Loss of HMGCS2 Enhances Lipogenesis and Attenuates the Protective Effect of the Ketogenic Diet in Liver Cancer


Abstract
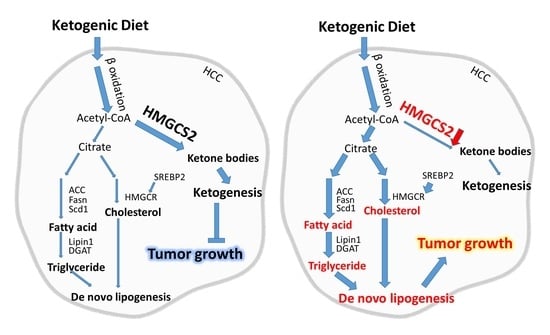
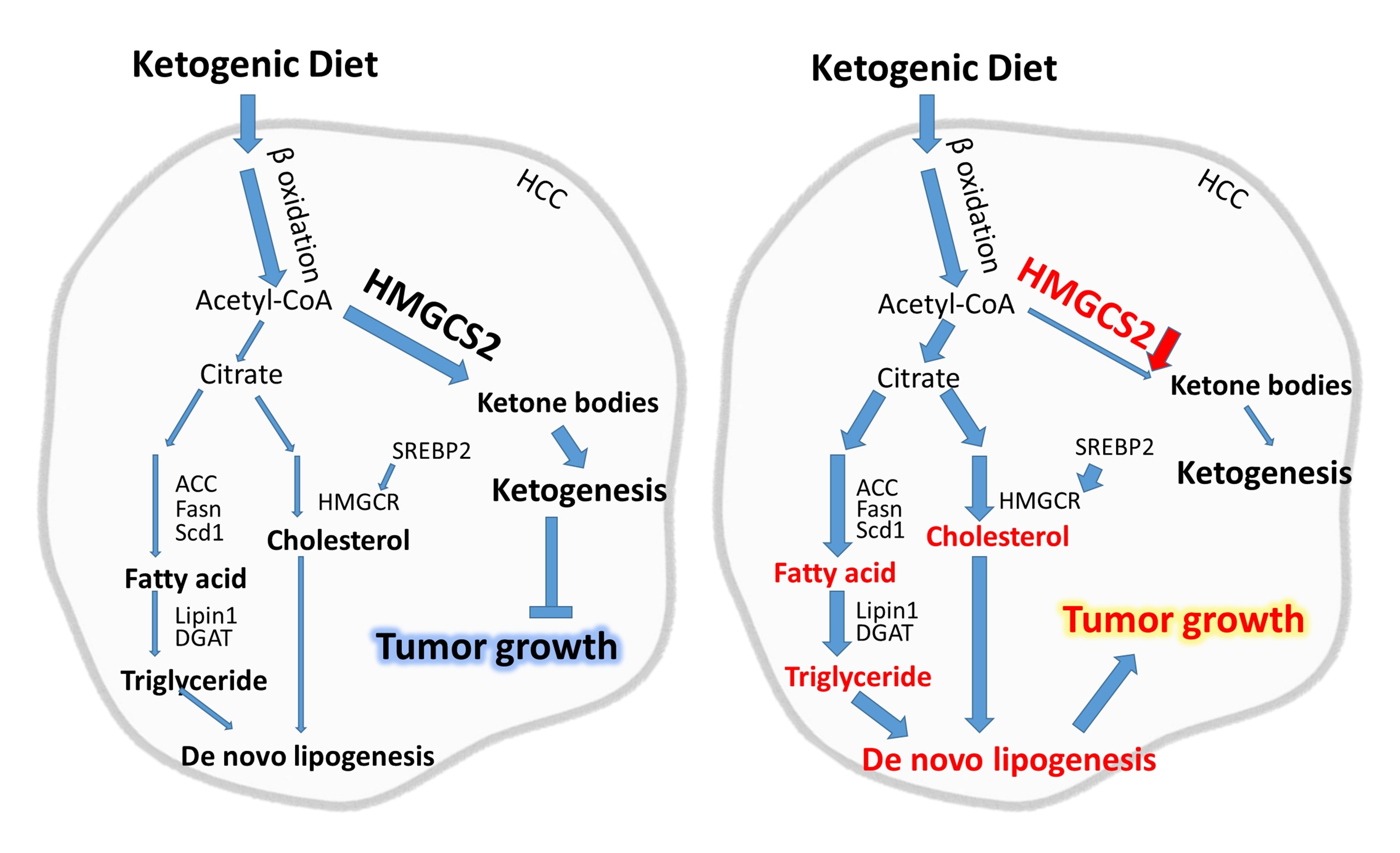{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
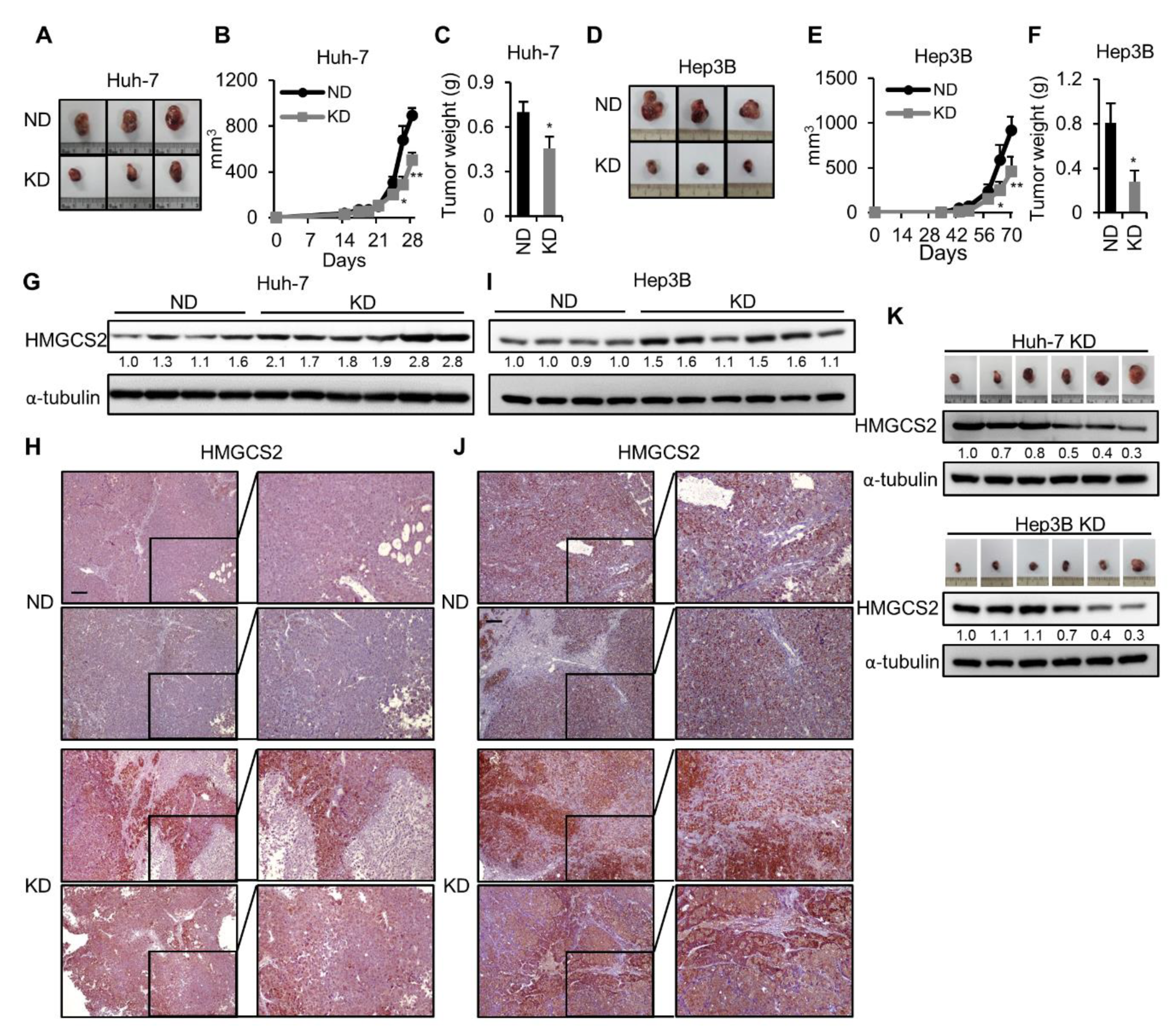
2.1. Ketogenic Diet Feeding Inhibits HCC Tumor Growth by Increasing the Protein Expression of HMGCS2
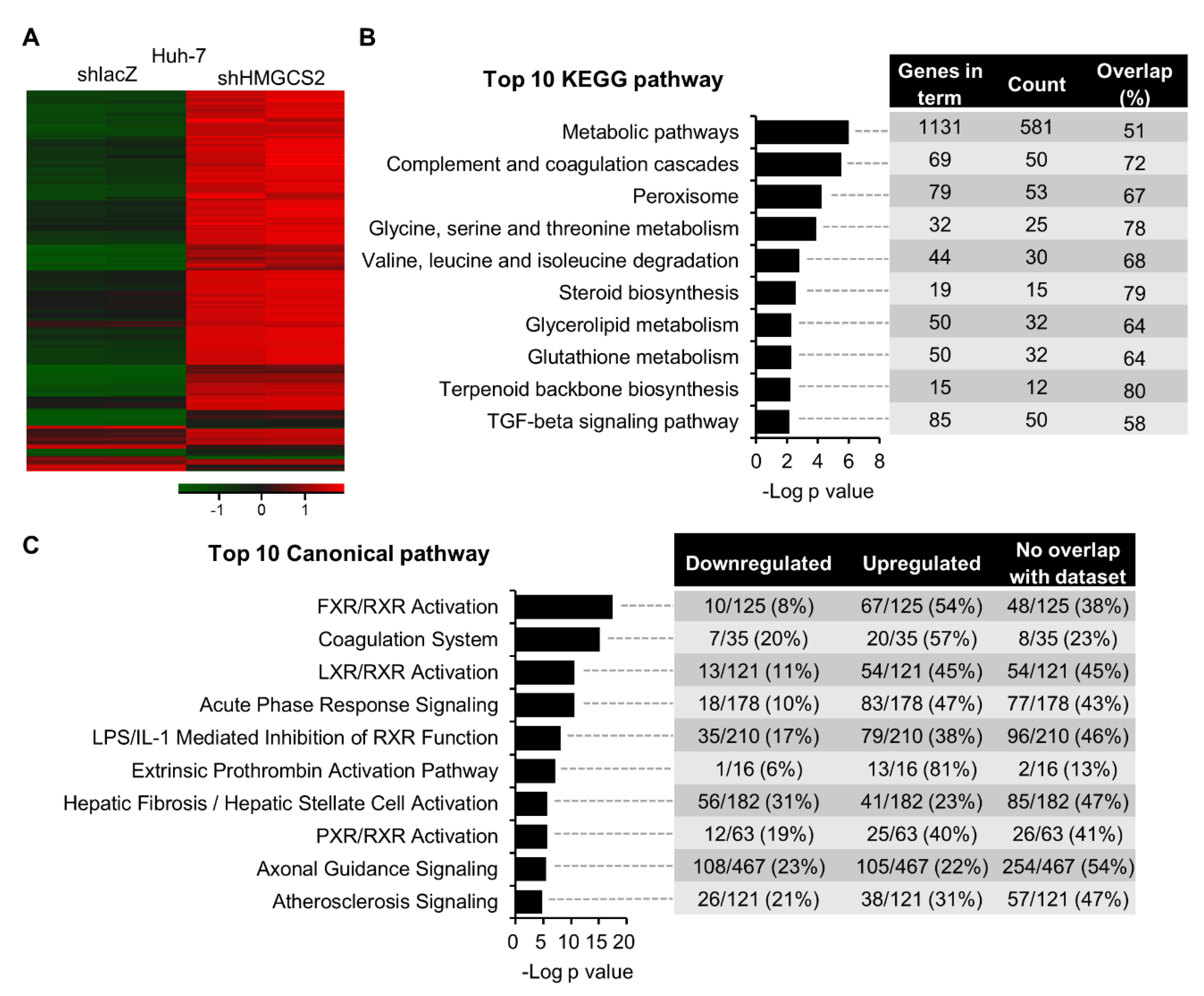
2.2. Gene Expression Profiling of HMGCS2 Knockdown HCC Cells
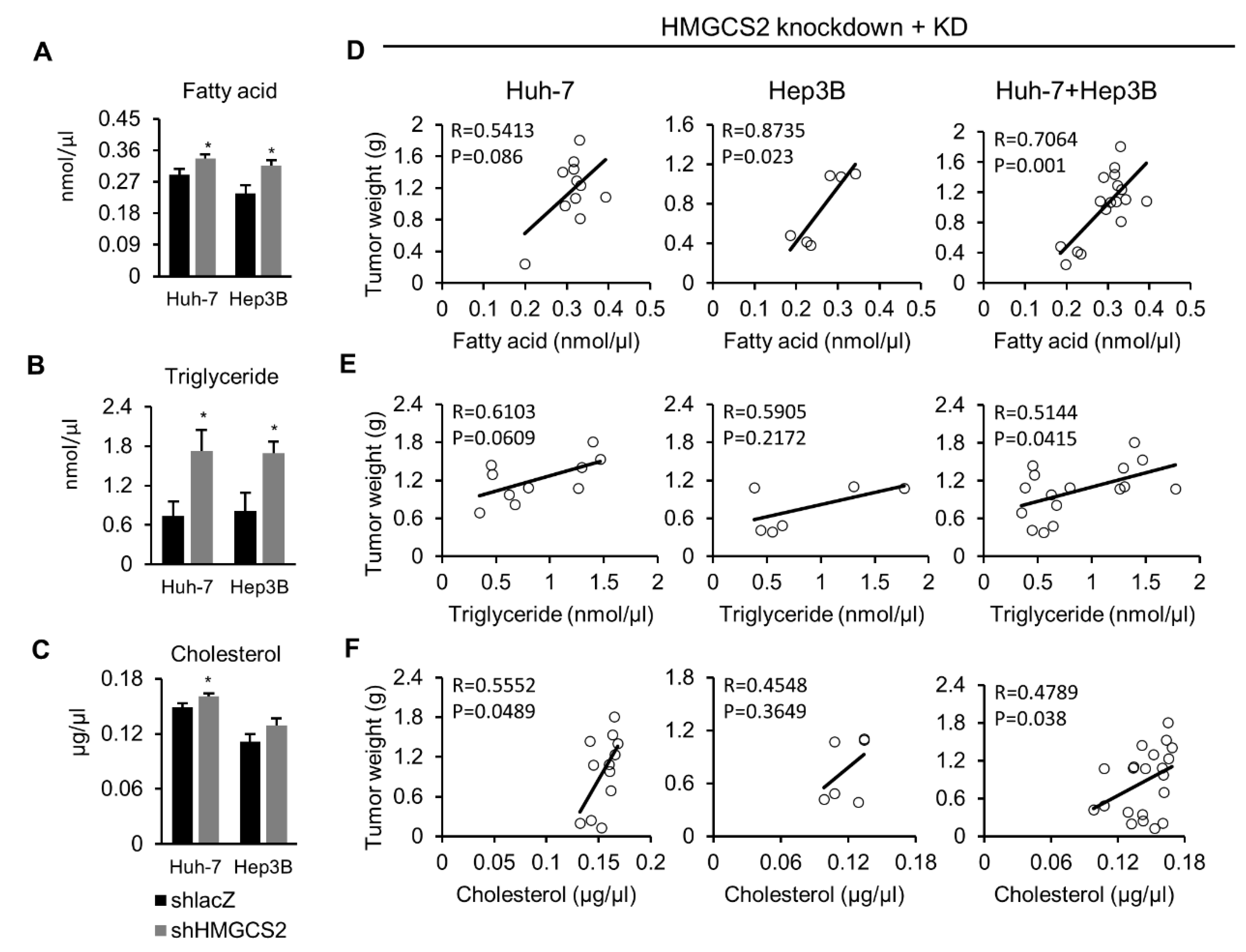
2.3. Knockdown of HMGCS2 Enhanced Fatty Acid, Triglyceride, and Cholesterol Synthesis in HCC Cells
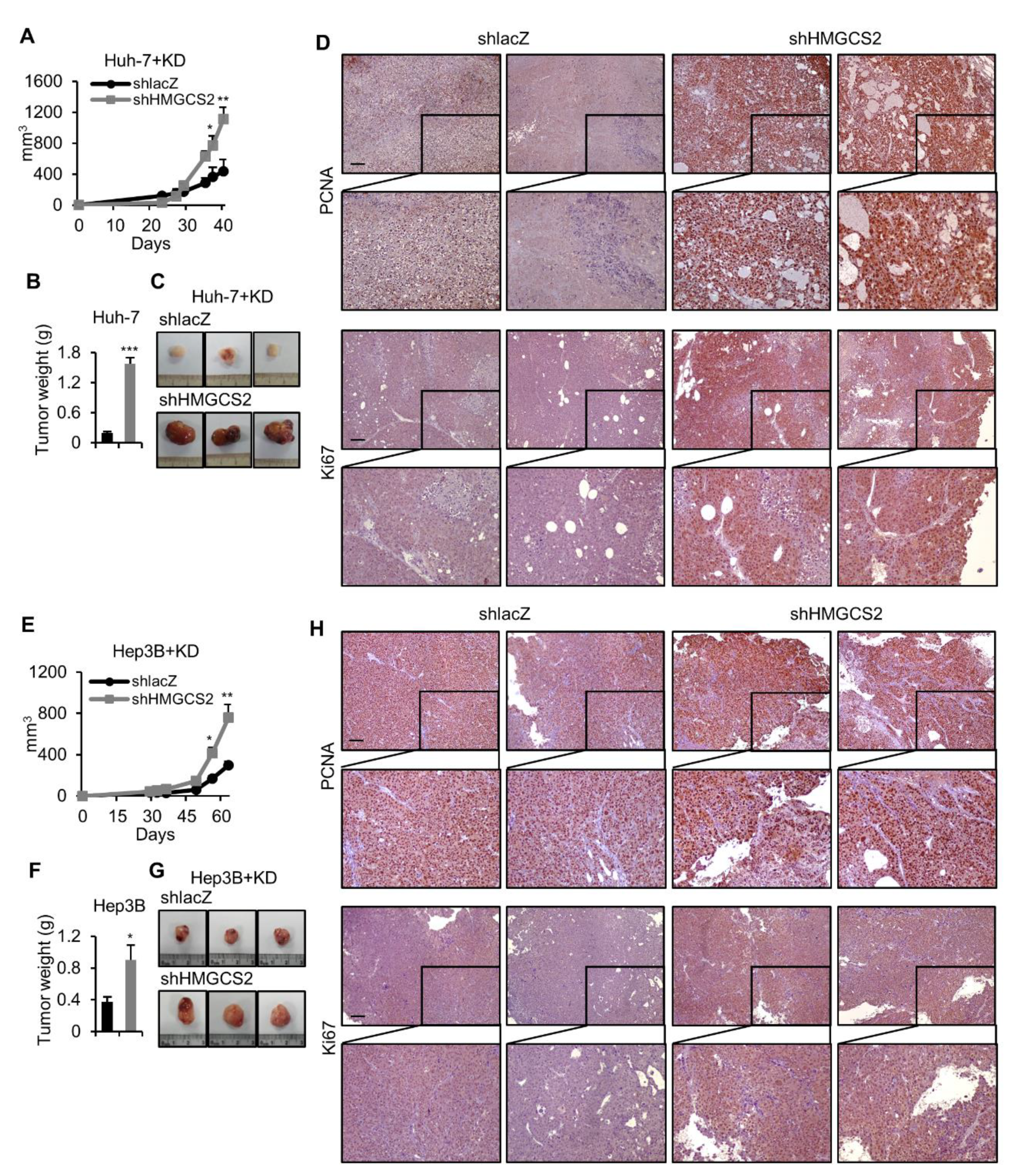
2.4. HMGCS2 Knockdown HCC Cells Possessed Increased Tumor Growth Ability under Ketogenic Diet Feeding
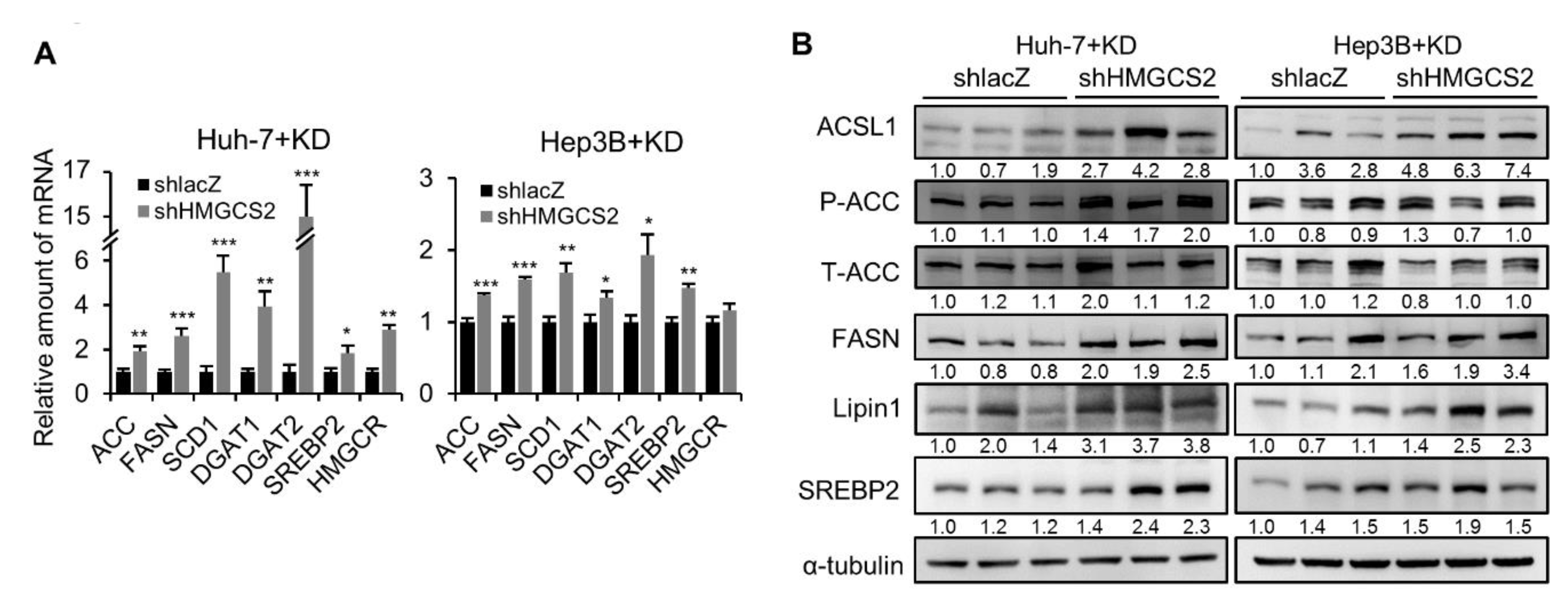
2.5. Tumors from HMGCS2 Knockdown HCC Cells Showed Increased Lipid Synthesis-related mRNA and Protein Markers under the Ketogenic Diet
2.6. Tumors from HMGCS2 Knockdown Cells Showed Increased Lipids Amount (Fatty Acids, Triglycerides, and Cholesterol) and That Was Correlated with the Tumor Weight
3. Discussion
4. Materials and Methods
4.1. Animal Study
4.2. Immunohistochemistry (IHC) Staining and Biochemical Blood Parameters
