Merkel Cell Polyomavirus and Merkel Cell Carcinoma


Abstract
1. Introduction
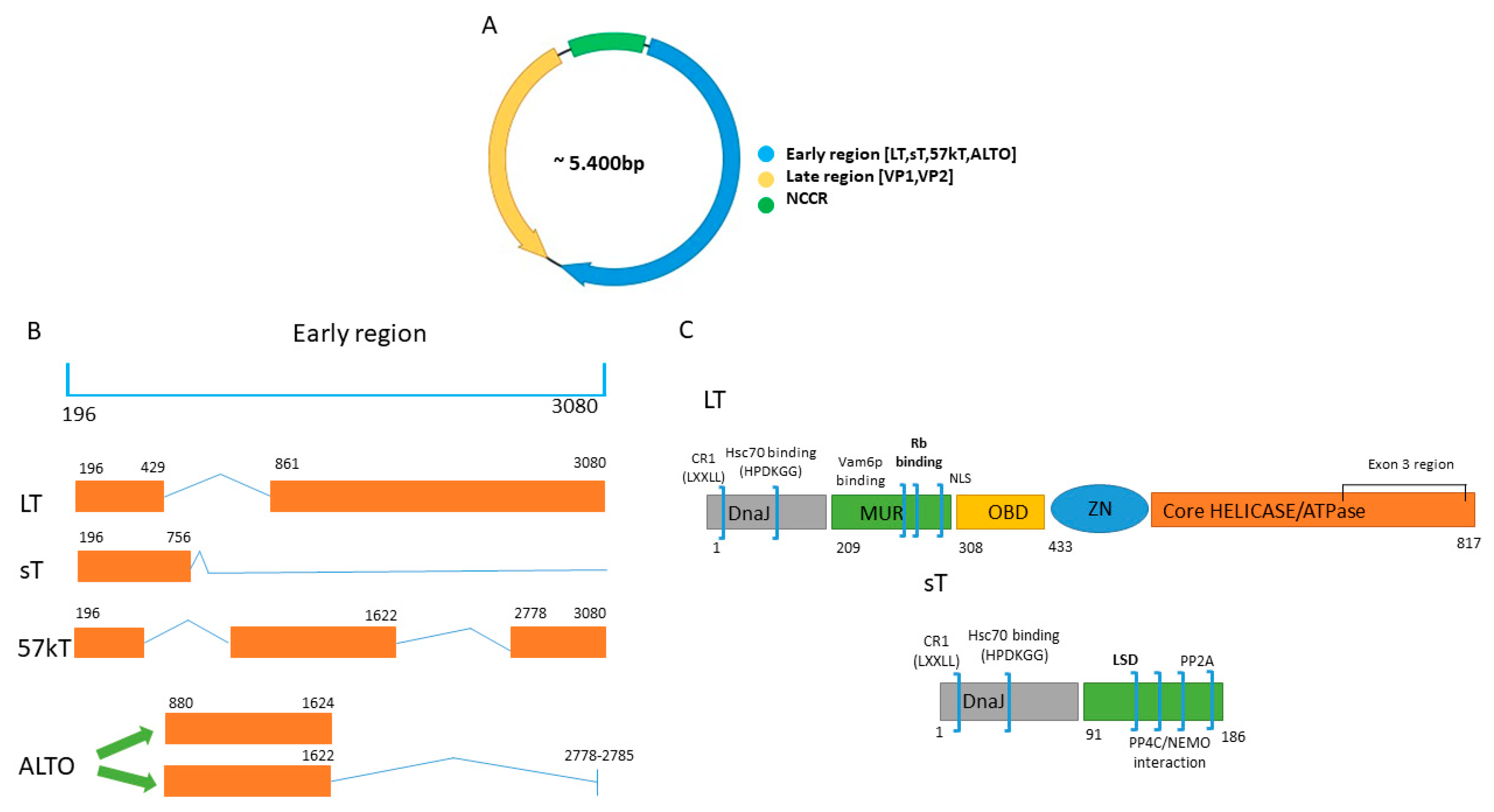
1.1. Genome MCPyV
1.2. Seroprevalence
1.3. Cell Tropism: Skin; Replication in Dermal Fibroblasts
2. MCPyV and MCC
3. Cells of Origin of MCC
4. The Oncogenic Mechanisms of MCPyV T Antigens
4.1. MCC Cell Growth Depends on LT But Not sT
4.2. Oncogenic Properties of LT
4.2.1. LT and p53
4.2.2. LT and Retinoblastoma (RB) Family
4.2.3. LT and HSC70
4.2.4. LT and VPS39 Subunit of HOPS Complex/Vam6p
4.2.5. LT and ATOH1
4.2.6. LT and Ubiquitin-Specific Protease 7 (Usp7)
4.2.7. LT and Other Interacting Proteins
4.3. The Role of 57kT and ALTO in VP-MCC
4.4. The Oncogenic Properties of sT
4.4.1. sT and Transgenic Mice
4.4.2. sT and Eukaryotic Translation Initiation Factor 4E Binding Protein (4E-BP1)
4.4.3. sT and E3 Ubiquitin Ligases
4.4.4. sT and N-myc Downstream Regulated Gene-1 (NDRG1)
4.4.5. sT and p53
4.4.6. sT and Protein Phosphatases
4.4.7. sT and Sheddases
4.4.8. sT and Metabolism
4.4.9. sT and Other Interaction Partners
4.5. Effect of MCPyV on Signaling Pathways in MCC
4.5.1. The Phosphatidyl-3-Kinase/AKT/Mammalian Target of the Rapamycin (PI3K/AKT/mTOR) Pathway
4.5.2. Protein Kinase C Pathway
4.5.3. Notch Pathway
4.5.4. Hedgehog Signaling Pathway
4.5.5. Apoptotic Pathway
5. Immune Evasion of VP-MCC
6. Specific Biomarker for VP-MCC
MicroRNA as VP-MCC Biomarkers
7. VP-MCC Specific Therapy
7.1. Vaccines
7.2. CRISPR/Cas9-Based Methodology
7.3. RNA Interference Based Treatment
7.4. Anti-Viral Drugs
8. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALTO | Alternate frame of the LT open reading frame |
| HLA-1 | Major histocompatibility complex class 1 |
| HPyV | Human polyomavirus |
| LSD | Large T antigen stabilization domain |
| LT | Large T antigen |
| MC | Merkel cell |
| MCC | Merkel cell carcinoma |
| MCPyV | Merkel cell polyomavirus |
| miR | MicroRNA |
| MUR | MCPyV T antigen unique region |
| NCCR | Non-coding control region |
| NLS | Nuclear localization signal |
| OBD | Origin binding domain |
| ORI | Origin of replication |
| ORR | Objective response rate |
| PD-1 | Programmed death 1 |
| PD-L1 | Programmed death ligand 1 |
| PP | Protein phosphatase |
| PyV | Polyomavirus |
| RB | Retinoblastoma protein |
| sT | Small t antigen |
| TLR | Toll-like receptor |
| tLT | Truncated LT |
| VN-MCC | Virus-negative Merkel cell carcinoma |
