] [CrossRef]
- Hanna, J.; Bechtel, P.; Zhai, Y.; Youssef, F.; McLachlan, K.; Mandelboim, O. Novel insights on human NK cells' immunological modalities revealed by gene expression profiling. J. Immunol. 2004, 173, 6547–6563. [Google Scholar] [CrossRef]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef]
- Ronchetti, S.; Zollo, O.; Bruscoli, S.; Agostini, M.; Bianchini, R.; Nocentini, G.; Ayroldi, E.; Riccardi, C. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur. J. Immunol. 2004, 34, 613–622. [Google Scholar] [CrossRef]
- Nocentini, G.; Bartoli, A.; Ronchetti, S.; Giunchi, L.; Cupelli, A.; Delfino, D.; Migliorati, G.; Riccardi, C. Gene structure and chromosomal assignment of mouse GITR, a member of the tumor necrosis factor/nerve growth factor receptor family. DNA Cell Biol. 2000, 19, 205–217. [Google Scholar] [CrossRef]
- Nocentini, G.; Riccardi, C. GITR: A multifaceted regulator of immunity belonging to the tumor necrosis factor receptor superfamily. Eur. J. Immunol. 2005, 35, 1016–1022. [Google Scholar] [CrossRef]
- Hurtado, J.C.; Kim, Y.J.; Kwon, B.S. Signals through 4-1BB are costimulatory to previously activated splenic T cells and inhibit activation-induced cell death. J. Immunol. 1997, 158, 2600–2609. [Google Scholar]
- Ji, H.B.; Liao, G.; Faubion, W.A.; Abadia-Molina, A.C.; Cozzo, C.; Laroux, F.S.; Caton, A.; Terhorst, C. Cutting edge: The natural ligand for glucocorticoid-induced TNF receptor-related protein abrogates regulatory T cell suppression. J. Immunol. 2004, 172, 5823–5827. [Google Scholar] [CrossRef]
- Li, Z.; Mahesh, S.P.; Kim, B.J.; Buggage, R.R.; Nussenblatt, R.B. Expression of glucocorticoid induced TNF receptor family related protein (GITR) on peripheral T cells from normal human donors and patients with non-infectious uveitis. J. Autoimmun. 2003, 21, 83–92. [Google Scholar] [CrossRef]
- Shin, H.H.; Lee, M.H.; Kim, S.G.; Lee, Y.H.; Kwon, B.S.; Choi, H.S. Recombinant glucocorticoid induced tumor necrosis factor receptor (rGITR) induces NOS in murine macrophage. FEBS Lett. 2002, 514, 275–280. [Google Scholar] [CrossRef]
- Tian, J.; Ma, J.; Ma, K.; Ma, B.; Tang, X.; Baidoo, S.E.; Tong, J.; Yan, J.; Lu, L.; Xu, H.; et al. Up-regulation of GITRL on dendritic cells by WGP improves anti-tumor immunity in murine Lewis lung carcinoma. PLoS ONE 2012, 7, e46936. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, Z.; Mahesh, S.P.; Pantanelli, S.; Hwang, F.S.; Siu, W.O.; Nussenblatt, R.B. Glucocorticoid-induced tumor necrosis factor receptor negatively regulates activation of human primary natural killer (NK) cells by blocking proliferative signals and increasing NK cell apoptosis. J. Biol. Chem. 2008, 283, 8202–8210. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M.; Stephens, G.L. The GITR-GITRL interaction: Co-stimulation or contrasuppression of regulatory activity? Nat. Rev. Immunol. 2006, 6, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Snell, L.M.; Lin, G.H.; McPherson, A.J.; Moraes, T.J.; Watts, T.H. T-cell intrinsic effects of GITR and 4-1BB during viral infection and cancer immunotherapy. Immunol. Rev. 2011, 244, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Hanabuchi, S.; Watanabe, N.; Wang, Y.H.; Wang, Y.H.; Ito, T.; Shaw, J.; Cao, W.; Qin, F.X.; Liu, Y.J. Human plasmacytoid predendritic cells activate NK cells through glucocorticoid-induced tumor necrosis factor receptor-ligand (GITRL). Blood 2006, 107, 3617–3623. [Google Scholar] [CrossRef]
- Ko, K.; Yamazaki, S.; Nakamura, K.; Nishioka, T.; Hirota, K.; Yamaguchi, T.; Shimizu, J.; Nomura, T.; Chiba, T.; Sakaguchi, S. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J. Exp. Med. 2005, 202, 885–891. [Google Scholar] [CrossRef]
- Cohen, A.D.; Schaer, D.A.; Liu, C.; Li, Y.; Hirschhorn-Cymmerman, D.; Kim, S.C.; Diab, A.; Rizzuto, G.; Duan, F.; Perales, M.A.; et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS ONE 2010, 5, e10436. [Google Scholar] [CrossRef] [PubMed]
- Turk, M.J.; Guevara-Patino, J.A.; Rizzuto, G.A.; Engelhorn, M.E.; Sakaguchi, S.; Houghton, A.N. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J. Exp. Med. 2004, 200, 771–782. [Google Scholar] [CrossRef]
- Zappasodi, R.; Sirard, C.; Li, Y.; Budhu, S.; Abu-Akeel, M.; Liu, C.; Yang, X.; Zhong, H.; Newman, W.; Qi, J.; et al. Rational design of anti-GITR-based combination immunotherapy. Nat. Med. 2019, 25, 759–766. [Google Scholar] [CrossRef]
- Marhelava, K.; Pilch, Z.; Bajor, M.; Graczyk-Jarzynka, A.; Zagozdzon, R. Targeting Negative and Positive Immune Checkpoints with Monoclonal Antibodies in Therapy of Cancer. Cancers 2019, 11, 1756. [Google Scholar] [CrossRef]
- Harjunpaa, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, H.; Ravens, I.; Papadogianni, G.; Bernhardt, G. Coming of Age: CD96 Emerges as Modulator of Immune Responses. Front. Immunol. 2018, 9, 1072. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar]
- Pesce, S.; Greppi, M.; Grossi, F.; Del Zotto, G.; Moretta, L.; Sivori, S.; Genova, C.; Marcenaro, E. PD/1-PD-Ls Checkpoint: Insight on the Potential Role of NK Cells. Front. Immunol. 2019, 10, 1242. [Google Scholar] [CrossRef]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef]
- Daly, J.; Carlsten, M.; O'Dwyer, M. Sugar Free: Novel Immunotherapeutic Approaches Targeting Siglecs and Sialic Acids to Enhance Natural Killer Cell Cytotoxicity Against Cancer. Front. Immunol. 2019, 10, 1047. [Google Scholar] [CrossRef]
- Borrego, F.; Masilamani, M.; Kabat, J.; Sanni, T.B.; Coligan, J.E. The cell biology of the human natural killer cell CD94/NKG2A inhibitory receptor. Mol. Immunol. 2005, 42, 485–488. [Google Scholar] [CrossRef]
- Rodriguez, J.A. HLA-mediated tumor escape mechanisms that may impair immunotherapy clinical outcomes via T-cell activation. Oncol. Lett 2017, 14, 4415–4427. [Google Scholar] [CrossRef] [PubMed]
- Malmberg, K.J.; Levitsky, V.; Norell, H.; de Matos, C.T.; Carlsten, M.; Schedvins, K.; Rabbani, H.; Moretta, A.; Soderstrom, K.; Levitskaya, J.; et al. IFN-gamma protects short-term ovarian carcinoma cell lines from CTL lysis via a CD94/NKG2A-dependent mechanism. J. Clin. Invest. 2002, 110, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Talebian Yazdi, M.; van Riet, S.; van Schadewijk, A.; Fiocco, M.; van Hall, T.; Taube, C.; Hiemstra, P.S.; van der Burg, S.H. The positive prognostic effect of stromal CD8+ tumor-infiltrating T cells is restrained by the expression of HLA-E in non-small cell lung carcinoma. Oncotarget 2016, 7, 3477–3488. [Google Scholar] [CrossRef]
- Borrego, F.; Ulbrecht, M.; Weiss, E.H.; Coligan, J.E.; Brooks, A.G. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J. Exp. Med. 1998, 187, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Llano, M.; Carretero, M.; Ishitani, A.; Navarro, F.; Lopez-Botet, M.; Geraghty, D.E. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc. Natl. Acad. Sci USA 1998, 95, 5199–5204. [Google Scholar] [CrossRef]