4.3. Cell Culture and Viral Infection
4.4. Gene Expression Profiling
4.5. Colorimetric Free Fatty Acid Assay
4.6. Colorimetric Triglyceride Assay
4.7. Colorimetric Total Cholesterol Assay
4.8. Colorimetric β-Hydroxybutyrate Assay
4.9. Western Blotting (WB)
4.10. RNA Extraction and Quantitative RT-PCR
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.M.; Howlader, N.; Henley, S.J.; Anderson, R.N.; Firth, A.U.; Ma, J.; et al. Annual report to the nation on the status of cancer, part i: National cancer statistics. Cancer 2018, 124, 2785–2800. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.A.; Priolli, D.G.; Tralhao, J.G.; Botelho, M.F. Hepatocellular carcinoma: Epidemiology, biology, diagnosis, and therapies. Rev. Assoc. Med. Bras. 2013, 59, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Amann, T.; Hellerbrand, C. Glut1 as a therapeutic target in hepatocellular carcinoma. Expert Opin. Ther. Tar. 2009, 13, 1411–1427. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef]
- Zarrinpar, A. Metabolic pathway inhibition in liver cancer. SLAS Technol. 2017, 22, 237–244. [Google Scholar] [CrossRef][Green Version]
- Wilder, R.M. The effects of ketonemia on the course of epilepsy. Mayo Clin. Proc. 1921, 2, 307–308. [Google Scholar]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Oliveira, C.L.P.; Mattingly, S.; Schirrmacher, R.; Sawyer, M.B.; Fine, E.J.; Prado, C.M. A nutritional perspective of ketogenic diet in cancer: A narrative review. J. Acad. Nutr. Diet. 2018, 118, 668–688. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J.; Brehm, N.; Sweeney, R.A. Ketogenic diets in medical oncology: A systematic review with focus on clinical outcomes. Med. Oncol. 2020, 37, 14. [Google Scholar] [CrossRef]
- Klement, R.J. The emerging role of ketogenic diets in cancer treatment. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Kaemmerer, U.; Illert, B.; Muehling, B.; Pfetzer, N.; Wittig, R.; Voelker, H.U.; Thiede, A.; Coy, J.F. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with omega-3 fatty acids and medium-chain triglycerides. BMC Cancer 2008, 8, 122. [Google Scholar] [CrossRef]
- Klement, R.J. Fasting, fats, and physics: Combining ketogenic and radiation therapy against cancer. Complement. Med. Res. 2018, 25, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J. The influence of ketogenic therapy on the 5 r’s of radiobiology. Int. J. Radiat. Biol. 2019, 95, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh-Gohari, S.; Feichtinger, R.G.; Vidali, S.; Locker, F.; Rutherford, T.; O’Donnel, M.; Stoger-Kleiber, A.; Mayr, J.A.; Sperl, W.; Kofler, B. A ketogenic diet supplemented with medium-chain triglycerides enhances the anti-tumor and anti-angiogenic efficacy of chemotherapy on neuroblastoma xenografts in a cd1-nu mouse model. Oncotarget 2017, 8, 64728–64744. [Google Scholar] [CrossRef]
- Erickson, N.; Boscheri, A.; Linke, B.; Huebner, J. Systematic review: Isocaloric ketogenic dietary regimes for cancer patients. Med. Oncol. 2017, 34, 72. [Google Scholar] [CrossRef]
- Kani, A.H.; Alavian, S.M.; Esmaillzadeh, A.; Adibi, P.; Azadbakht, L. Effects of a novel therapeutic diet on liver enzymes and coagulating factors in patients with non-alcoholic fatty liver disease: A parallel randomized trial. Nutrition 2014, 30, 814–821. [Google Scholar] [CrossRef]
- Schugar, R.C.; Crawford, P.A. Low-carbohydrate ketogenic diets, glucose homeostasis, and nonalcoholic fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 374–380. [Google Scholar] [CrossRef]
- Healy, M.E.; Chow, J.D.; Byrne, F.L.; Breen, D.S.; Leitinger, N.; Li, C.; Lackner, C.; Caldwell, S.H.; Hoehn, K.L. Dietary effects on liver tumor burden in mice treated with the hepatocellular carcinogen diethylnitrosamine. J. Hepatol. 