- Llano, M.; Lee, N.; Navarro, F.; Garcia, P.; Albar, J.P.; Geraghty, D.E.; Lopez-Botet, M. HLA-E-bound peptides influence recognition by inhibitory and triggering CD94/NKG2 receptors: Preferential response to an HLA-G-derived nonamer. Eur. J. Immunol. 1998, 28, 2854–2863. [Google Scholar] [CrossRef]
- Mamessier, E.; Sylvain, A.; Thibult, M.L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Goncalves, A.; Andre, P.; Romagne, F.; Thibault, G.; et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Invest. 2011, 121, 3609–3622. [Google Scholar] [CrossRef]
- Sheu, B.C.; Chiou, S.H.; Lin, H.H.; Chow, S.N.; Huang, S.C.; Ho, H.N.; Hsu, S.M. Up-regulation of inhibitory natural killer receptors CD94/NKG2A with suppressed intracellular perforin expression of tumor-infiltrating CD8+ T lymphocytes in human cervical carcinoma. Cancer Res. 2005, 65, 2921–2929. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Vitale, M.; Sivori, S.; Bottino, C.; Morelli, L.; Augugliaro, R.; Barbaresi, M.; Pende, D.; Ciccone, E.; Lopez-Botet, M.; et al. Human natural killer cell receptors for HLA-class I molecules. Evidence that the Kp43 (CD94) molecule functions as receptor for HLA-B alleles. J. Exp. Med. 1994, 180, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Urbani, E.; Andre, P.; Mancusi, A.; Tosti, A.; Topini, F.; Blery, M.; Animobono, L.; Romagne, F.; Wagtmann, N.; et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica 2016, 101, 626–633. [Google Scholar] [CrossRef]
- McWilliams, E.M.; Mele, J.M.; Cheney, C.; Timmerman, E.A.; Fiazuddin, F.; Strattan, E.J.; Mo, X.; Byrd, J.C.; Muthusamy, N.; Awan, F.T. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. OncoImmunology 2016, 5, e1226720. [Google Scholar] [CrossRef]
- Moretta, L.; Bottino, C.; Pende, D.; Vitale, M.; Mingari, M.C.; Moretta, A. Different checkpoints in human NK-cell activation. Trends Immunol. 2004, 25, 670–676. [Google Scholar] [CrossRef]
- van Hall, T.; Andre, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Moretta, A.; Biassoni, R.; Bottino, C.; Pende, D.; Vitale, M.; Poggi, A.; Mingari, M.C.; Moretta, L. Major histocompatibility complex class I-specific receptors on human natural killer and T lymphocytes. Immunol. Rev. 1997, 155, 105–117. [Google Scholar] [CrossRef]
- Jamil, K.M.; Khakoo, S.I. KIR/HLA interactions and pathogen immunity. J. Biomed. Biotechnol. 2011, 2011, 298348. [Google Scholar] [CrossRef]
- Long, E.O.; Barber, D.F.; Burshtyn, D.N.; Faure, M.; Peterson, M.; Rajagopalan, S.; Renard, V.; Sandusky, M.; Stebbins, C.C.; Wagtmann, N.; et al. Inhibition of natural killer cell activation signals by killer cell immunoglobulin-like receptors (CD158). Immunol. Rev. 2001, 181, 223–233. [Google Scholar] [CrossRef]
- Yusa, S.; Campbell, K.S. Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2) can play a direct role in the inhibitory function of killer cell Ig-like receptors in human NK cells. J. Immunol. 2003, 170, 4539–4547. [Google Scholar] [CrossRef] [PubMed]
- Uhrberg, M.; Valiante, N.M.; Shum, B.P.; Shilling, H.G.; Lienert-Weidenbach, K.; Corliss, B.; Tyan, D.; Lanier, L.L.; Parham, P. Human diversity in killer cell inhibitory receptor genes. Immunity 1997, 7, 753–763. [Google Scholar] [CrossRef]
- Purdy, A.K.; Campbell, K.S. Natural killer cells and cancer: Regulation by the killer cell Ig-like receptors (KIR). Cancer Biol. Ther. 2009, 8, 2211–2220. [Google Scholar] [CrossRef]
- Yawata, M.; Yawata, N.; Draghi, M.; Little, A.M.; Partheniou, F.; Parham, P. Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J. Exp. Med. 2006, 203, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Velardi, A.; Ruggeri, L.; Moretta, L. NK cells: A lesson from mismatched hematopoietic transplantation. Trends Immunol. 2002, 23, 438–444. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Capanni, M.; Casucci, M.; Volpi, I.; Tosti, A.; Perruccio, K.; Urbani, E.; Negrin, R.S.; Martelli, M.F.; Velardi, A. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood 1999, 94, 333–339. [Google Scholar] [CrossRef]
- Shi, J.; Tricot, G.; Szmania, S.; Rosen, N.; Garg, T.K.; Malaviarachchi, P.A.; Moreno, A.; Dupont, B.; Hsu, K.C.; Baxter-Lowe, L.A.; et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br. J. Haematol. 2008, 143, 641–653. [Google Scholar] [CrossRef]
- Kohrt, H.E.; Thielens, A.; Marabelle, A.; Sagiv-Barfi, I.; Sola, C.; Chanuc, F.; Fuseri, N.; Bonnafous, C.; Czerwinski, D.; Rajapaksa, A.; et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 2014, 123, 678–686. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Cohen, A.D.; Jagannath, S.; Munshi, N.C.; Spitzer, G.; Hofmeister, C.C.; Efebera, Y.A.; Andre, P.; Zerbib, R.; Caligiuri, M.A. A Phase I Trial of the Anti-KIR Antibody IPH2101 and Lenalidomide in Patients with Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4055–4061. [Google Scholar] [CrossRef]
- Vey, N.; Karlin, L.; Sadot-Lebouvier, S.; Broussais, F.; Berton-Rigaud, D.; Rey, J.; Charbonnier, A.; Marie, D.; Andre, P.; Paturel, C.; et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 2018, 9, 17675–17688. [Google Scholar] [CrossRef] [PubMed]
- Bagot, M.; Porcu, P.; Marie-Cardine, A.; Battistella, M.; William, B.M.; Vermeer, M.; Whittaker, S.; Rotolo, F.; Ram-Wolff, C.; Khodadoust, M.S.; et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: An international, first-in-human, open-label, phase 1 trial. Lancet Oncol. 2019, 20, 1160–1170. [Google Scholar] [CrossRef]
- Vey, N.; Bourhis, J.H.; Boissel, N.; Bordessoule, D.; Prebet, T.; Charbonnier, A.; Etienne, A.; Andre, P.; Romagne, F.; Benson, D.; et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood 2012, 120, 4317–4323. [Google Scholar] [CrossRef]
- Perez-Santos, M.; Guerrero-Gonzalez, T.; Gomez-Conde, E.; Cebada, J.; Flores, A.; Villa-Ruano, N. Treatment of cancer with an anti-KIR antibody: A patent evaluation of US9879082 and US2018208652. Expert. Opin. Ther. Pat. 2020, 30, 159–162. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Deuss, F.A.; Watson, G.M.; Fu, Z.; Rossjohn, J.; Berry, R. Structural Basis for CD96 Immune Receptor Recognition of Nectin-like Protein-5, CD155. Structure 2019, 27, 219–228. [Google Scholar] [CrossRef]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.R.; Readler, J.M.; Sharma, P.; Excoffon, K. Poliovirus Receptor: More than a simple viral receptor. Virus Res. 2017, 242, 1–6. [Google Scholar] [CrossRef]
- Kucan Brlic, P.; Lenac Rovis, T.; Cinamon, G.; Tsukerman, P.; Mandelboim, O.; Jonjic, S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell. Mol. Immunol. 2019, 16, 40–52. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xia, P.; Du, Y.; Liu, S.; Huang, G.; Chen, J.; Zhang, H.; Hou, N.; Cheng, X.; Zhou, L.; et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-gamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J. Biol. Chem. 2014, 289, 17647–17657. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.L.; Garrido-Laguna, I. TIGIT: A novel immunotherapy target moving from bench to bedside. Cancer Immunol. Immunother. 2018, 67, 1659–1667. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef]
- Hattori, N.; Kawaguchi, Y.; Sasaki, Y.; Shimada, S.; Murai, S.; Abe, M.; Baba, Y.; Watanuki, M.; Fujiwara, S.; Arai, N.; et al. Monitoring TIGIT/DNAM-1 and PVR/PVRL2 Immune Checkpoint Expression Levels in Allogeneic Stem Cell Transplantation for Acute Myeloid Leukemia. Biol. Blood Marrow. Transpl. 2019, 25, 861–867. [Google Scholar] [CrossRef]
- Kurtulus, S.; Sakuishi, K.; Ngiow, S.F.; Joller, N.; Tan, D.J.; Teng, M.W.; Smyth, M.J.; Kuchroo, V.K.; Anderson, A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Invest. 2015, 125, 4053–4062. [Google Scholar] [CrossRef]
- Blake, S.J.; Dougall, W.C.; Miles, J.J.; Teng, M.W.; Smyth, M.J. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 5183–5188. [Google Scholar] [CrossRef] [PubMed]
- Perez-Santos, M.; Anaya-Ruiz, M.; Herrera-Camacho, I.; Rosas-Murrieta, N.H.; Millan-Perez Pena, L. Cancer combinatorial immunotherapy using etigilimab and nivolumab: A patent evaluation of WO2018102536. Expert Opin. Ther. Pat. 2020, 30, 83–86. [Google Scholar] [CrossRef]
- Macauley, M.S.; Crocker, P.R.; Paulson, J.C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, G.; Ni, J.; Liu, D.; Klenerman, P.; Munday, J.; Dubock, S.; Mattei, M.G.; Crocker, P.R. Identification and characterization of a novel siglec, siglec-7, expressed by human natural killer cells and monocytes. J. Biol. Chem. 1999, 274, 34089–34095. [Google Scholar] [CrossRef]
- Jandus, C.; Boligan, K.F.; Chijioke, O.; Liu, H.; Dahlhaus, M.; Demoulins, T.; Schneider, C.; Wehrli, M.; Hunger, R.E.; Baerlocher, G.M.; et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J. Clin. Invest. 2014, 124, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Von Gunten, S.; Simon, H.U. Natural anti-Siglec autoantibodies mediate potential immunoregulatory mechanisms: Implications for the clinical use of intravenous immunoglobulins (IVIg). Autoimmun. Rev. 2008, 7, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Bull, C.; den Brok, M.H.; Adema, G.J. Sweet escape: Sialic acids in tumor immune evasion. Biochim. Biophys. Acta 2014, 1846, 238–246. [Google Scholar] [CrossRef]
- Fuster, M.M.; Esko, J.D. The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat. Rev. Cancer 2005, 5, 526–542. [Google Scholar] [CrossRef]
- Hudak, J.E.; Canham, S.M.; Bertozzi, C.R. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat. Chem. Biol. 2014, 10, 69–75. [Google Scholar] [CrossRef]
- Xiao, H.; Woods, E.C.; Vukojicic, P.; Bertozzi, C.R. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2016, 113, 10304–10309. [Google Scholar] [CrossRef]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef]
- Workman, C.J.; Dugger, K.J.; Vignali, D.A. Cutting edge: Molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol. 2002, 169, 5392–5395. [Google Scholar] [CrossRef] [PubMed]
- Demeure, C.E.; Wolfers, J.; Martin-Garcia, N.; Gaulard, P.; Triebel, F. T Lymphocytes infiltrating various tumour types express the MHC class II ligand lymphocyte activation gene-3 (LAG-3): Role of LAG-3/MHC class II interactions in cell-cell contacts. Eur. J. Cancer 2001, 37, 1709–1718. [Google Scholar] [CrossRef]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef]
- Kisielow, M.; Kisielow, J.; Capoferri-Sollami, G.; Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 2005, 35, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Andreae, S.; Buisson, S.; Triebel, F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood 2003, 102, 2130–2137. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tang, L.; Zhang, G.; Wei, H.; Cui, Y.; Guo, L.; Gou, Z.; Chen, X.; Jiang, D.; Zhu, Y.; et al. Characterization of a novel C-type lectin-like gene, LSECtin: Demonstration of carbohydrate binding and expression in sinusoidal endothelial cells of liver and lymph node. J. Biol. Chem. 2004, 279, 18748–18758. [Google Scholar] [CrossRef]
- Workman, C.J.; Vignali, D.A. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J. Immunol. 2005, 174, 688–695. [Google Scholar] [CrossRef]
- Macon-Lemaitre, L.; Triebel, F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology 2005, 115, 170–178. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Miyazaki, T.; Dierich, A.; Benoist, C.; Mathis, D. Independent modes of natural killing distinguished in mice lacking Lag3. Science 1996, 272, 405–408. [Google Scholar] [CrossRef]
- Huard, B.; Tournier, M.; Triebel, F. LAG-3 does not define a specific mode of natural killing in human. Immunol. Lett. 1998, 61, 109–112. [Google Scholar] [CrossRef]
- Brignone, C.; Grygar, C.; Marcu, M.; Schakel, K.; Triebel, F. A soluble form of lymphocyte activation gene-3 (IMP321) induces activation of a large range of human effector cytotoxic cells. J. Immunol. 2007, 17, 4202–4211. [Google Scholar] [CrossRef]
- He, Y.; Rivard, C.J.; Rozeboom, L.; Yu, H.; Ellison, K.; Kowalewski, A.; Zhou, C.; Hirsch, F.R. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. 2016, 107, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, A.; Addeo, A.; Banna, G. New emerging targets in cancer immunotherapy: The role of TIM3. Esmo Open 2019, 4 (Suppl. S3), e000497. [Google Scholar] [CrossRef]
- Gautron, A.S.; Dominguez-Villar, M.; de Marcken, M.; Hafler, D.A. Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur. J. Immunol. 2014, 44, 2703–2711. [Google Scholar] [CrossRef] [PubMed]
- Avery, L.; Filderman, J.; Szymczak-Workman, A.L.; Kane, L.P. Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc. Natl. Acad. Sci. USA 2018, 115, 2455–2460. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef]
- So, E.C.; Khaladj-Ghom, A.; Ji, Y.; Amin, J.; Song, Y.; Burch, E.; Zhou, H.; Sun, H.; Chen, S.; Bentzen, S.; et al. NK cell expression of Tim-3: First impressions matter. Immunobiology 2019, 224, 362–370. [Google Scholar] [CrossRef]
- Ndhlovu, L.C.; Lopez-Verges, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, J.; Gu, H.; Yuan, Y.; Zhang, B.; Zhu, D.; Zhou, J.; Zhu, Y.; Chen, W. The Clinical Significance of Abnormal Tim-3 Expression on NK Cells from Patients with Gastric Cancer. Immunol. Invest. 2015, 44, 578–589. [Google Scholar] [CrossRef]