2015, 62, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Healy, M.E.; Lahiri, S.; Hargett, S.R.; Chow, J.D.; Byrne, F.L.; Breen, D.S.; Kenwood, B.M.; Taddeo, E.P.; Lackner, C.; Caldwell, S.H.; et al. Dietary sugar intake increases liver tumor incidence in female mice. Sci. Rep. 2016, 6, 22292. [Google Scholar] [CrossRef]
- Byrne, F.L.; Hargett, S.R.; Lahiri, S.; Roy, R.J.; Berr, S.S.; Caldwell, S.H.; Hoehn, K.L. Serial mri imaging reveals minimal impact of ketogenic diet on established liver tumor growth. Cancers 2018, 10, 312. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of ketone body metabolism and the role of pparalpha. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, P.; Crawford, P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Shafqat, N.; Turnbull, A.; Zschocke, J.; Oppermann, U.; Yue, W.W. Crystal structures of human hmg-coa synthase isoforms provide insights into inherited ketogenesis disorders and inhibitor design. J. Mol. Biol. 2010, 398, 497–506. [Google Scholar] [CrossRef]
- Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; d’Avignon, D.A.; Graham, M.J.; Dietzen, D.J.; Brunt, E.M.; Patti, G.J.; Crawford, P.A. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef]
- Wang, Y.H.; Liu, C.L.; Chiu, W.C.; Twu, Y.C.; Liao, Y.J. Hmgcs2 mediates ketone production and regulates the proliferation and metastasis of hepatocellular carcinoma. Cancers 2019, 11, 1876. [Google Scholar] [CrossRef]
- Yu, L.X.; Ling, Y.; Wang, H.Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2018, 2, 1–10. [Google Scholar] [CrossRef]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Model. Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef]
- Pope, E.D., 3rd; Kimbrough, E.O.; Vemireddy, L.P.; Surapaneni, P.K.; Copland, J.A., 3rd; Mody, K. Aberrant lipid metabolism as a therapeutic target in liver cancer. Expert Opin. Ther. Targets 2019, 23, 473–483. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Wendel, A.A.; Lewin, T.M.; Coleman, R.A. Glycerol-3-phosphate acyltransferases: Rate limiting enzymes of triacylglycerol biosynthesis. Biochim. Biophys. Acta 2009, 1791, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J. Beneficial effects of ketogenic diets for cancer patients: A realist review with focus on evidence and confirmation. Med. Oncol. 2017, 34, 132. [Google Scholar] [CrossRef] [PubMed]
- De Feyter, H.M.; Behar, K.L.; Rao, J.U.; Madden-Hennessey, K.; Ip, K.L.; Hyder, F.; Drewes, L.R.; Geschwind, J.F.; de Graaf, R.A.; Rothman, D.L. A ketogenic diet increases transport and oxidation of ketone bodies in rg2 and 9l gliomas without affecting tumor growth. Neuro Oncol. 