- Xu, L.; Huang, Y.; Tan, L.; Yu, W.; Chen, D.; Lu, C.; He, J.; Wu, G.; Liu, X.; Zhang, Y. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int. Immunopharmacol. 2015, 29, 635–641. [Google Scholar] [CrossRef]
- Da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; McMahan, R.H.; Strong, M.; Reisdorph, R.; Mahaffey, S.; Palmer, B.E.; Cheng, L.; Kulesza, C.; Hirashima, M.; Niki, T.; et al. Galectin-9 functionally impairs natural killer cells in humans and mice. J. Virol. 2013, 87, 4835–4845. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef]
- Lougaris, V.; Tabellini, G.; Baronio, M.; Patrizi, O.; Gazzurelli, L.; Mitsuiki, N.; Pozzi, M.R.; Grimbacher, B.; Parolini, S.; Plebani, A. CTLA-4 regulates human Natural Killer cell effector functions. Clin. Immunol. 2018, 194, 43–45. [Google Scholar] [CrossRef]
- Stojanovic, A.; Fiegler, N.; Brunner-Weinzierl, M.; Cerwenka, A. CTLA-4 is expressed by activated mouse NK cells and inhibits NK Cell IFN-gamma production in response to mature dendritic cells. J. Immunol. 2014, 192, 4184–4191. [Google Scholar] [CrossRef] [PubMed]
- Beldi-Ferchiou, A.; Caillat-Zucman, S. Control of NK Cell Activation by Immune Checkpoint Molecules. Int. J. Mol. Sci. 2017, 18, 2129. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D.; Bode, C.; Kenefeck, R.; Hou, T.Z.; Wing, J.B.; Kennedy, A.; Bulashevska, A.; Petersen, B.S.; Schaffer, A.A.; Gruning, B.A.; et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med. 2014, 20, 1410–1416. [Google Scholar] [CrossRef]
- Kuehn, H.S.; Ouyang, W.; Lo, B.; Deenick, E.K.; Niemela, J.E.; Avery, D.T.; Schickel, J.N.; Tran, D.Q.; Stoddard, J.; Zhang, Y.; et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 2014, 345, 1623–1627. [Google Scholar] [CrossRef]
- Schwab, C.; Gabrysch, A.; Olbrich, P.; Patino, V.; Warnatz, K.; Wolff, D.; Hoshino, A.; Kobayashi, M.; Imai, K.; Takagi, M.; et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J. Allergy Clin. Immunol. 2018, 142, 1932–1946. [Google Scholar] [CrossRef]
- Pentcheva-Hoang, T.; Egen, J.G.; Wojnoonski, K.; Allison, J.P. B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity 2004, 21, 401–413. [Google Scholar] [CrossRef]
- Yokosuka, T.; Kobayashi, W.; Takamatsu, M.; Sakata-Sogawa, K.; Zeng, H.; Hashimoto-Tane, A.; Yagita, H.; Tokunaga, M.; Saito, T. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity 2010, 33, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Tocut, M.; Brenner, R.; Zandman-Goddard, G. Autoimmune phenomena and disease in cancer patients treated with immune checkpoint inhibitors. Autoimmun. Rev. 2018, 17, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Rapoport, B.L. Immune Dysregulation in Cancer Patients Undergoing Immune Checkpoint Inhibitor Treatment and Potential Predictive Strategies for Future Clinical Practice. Front. Oncol. 2018, 8, 80. [Google Scholar] [CrossRef]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef]
- Jie, H.B.; Schuler, P.J.; Lee, S.C.; Srivastava, R.M.; Argiris, A.; Ferrone, S.; Whiteside, T.L.; Ferris, R.L. CTLA-4(+) Regulatory T Cells Increased in Cetuximab-Treated Head and Neck Cancer Patients Suppress NK Cell Cytotoxicity and Correlate with Poor Prognosis. Cancer Res. 2015, 75, 2200–2210. [Google Scholar] [CrossRef]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef]
- Lotze, M.T.; Matory, Y.L.; Ettinghausen, S.E.; Rayner, A.A.; Sharrow, S.O.; Seipp, C.A.; Custer, M.C.; Rosenberg, S.A. In vivo administration of purified human interleukin 2. II. Half life, immunologic effects, and expansion of peripheral lymphoid cells in vivo with recombinant IL 2. J. Immunol. 1985, 135, 2865–2875. [Google Scholar]
- Hannani, D.; Vetizou, M.; Enot, D.; Rusakiewicz, S.; Chaput, N.; Klatzmann, D.; Desbois, M.; Jacquelot, N.; Vimond, N.; Chouaib, S.; et al. Anticancer immunotherapy by CTLA-4 blockade: Obligatory contribution of IL-2 receptors and negative prognostic impact of soluble CD25. Cell Res. 2015, 25, 208–224. [Google Scholar] [CrossRef]
- Sanseviero, E.; O’Brien, E.M.; Karras, J.R.; Shabaneh, T.B.; Aksoy, B.A.; Xu, W.; Zheng, C.; Yin, X.; Xu, X.; Karakousis, G.C.; et al. Anti-CTLA-4 Activates Intratumoral NK Cells and Combined with IL15/IL15Ralpha Complexes Enhances Tumor Control. Cancer Immunol. Res. 2019, 7, 1371–1380. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Lorigan, P.; Hersey, P.; Hauschild, A.; Robert, C.; McDermott, D.; Marshall, M.A.; Gomez-Navarro, J.; Liang, J.Q.; Bulanhagui, C.A. Phase II trial of tremelimumab (CP-675,206) in patients with advanced refractory or relapsed melanoma. Clin. Cancer Res. 2010, 16, 1042–1048. [Google Scholar] [CrossRef]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, F.R.; Petrini, S.; Ingegnere, T.; Tumino, N.; Besi, F.; Scordamaglia, F.; Munari, E.; Pesce, S.; Marcenaro, E.; Moretta, A.; et al. PD-1 in human NK cells: Evidence of cytoplasmic mRNA and protein expression. OncoImmunology 2019, 8, 1557030. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017, 36, 6143–6153. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Pesce, S.; Muccio, L.; Carlomagno, S.; Sivori, S.; Moretta, A.; Marcenaro, E. Features of Memory-Like and PD-1(+) Human NK Cell Subsets. Front. Immunol. 2016, 7, 351. [Google Scholar] [CrossRef]
- Hicks, K.C.; Fantini, M.; Donahue, R.N.; Schwab, A.; Knudson, K.M.; Tritsch, S.R.; Jochems, C.; Clavijo, P.E.; Allen, C.T.; Hodge, J.W.; et al. Epigenetic priming of both tumor and NK cells augments antibody-dependent cellular cytotoxicity elicited by the anti-PD-L1 antibody avelumab against multiple carcinoma cell types. OncoImmunology 2018, 7, e1466018. [Google Scholar] [CrossRef]
- Julia, E.P.; Amante, A.; Pampena, M.B.; Mordoh, J.; Levy, E.M. Avelumab, an IgG1 anti-PD-L1 Immune Checkpoint Inhibitor, Triggers NK Cell-Mediated Cytotoxicity and Cytokine Production Against Triple Negative Breast Cancer Cells. Front. Immunol. 2018, 9, 2140. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Rotem-Yehudar, R.; Slama, G.; Landes, S.; Kneller, A.; Leiba, M.; Koren-Michowitz, M.; Shimoni, A.; Nagler, A. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2008, 14, 3044–3051. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Powderly, J.D.; Burris, H.A.; Kittaneh, M.; Grice, J.H.; Smothers, J.F.; Brett, S.; Fleming, M.E.; May, R.; Marshall, S.; et al. Clinical and pharmacodynamic (PD) results of a phase I trial with AMP-224 (B7-DC Fc) that binds to the PD-1 receptor. J. Clin. Oncol. 2013, 31 (Suppl. S15), 3044. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M. Chimeric antigen receptors: Driving immunology towards synthetic biology. Curr. Opin. Immunol. 2016, 41, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Ping, N.; Kang, L.; Liu, H.; Qin, S.; Wu, Q.; Chen, X.; Zhou, M.; Xia, F.; Ye, A.; et al. Radiation Priming Chimeric Antigen Receptor T-Cell Therapy in Relapsed/Refractory Diffuse Large B-Cell Lymphoma With High Tumor Burden. J. Immunother. 2020, 43, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.; Hewitson, R.; Pagliuca, A.; Benjamin, R. Cancer immunotherapy with CAR-T cells-behold the future. Clin. Med. 2018, 18, 324–328. [Google Scholar] [CrossRef]
- Daher, M.; Rezvani, K. Next generation natural killer cells for cancer immunotherapy: The promise of genetic engineering. Curr. Opin. Immunol. 2018, 51, 146–153. [Google Scholar] [CrossRef]
- Lang, S.; Vujanovic, N.L.; Wollenberg, B.; Whiteside, T.L. Absence of B7.1-CD28/CTLA-4-mediated co-stimulation in human NK cells. Eur. J. Immunol. 1998, 28, 780–786. [Google Scholar] [CrossRef]
- Pule, M.A.; Straathof, K.C.; Dotti, G.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 2005, 12, 933–941. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Receptor/Target Antigen | Drugs /Interventions | Combinations (Drug, Biologics) | Disease | Clinical Trials, Allocation | Study Title | Participants | Status | Sponsors | Clinical Trials Identifier |
|---|---|---|---|---|---|---|---|---|---|
| 4-1BB | PF-04518600 (4-1BB agonist) | Utomilumab (PF-05082566) | Neoplasms | Phase 1, Nonrandomized | Study of OX40 Agonist PF-04518600 Alone And In Combination With4-1BB Agonist PF-05082566 | 176 | Active, not recruiting | Pfizer | NCT02315066 |
| CAR-T cells (PD-L1 CAR gene composed of CD137/4-1BB and CD3ζ and PD-L1 single-chain variable/ scFv fragment) | Chemotherapy | Advanced Lung Cancer | Phase 1, Not available (N/A) | CAR-T cell immunotherapy for advanced lung cancer | 22 | Recruiting | Sun Yat-sen University | NCT03330834 | |
| ATOR-1017 (human mAb) | Single agent | Solid tumor, Neoplasms | Phase 1, N/A | ATOR-1017 First-in-human study | 50 | Recruiting | Alligator Bioscience | NCT04144842 | |
| BMS-663513 (anti-41BB agonistic antibody) | Nivolumab, Cyclophosphamide, Fludarabine, Interleukin-2 | Melanoma | Phase 1, N/A | Combining PD-1 blockade, CD137 Agonism and adoptive cell therapy for metastatic melanoma | 11 | Active, not recruiting | H. Lee Moffitt Cancer Center and Research Institute | NCT02652455 | |
| PRS-343 (4-1BB/HER2 bispecific antibody) | Atezolizumab | HER2-positive Breast, Gastric, Bladder, Solid tumor | Phase 1, N/A | PRS-343 in combination with Atezolizumab in HER2-positive solid tumor | 70 | Recruiting | Pieris Pharmaceuticals, Inc. | NCT03650348 | |
| Anti-CD137 (4-1BB) (BMS-663513) | Single agent | Melanoma | Phase 2, Randomized | Phase II, 2nd Line Melanoma—RAND Monotherapy | 158 | Completed | Bristol-Myers Squibb | NCT00612664 | |
| OX40 (CD134) | Anti-OX40 Antibody BMS 986178 | TLR9 Agonist SD-101 | Neoplasms | Phase 1, N/A | SD-101 and BMS-986178 in Treating Patients With Advanced or Metastatic Solid Malignancies | 27 | Recruiting | Ronald Levy | NCT03831295 |
| anti-OX40 murine monoclonal antibody, MEDI 6469 | Single agent | Head and Neck Cancer | Phase 1, Nonrandomized | Anti-OX40 Antibody in Head and Neck Cancer Patients | 17 | Active, not recruiting | Providence Health & Services | NCT02274155 | |
| Anti-OX40 Antibody PF-04518600 | Avelumab, Binimetinib, Utomilumab | Breast Carcinoma | Phase 2, Randomized | Avelumab With Binimetinib, Utomilumab, or Anti-OX40 Antibody PF-04518600 in Treating Triple Negative Breast Cancer | 150 | Recruiting | Hope Rugo, MD | NCT03971409 | |
| GSK3174998 | Pembrolizumab | Neoplasms | Phase 1, Nonrandomized | GSK3174998 Alone or With Pembrolizumab in Subjects With Advanced Solid Tumors (ENGAGE-1) | 142 | Completed | GlaxoSmithKline | NCT02528357 | |
| MOXR0916 | Single agent | Neoplasms | Phase 1, Nonrandomized | A Study to Assess Safety and Pharmacokinetics of MOXR0916 in Participants With Locally Advanced or Metastatic Solid Tumors | 174 | Completed | Genentech, Inc. | NCT02219724 | |
| anti-OX40 | Radiation: Radiation, Cyclophosphamide | Prostate Cancer | Phase 1, Nonrandomized | Anti-OX40, Cyclophosphamide (CTX) and Radiation in Patients With Progressive Metastatic Prostate Cancer | 13 | Completed | Providence Health & Services | NCT01303705 | |
| anti-OX40 | Biological: Tetanus vaccine Biological: KLH | Advanced Cancer | Phase 1, Randomized | Phase 1 Study of anti-OX40 in Patients With Advanced Cancer | 30 | Completed | Providence Health & Services | NCT01644968 | |
| GITR/GITRL | Biological: TRX518 | N/A | Unresectable Stage III or Stage IV Malignant Melanoma or Other Solid tumor malignancies | Phase 1, N/A | Trial of TRX518 (Anti-GITR mAb) in Stage III or IV Malignant Melanoma or Other Solid Tumors (TRX518-001) | 10 | Completed | Leap Therapeutics, Inc. | NCT01239134 |
| Anti-GITR Agonistic Monoclonal Antibody BMS-986156 | Ipilimumab, Nivolumab, Radiation: Stereotactic Body, Radiation Therapy | Neoplasms & Lung carcinoma | Phase 1, 2, Nonrandomized | BMS-986156, Ipilimumab, and Nivolumab With or Without Stereotactic Body Radiation Therapy in Treating Patients With Advanced or Metastatic Lung/Chest or Liver Cancers | 60 | Recruiting | M.D. Anderson Cancer Center | NCT04021043 | |
| MEDI1873 | Single agent | Advanced solid tumor | Phase 1, N/A | A Study in Adult Subjects With Select Advanced Solid Tumors | 40 | Completed | MedImmune LLC | NCT02583165 | |
| anti-GITR agonistic monoclonal antibody ASP1951 | Pembrolizumab | Advanced solid tumor | Phase 1, Nonrandomized | A Study of ASP1951 in Subjects With Advanced Solid Tumors | 435 | Recruiting | Astellas Pharma Global Development, Inc. | NCT03799003 | |
| GWN323 (Anti-GITR) | PDR001 | Solid tumor | Phase 1, Nonrandomized | Phase I/Ib Study of GWN323 Alone and in Combination With PDR001 in Patients With Advanced Malignancies and Lymphomas | 92 | Completed | Novartis Pharmaceuticals | NCT02740270 | |
| BMS-986156 | Nivolumab | Solid tumor | Phase 1, 2, Nonrandomized | An Investigational Immuno-therapy Study of Experimental Medication BMS-986156, Given by Itself or in Combination With Nivolumab in Patients With Solid Cancers or Cancers That Have Spread. | 331 | Active, not recruiting | Bristol-Myers Squibb | NCT02598960 | |
| NKG2A | Monalizumab (IPH2201) | Single agent | Gynecologic Cancer | Phase 1, N/A | A Dose-Ranging Study of IPH2201 in Patients With Gynecologic Malignancies | 59 | Completed | Canadian Cancer Trials Group | NCT02459301 |
| Single agent | Hematologic Malignancies | Phase 1, N/A | Study of a Humanized Antibody Initiated 2 Months After an HLA Matched Allogenic Stem Cell Transplantation (PIRAT) | 18 | Recruiting | Institut Paoli-Calmettes | NCT02921685 | ||
| Cetuximab, Anti-PD-L1 | Head and Neck Neoplasms | Phase 1, 2, Nonrandomized | Study of Monalizumab and Cetuximab in Patients With Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck | 140 | Recruiting | Innate Pharma | NCT02643550 | ||