2016, 18, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.T.; Wehrli, S.; Dang, C.V.; Curran, T. The ketogenic diet does not affect growth of hedgehog pathway medulloblastoma in mice. PLoS ONE 2015, 10, e0133633. [Google Scholar] [CrossRef] [PubMed]
- Liskiewicz, A.D.; Kasprowska, D.; Wojakowska, A.; Polanski, K.; Lewin-Kowalik, J.; Kotulska, K.; Jedrzejowska-Szypulka, H. Long-term high fat ketogenic diet promotes renal tumor growth in a rat model of tuberous sclerosis. Sci. Rep. 2016, 6, 21807. [Google Scholar] [CrossRef] [PubMed]
- Sikder, K.; Shukla, S.K.; Patel, N.; Singh, H.; Rafiq, K. High fat diet upregulates fatty acid oxidation and ketogenesis via intervention of ppar-gamma. Cell Physiol. Biochem. 2018, 48, 1317–1331. [Google Scholar] [CrossRef]
- Meertens, L.M.; Miyata, K.S.; Cechetto, J.D.; Rachubinski, R.A.; Capone, J.P. A mitochondrial ketogenic enzyme regulates its gene expression by association with the nuclear hormone receptor pparalpha. EMBO J. 1998, 17, 6972–6978. [Google Scholar] [CrossRef]
- Kostiuk, M.A.; Keller, B.O.; Berthiaume, L.G. Palmitoylation of ketogenic enzyme hmgcs2 enhances its interaction with pparalpha and transcription at the hmgcs2 ppre. FASEB J. 2010, 24, 1914–1924. [Google Scholar] [CrossRef]