| Durvalumab (MEDI4736) | Advanced solid tumor | Phase 1, 2, Nonrandomized | A Study of Durvalumab (MEDI4736) and Monalizumab in Solid Tumors | 383 | Active, not recruiting | MedImmune LLC | NCT02671435 | ||
| Afatinib, Palbociclib, standard of care,IPH2201, Durvalumab, Niraparib, BAY1163877 | Carcinoma, Squamous Cell of Head and Neck | Phase 2, Nonrandomized | Biomarker-based Study in recurrent or metastatic squamous cell carcinoma of the head and neck (UPSTREAM) | 340 | Recruiting | European Organisation for Research and Treatment of Cancer - EORTC | NCT03088059 | ||
| Durvalumab + Oleclumab | Stage III Non-small Cell Lung Cancer Unresectable | Phase 2, Randomized | Durvalumab Alone or in Combination With Novel Agents in Subjects With NSCLC (COAST) | 300 | Recruiting | MedImmune LLC | NCT03822351 | ||
| KIR | Anti-KIR (1-7F9) | Single agent | Multiple Myeloma | Phase 1, N/A | An Open-label, Dose-escalation Safety and Tolerability Trial Assessing Anti-KIR (1-7F9) in Subjects With Multiple Myeloma | 32 | Completed | Innate Pharma | NCT00552396 |
| IPH2101, a human monoclonal anti-KIR antibody | Single agent | Multiple Myeloma | Phase 2, Randomized | Evaluation of Activity, Safety and Pharmacology of IPH2101 a Human Monoclonal Antibody in Patients With Multiple Myeloma (REMYKIR) | 27 | Completed | Innate Pharma | NCT00999830 | |
| Single agent | Acute myeloid leukemia | Phase 1, Nonrandomized | A Safety and Tolerability Extension Trial Assessing Repeated Dosing of Anti-KIR (1-7F9) Human Monoclonal Antibody in Patients With Acute Myeloid Leukaemia | 21 | Completed | Innate Pharma | NCT01256073 | ||
| Single agent | Smoldering Multiple Myeloma | Phase 2, Randomized | Study on the Anti-tumor Activity, Safety and Pharmacology of IPH2101 in Patients With Smoldering Multiple Myeloma (KIRMONO) | 30 | Completed | Innate Pharma | NCT01222286 | ||
| Lenalidomide | Relapsed Multiple Myeloma | Phase 1, Nonrandomized | Study on the Safety, Anti-tumor Activity and Pharmacology of IPH2101 Combined With Lenalidomide in Patients With Multiple Myeloma Experiencing a First or Second Relapse (KIRIMID) | 15 | Completed | Innate Pharma | NCT01217203 | ||
| Lirilumab (IPH2102/BMS-986015) | Placebo | Acute myeloid leukemia | Phase 2, Randomized | Efficacy Study of Anti-KIR Monoclonal Antibody as Maintenance Treatment in Acute Myeloid Leukemia (EFFIKIR) (EFFIKIR) | 152 | Completed | Innate Pharma | NCT01687387 | |
| Lirilumab (BMS-986015) | Ipilimumab | Cancer, (not-otherwise specified) NOS | Phase 1, Nonrandomized | Safety Study of BMS-986015 (Anti-KIR) in Combination With Ipilimumab in Subjects With Selected Advanced Tumor | 22 | Completed | Bristol-Myers Squibb | NCT01750580 | |
| Elotuzumab, Urelumab | Multiple Myeloma | Phase 1, Randomized | A Phase I Open Label Study of the Safety and Tolerability of Elotuzumab (BMS-901608) Administered in Combination With Either Lirilumab (BMS-986015) or Urelumab (BMS-663513) in Subjects With Multiple Myeloma | 44 | Completed | Bristol-Myers Squibb | NCT02252263 | ||
| Rituximab | Leukemia | Phase 2, Nonrandomized | Lirilumab With Rituximab for Relapsed, Refractory or High-risk Untreated Chronic Lymphocytic Leukemia (CLL) Patients | 7 | Completed | M.D. Anderson Cancer Center | NCT02481297 | ||
| Nivolumab, Ipilimumab | Advanced Cancer | Phase 1, Nonrandomized | A Safety Study of Lirilumab in Combination With Nivolumab or in Combination With Nivolumab and Ipilimumab in Advanced and/or Metastatic Solid Tumors | 21 | Active, not recruiting | Bristol-Myers Squibb | NCT03203876 | ||
| Nivolumab, Ipilimumab | Cancer, NOS | Phase 1, 2 Randomized | A Study of an Anti-KIR Antibody Lirilumab in Combination With an Anti-PD-1 Antibody Nivolumab and Nivolumab Plus an Anti-CTLA-4 Ipilimumab Antibody in Patients With Advanced Solid Tumors | 337 | Completed | Bristol-Myers Squibb | NCT01714739 | ||
| IPH4102 | Single agent | Cutaneous T cell Lymphoma | Phase 1, N/A | Study of IPH4102 in Patients With Relapsed/Refractory Cutaneous T cell Lymphomas (CTCL) | 60 | Active, not recruiting | Innate Pharma | NCT02593045 | |
| Gemcitabine + Oxaliplatin | T cell lymphoma | Phase 2, Nonrandomized | IPH4102 Alone or in Combination With Chemotherapy in Patients With Advanced T Cell Lymphoma (TELLOMAK) | 250 | Recruiting | Innate Pharma | NCT03902184 | ||
| TIGIT | MTIG7192A (anti-TIGIT mAb) | Atezolizumab, Carboplatin, Cisplatin, Pemetrexed, Paclitaxel, Etoposide | Advanced/Metastatic Tumors | Phase 1, Nonrandomized | Safety and Pharmacokinetics (PK) of Escalating Doses of MTIG7192A as a Single Agent and in Combination With Atezolizumab With and Without Chemotherapy in Locally Advanced or Metastatic Tumors | 400 | Recruiting | Genentech, Inc. | NCT02794571 |
| Atezolizumab, Placebo | Non-small Cell Lung Cancer | Phase 2, Randomized | A Study of MTIG7192A in Combination With Atezolizumab in Chemotherapy-Naïve Patients With Locally Advanced or Metastatic Non-Small Cell Lung Cancer | 135 | Active, not recruiting | Genentech, Inc. | NCT03563716 | ||
| Tiragolumab | Atezolizumab, Carboplatin, Etoposide, Placebo | Small cell lung cancer | Phase 3, Randomized | A Study of Atezolizumab Plus Carboplatin and Etoposide With or Without Tiragolumab in Patients With Untreated Extensive-Stage Small Cell Lung Cancer (SKYSCRAPER-02) | 400 | Recruiting | Hoffmann-La Roche | NCT04256421 | |
| Atezolizumab, Matching Placebo | Non-small Cell Lung Cancer | Phase 3, Randomized | A Study of Tiragolumab in Combination With Atezolizumab Compared With Placebo in Combination With Atezolizumab in Patients With Previously Untreated Locally Advanced Unresectable or Metastatic PD-L1-Selected Non-Small Cell Lung Cancer (SKYSCRAPER-01) | 500 | Recruiting | Hoffmann-La Roche | NCT04294810 | ||
| AB154 (anti-TIGIT mAb) | AB122 | Solid Tumor | Phase 1, Nonrandomized | A Study to Evaluate the Safety and Tolerability of AB154 in Participants With Advanced Malignancies | 66 | Recruiting | Arcus Biosciences, Inc. | NCT03628677 | |
| LAG-3 | Sym022 | Single agent | Metastatic Cancer, Solid Tumor, Lymphoma | Phase 1, N/A | Sym022 (Anti-LAG-3) in Patients With Advanced Solid Tumor Malignancies or Lymphomas | 15 | Completed | Symphogen A/S | NCT03489369 |
| Sym021 (anti-PD-1 mAb), Sym023 (anti-TIM-3 mAb) | Metastatic Cancer, Solid Tumor, Lymphoma | Phase 1, Nonrandomized | Sym021 Monotherapy and in Combination With Sym022 or Sym023 in Patients With Advanced Solid Tumor Malignancies or Lymphomas | 102 | Recruiting | Symphogen A/S | NCT03311412 | ||
| BMS-986213 | Relatlimab, Nivolumab | Neoplasms | Phase 1, 2, Randomized | An Investigational Immuno-therapy Study to Assess the Safety, Tolerability and Effectiveness of Anti-LAG-3 With and Without Anti-PD-1 in the Treatment of Solid Tumors | 1500 | Recruiting | Bristol-Myers Squibb | NCT01968109 | |
| BMS-986016 (Relatlimab) | BMS-936558 | Hematologic Neoplasms | Phase 1, 2 Nonrandomized | Safety Study of Anti-LAG-3 in Relapsed or Refractory Hematologic Malignancies | 109 | Active, not recruiting | Bristol-Myers Squibb | NCT02061761 | |
| Anti-PD-1, Anti-CD137 | Glioblastoma, Gliosarcoma, Recurrent Brain Neoplasm | Phase 1, Nonrandomized | Anti-LAG-3 Alone & in Combination with Nivolumab Treating Patients with Recurrent glioblastoma multiforme (GBM) (Anti-CD137 Arm Closed 10/16/18) | 63 | Active, not recruiting | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins | NCT02658981 | ||
| Nivolumab | Cancer | Phase 1, Nonrandomized | Safety Study of BMS-986016 With or Without Nivolumab in Patients With Advanced Solid Tumors | 45 | Active, not recruiting | Bristol-Myers Squibb | NCT02966548 | ||
| Nivolumab, Carboplatin, Paclitaxel Radiation: Radiation | Gastric Cancer, Esophageal Cancer, Gastro Esophageal Cancer | Phase 1, Nonrandomized | Nivolumab or Nivolumab/Relatlimab Prior to Chemoradiation With II/III Gastro/Esophageal Cancer | 25 | Recruiting | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins | NCT03044613 | ||
| Drug: Nivolumab | Glioblastoma | Phase 1, N/A | Cytokine Microdialysis for Real-Time Immune Monitoring in Glioblastoma Patients Undergoing Checkpoint Blockade | 25 | Recruiting | National Institute of Neurological Disorders and Stroke (NINDS) | NCT03493932 | ||
| Nivolumab | Chordoma, Locally Advanced Chordoma, Metastatic Chordoma, Unresectable Chordoma | Phase 2, N/A | Nivolumab and Relatlimab in Treating Participants With Advanced Chordoma | 20 | Recruiting | Jonsson Comprehensive Cancer Center | NCT03623854 | ||
| Nivolumab | Melanoma | Phase 2, Randomized | Nivolumab, BMS-936558 in Combination With Relatlimab, BMS-986016 in Patients With Metastatic Melanoma Naïve to Prior Immunotherapy in the Metastatic Setting | 42 | Recruiting | John Kirkwood | NCT03743766 | ||
| IMP321 (eftilagimod alpha) | Placebo, Paclitaxel | Adenocarcinoma Breast Stage IV | Phase 2, Randomized | IMP321 (Eftilagimod Alpha) as Adjunctive to a Standard Chemotherapy Paclitaxel Metastatic Breast Carcinoma | 241 | Active, not recruiting | Immutep S.A. | NCT02614833 | |
| Pembrolizumab | Stage III, IV Melanoma | Phase 1 | Phase 1 Study of IMP321 (Eftilagimod Alpha) Adjuvant to Anti-programmed cell death protein -1 (PD-1) Therapy in Unresectable or Metastatic Melanoma | 24 | Completed | Immutep Australia Pty. Ltd. | NCT02676869 | ||
| Avelumab | Solid Tumors, Peritoneal Carcinomatosis | Phase 1, Nonrandomized | Feasibility and Safety of IMP321 for Advanced Stage Solid Tumors | 26 | Active, not recruiting | InstitutfürKlinischeKrebsforschung IKF GmbH at KrankenhausNordwest | NCT03252938 | ||
| Pembrolizumab | Non-small-cell lung carcinoma (NSCLC), Head and Neck squamous cell carcinoma (HNSCC) | Phase 2, Nonrandomized | Combination Study With Soluble LAG-3 Fusion Protein Eftilagimod Alpha (IMP321) and Pembrolizumab in Patients With Previously Untreated Unresectable or Metastatic NSCLC, or Recurrent PD-X Refractory NSCLC or With Recurrent or Metastatic HNSCC (TACTI-002) | 109 | Recruiting | Immutep S.A. | NCT03625323 | ||
| TIM-3 | Sym023 (anti-TIM-3) | Single agent | Metastatic Cancer Solid Tumor Lymphoma | Phase 1, N/A | Sym023 (Anti-TIM-3) in Patients With Advanced Solid Tumor Malignancies or Lymphomas | 24 | Active, not recruiting | Symphogen A/S | NCT03489343 |
| Drug: TSR-022 (anti-TIM-3 mAb) | TSR-042 (an anti-PD-1 antibody), TSR-033 (an anti-LAG-3 antibody) | Advanced or Metastatic Solid Tumors | Phase 1, Nonrandomized | A Phase 1 Study of TSR-022, an Anti-TIM-3 Monoclonal Antibody, in Patients With Advanced Solid Tumors (AMBER) | 873 | Recruiting | Tesaro, Inc. | NCT02817633 | |
| TSR-042 | Adult Primary Liver Cancer Advanced Adult Primary Liver Cancer Localized Unresectable Adult Primary Liver Cancer | Phase 2, N/A | TSR-022 (Anti-TIM-3 Antibody) and TSR-042 (Anti-PD-1 Antibody) in Patients With Liver Cancer | 42 | Recruiting | University of Hawaii | NCT03680508 | ||
| Dostarlimab (TSR-042) (singly) | Melanoma Stage III, IV | Phase 2, Randomized | Neoadjuvant PD-1 Inhibitor Dostarlimab (TSR-042) vs. Combination of Tim-3 Inhibitor TSR-022 and PD-1 Inhibitor Dostarlimab (TSR-042) in Melanoma | 56 | Recruiting | Diwakar Davar | NCT04139902 | ||
| LY3321367 (anti-TIM-3 mAb) | LY3300054 | Solid Tumor | Phase 1, Nonrandomized | A Study of LY3321367 Alone or With LY3300054 in Participants With Advanced Relapsed/Refractory Solid Tumors | 275 | Active, not recruiting | Eli Lilly and Company | NCT03099109 | |
| BGB-A425 (anti-TIM-3 mAb) | Tislelizumab | Locally Advanced or Metastatic Solid Tumors | Phase 1, 2, Nonrandomized | Study of BGB-A425 in Combination With Tislelizumab in Advanced Solid Tumors | 162 | Recruiting | BeiGene | NCT03744468 | |
| MBG453 (anti-TIM-3 hmAb) | PDR001 (anti-PD-1), Decitabine | Advanced Malignancies | Phase 1, 2, Nonrandomized | Phase I-Ib/II Study of MBG453 as Single Agent and in Combination With PDR001 in Patients With Advanced Malignancies | 269 | Recruiting | Novartis Pharmaceuticals | NCT02608268 | |
| Decitabine, PDR001 | Leukemia, Myeloid Leukemia, Acute Myeloid Leukemia, Myelodysplastic Syndromes, Preleukemia, Bone Marrow Diseases, Hematologic Diseases | Phase 1, Randomized | Study of PDR001 and/or MBG453 in Combination With Decitabine in Patients With AML or High Risk MDS | 235 | Recruiting | Novartis Pharmaceuticals | NCT03066648 | ||
| Spartalizumab (PDR001) | Glioblastoma, Multiforme | Phase 1, N/A | Trial of Anti-Tim-3 in Combination With Anti-PD-1 and Stereotactic radiosurgery (SRS) in Recurrent GBM | 15 | Active, not recruiting | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins | NCT03961971 | ||
| CTLA-4 | AGEN1181 (anti-CTLA-4 mAb) | AGEN2034 (anti-PD-1) | Advanced Cancer | Phase 1, Randomized | Fc-Engineered Anti-CTLA-4 Monoclonal Antibody in Advanced Cancer | 86 | Recruiting | Agenus Inc. | NCT03860272 |
| Tremelimumab (anti-CTLA-4 mAb) | Durvalumab, Fulvestrant | Breast Cancer | Phase 2, Nonrandomized | Anti PD-L1 Antibody + Anti CTLA-4 Antibody in Combination With Hormone Therapy in Patients With Hormone Receptor Positive HER2-negative Recurrent or Metastatic Breast Cancer | 33 | Current Unknown | Kyoto Breast Cancer Research Network | NCT03430466 | |
| Azacitidine, Durvalumab | Head and Neck Cancer | Phase 1, 2, N/A | Azacitidine, Durvalumab, and Tremelimumab in Recurrent and/or Metastatic Head and Neck Cancer Patients | 59 | Recruiting | Massachusetts General Hospital | NCT03019003 | ||