- Vila-Brau, A.; De Sousa-Coelho, A.L.; Mayordomo, C.; Haro, D.; Marrero, P.F. Human hmgcs2 regulates mitochondrial fatty acid oxidation and fgf21 expression in hepg2 cell line. J. Biol. Chem. 2011, 286, 20423–20430. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by akt-mtorc1-rps6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-H.; Suk, F.-M.; Liao, Y.-J. Loss of HMGCS2 Enhances Lipogenesis and Attenuates the Protective Effect of the Ketogenic Diet in Liver Cancer. Cancers 2020, 12, 1797. https://doi.org/10.3390/cancers12071797
Wang Y-H, Suk F-M, Liao Y-J. Loss of HMGCS2 Enhances Lipogenesis and Attenuates the Protective Effect of the Ketogenic Diet in Liver Cancer. Cancers. 2020; 12(7):1797. https://doi.org/10.3390/cancers12071797
Chicago/Turabian StyleWang, Yuan-Hsi, Fat-Moon Suk, and Yi-Jen Liao. 2020. "Loss of HMGCS2 Enhances Lipogenesis and Attenuates the Protective Effect of the Ketogenic Diet in Liver Cancer" Cancers 12, no. 7: 1797. https://doi.org/10.3390/cancers12071797
APA StyleWang, Y.-H., Suk, F.-M., & Liao, Y.-J. (2020). Loss of HMGCS2 Enhances Lipogenesis and Attenuates the Protective Effect of the Ketogenic Diet in Liver Cancer. Cancers, 12(7), 1797. https://doi.org/10.3390/cancers12071797