| Zalifrelimab (AGEN1884) (anti-CTLA-4) | Balstilimab (AGEN2034) (anti-PD-1) | Cervical Cancer | Phase 2, Randomized | Phase 2 Study of Anti-PD-1 Independently or in Combination With Anti-CTLA-4 in Second-Line Cervical Cancer | 200 | Recruiting | Agenus Inc. | NCT03894215 | |
| PD-L1/CTLA4 BsAb | FOLFIRINOX (chemo drug) | Locally Advanced and Metastatic Pancreatic Cancer | Phase 1, 2, Nonrandomized | Study of Immunotherapy Combined With Chemotherapy in Locally Advanced and Metastatic Pancreatic Cancer | 60 | Recruiting | Changhai Hospital | NCT04324307 | |
| AGEN1884 (anti-CTLA-4 mAb) | PD-1/PD-L-1 inhibitor | Advanced Solid Cancers Refractory to PD-1 | Phase 1, 2, N/A | AGEN1884, an Anti-CTLA-4 Human Monoclonal Antibody in Subjects With Advanced or Refractory Cancer and Who Have Progressed With PD-1/PD-L1 Inhibitor as Their Most Recent Therapy | 90 | Recruiting | Agenus Inc. | NCT02694822 | |
| Ipilimumab (MDX-010) (BMS-734016) (anti-CTLA-4 mAb) | Docetaxel | Prostate Cancer | Phase 2, Randomized | Comparison Study of MDX-010 (CTLA-4) Alone and Combined With Docetaxel in the Treatment of Patients With Hormone Refractory Prostate Cancer | N/A | Completed | Bristol-Myers Squibb | NCT00050596 | |
| Single agent | Melanoma | Phase 2, Randomized | Study of Ipilimumab (MDX-010) Monotherapy in Patients With Previously Treated Unresectable Stage III or IV Melanoma | 210 | Completed | Bristol-Myers Squibb | NCT00289640 | ||
| REGN2810, Pembrolizumab | Non-small Cell Lung Cancer | Phase 3, Randomized | REGN2810 (Anti-PD-1 Antibody), Platinum-based Doublet Chemotherapy, and Ipilimumab (Anti-CTLA-4 Antibody) Versus Pembrolizumab Monotherapy in Patients With Lung Cancer | 5 | Active, not recruiting | Regeneron Pharmaceuticals | NCT03515629 | ||
| SHR-1210 | Non-small Cell Lung Cancer | Phase 1, N/A | Anti-CTLA-4 Antibody Followed by Anti-PD-1 Antibody in Recurrent or Metastatic NSCLC (SEQUENCE) | 10 | Current Unknown | Sun Yat-sen University | NCT03527251 | ||
| AK104, a PD-1 and CTLA-4 bispecific antibody | Single agent | Advanced solid tumor | Phase 1, 2, N/A | Safety and Efficacy of AK104, a PD-1/CTLA-4 Bispecific Antibody, in Selected Advanced Solid Tumors | 120 | Not yet recruiting | Akeso | NCT04172454 | |
| Tremelimumab | Olaparib | Ovarian Cancer Fallopian Tube Cancer Peritoneal Neoplasms | Phase 1, 2, N/A | PARP-inhibition and CTLA-4 Blockade in BRCA-deficient Ovarian Cancer | 50 | Recruiting | New Mexico Cancer Care Alliance | NCT02571725 | |
| MGD019 (DART protein binding PD-1 and CTLA-4) | Single agent | Solid Tumor, Adult Advanced Cancer | Phase 1, N/A | MGD019 DART Protein in Unresectable/Metastatic Cancer | Recruiting | MacroGenics | NCT03761017 | ||
| CS1002 (anti-CTLA-4 mAb) | CS1003 (anti-PD-1 mAb) | Solid tumor, Adult | Phase 1, Randomized | A Study of CS1002 in Subjects With Advanced Solid Tumors | 108 | Recruiting | CStone Pharmaceuticals | NCT03523819 | |
| PD-1 | Pembrolizumab (MK-3475) (lambrolizumab) | Single agent | Anal Cancer | Phase 2, N/A | Pembrolizumab in Refractory Metastatic Anal Cancer | 32 | Recruiting | Dana-Farber Cancer Institute | NCT02919969 |
| Radiation: RT Boost | Breast Cancer | Phase 1, 2, N/A | Breast Cancer Study of Preoperative Pembrolizumab + Radiation | 60 | Recruiting | Stephen Shiao | NCT03366844 | ||
| Nivolumab (anti-PD-1 mAb) (Opdivo) | Single agent | Prostate Cancer | Phase 2, Nonrandomized | Nivolumab in Patients With High-Risk Biochemically Recurrent Prostate Cancer | 34 | Recruiting | Beth Israel Deaconess Medical Center | NCT03637543 | |
| DKN-01 | Biliary tract cancer | Phase 2, Nonrandomized | Study of the Combination of DKN-01 and Nivolumab in Previously Treated Patients With Advanced Biliary Tract Cancer (BTC) | 30 | Recruiting | Massachusetts General Hospital | NCT04057365 | ||
| Atezolizumab (anti-PD-1 mAb) (MPDL3280A) (RG7446) | Single agent | Non-small Cell Lung Cancer | Phase 2, N/A | Atezolizumab in Advanced Non-small Cell Lung Cancer With Rare Histologies (CHANCE Trial) (CHANCE) | 43 | Recruiting | GruppoOncologicoItaliano di RicercaClinica | NCT03976518 | |
| Single agent | Breast Cancer | Phase 2, N/A | A Study of Atezolizumab in Participants With Locally Advanced or Metastatic Urothelial Bladder Cancer (Cohort 1) | 119 | Active, not recruiting | Hoffmann-La Roche | NCT02951767 | ||
| Avelumab (MSB0010718C) | Single agent | Metastatic Colorectal Cancer | Phase 2, N/A | Avelumab for microsatellite instability-high (MSI-H) or POLE (i.e. Mutations in the exonuclease domain of the DNA polymerase epsilon (POLE) gene) Mutated Metastatic Colorectal Cancer | 33 | Active, not recruiting | Asan Medical Center | NCT03150706 | |
| Axitinib | Cervical Cancer | Not applicable | Avelumab With Axitinib in Persistent or Recurrent Cervical Cancer After Platinum-based Chemotherapy (ALARICE) | 23 | Recruiting | The University of Hong Kong | NCT03826589 | ||
| Durvalumab (MEDI4736) | Single agent | Bladder Cancer | Phase 2, N/A | Efficacy of Durvalumab in Non-muscle-invasive Bladder Cancer | 39 | Recruiting | Hellenic Genito Urinary Cancer Group | NCT03759496 | |
| Azacitidine, Tremelimumab | Head and Neck Cancer | Phase 1, 2, N/A | Azacitidine, Durvalumab, and Tremelimumab in Recurrent and/or Metastatic Head and Neck Cancer Patients | 59 | Recruiting | Massachusetts General Hospital | NCT03019003 | ||
| Cemiplimab (REGN-2810) (Libtayo) | Plerixafor | Metastatic Pancreatic Cancer | Phase 2, N/A | Plerixafor and Cemiplimab in Metastatic Pancreatic Cancer | 21 | Not yet recruiting | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins | NCT04177810 | |
| REGN5678 | Metastatic Castration-resistant Prostate Cancer | Phase 1, 2, Nonrandomized | Study of REGN5678 (Anti-PSMAxCD28) With Cemiplimab (Anti-PD-1) in Patients With Metastatic Castration-resistant Prostate Cancer | 123 | Recruiting | Regeneron Pharmaceuticals | NCT03972657 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chauhan, S.K.S.; Koehl, U.; Kloess, S. Harnessing NK Cell Checkpoint-Modulating Immunotherapies. Cancers 2020, 12, 1807. https://doi.org/10.3390/cancers12071807
Chauhan SKS, Koehl U, Kloess S. Harnessing NK Cell Checkpoint-Modulating Immunotherapies. Cancers. 2020; 12(7):1807. https://doi.org/10.3390/cancers12071807
Chicago/Turabian StyleChauhan, Sachin Kumar Singh, Ulrike Koehl, and Stephan Kloess. 2020. "Harnessing NK Cell Checkpoint-Modulating Immunotherapies" Cancers 12, no. 7: 1807. https://doi.org/10.3390/cancers12071807
APA StyleChauhan, S. K. S., Koehl, U., & Kloess, S. (2020). Harnessing NK Cell Checkpoint-Modulating Immunotherapies. Cancers, 12(7), 1807. https://doi.org/10.3390/cancers12071807