| VP-MCC | Virus-positive Merkel cell carcinoma |
References
- Calvignac-Spencer, S.; Feltkamp, M.C.; Daugherty, M.D.; Moens, U.; Ramqvist, T.; Johne, R.; Ehlers, B. A taxonomy update for the family Polyomaviridae. Arch. Virol. 2016, 161, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Gjoerup, O.; Chang, Y. Update on human polyomaviruses and cancer. Adv. Cancer Res. 2010, 106, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.J.; Daugherty, M.D.; Qi, X.; Bheda-Malge, A.; Wipf, G.C.; Robinson, K.; Roman, A.; Malik, H.S.; Galloway, D.A. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc. Natl. Acad. Sci. USA 2013, 110, 12744–12749. [Google Scholar] [CrossRef]
- Coursaget, P.; Samimi, M.; Nicol, J.T.; Gardair, C.; Touzé, A. Human Merkel cell polyomavirus: Virological background and clinical implications. APMIS 2013, 121, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Saenz Robles, M.T.; Pipas, J.M. Large T antigens of polyomaviruses: Amazing molecular machines. Annu. Rev. Microbiol. 2012, 66, 213–236. [Google Scholar] [CrossRef]
- Cotsiki, M.; Lock, R.L.; Cheng, Y.; Williams, G.L.; Zhao, J.; Perera, D.; Freire, R.; Entwistle, A.; Golemis, E.A.; Roberts, T.M.; et al. Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc. Natl. Acad. Sci. USA 2004, 101, 947–952. [Google Scholar] [CrossRef]
- Wendzicki, J.A.; Moore, P.S.; Chang, Y. Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol. 2015, 11, 38–43. [Google Scholar] [CrossRef]
- Nakamura, T.; Sato, Y.; Watanabe, D.; Ito, H.; Shimonohara, N.; Tsuji, T.; Nakajima, N.; Suzuki, Y.; Matsuo, K.; Nakagawa, H.; et al. Nuclear localization of Merkel cell polyomavirus large T antigen in Merkel cell carcinoma. Virology 2010, 398, 273–279. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015, 356, 197–203. [Google Scholar] [CrossRef]
- Van Ghelue, M.; Khan, M.T.; Ehlers, B.; Moens, U. Genome analysis of the new human polyomaviruses. Rev. Med. Virol. 2012, 22, 354–377. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Guastafierro, A.; Shuda, M.; Meinke, G.; Bohm, A.; Moore, P.S.; Chang, Y. The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J. Virol. 2009, 83, 12118–12128. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Moore, P.S. Merkel cell carcinoma: A virus-induced human cancer. Annu. Rev. Pathol. 2012, 7, 123–144. [Google Scholar] [CrossRef]
- Li, J.; Diaz, J.; Wang, X.; Tsang, S.H.; You, J. Phosphorylation of Merkel cell polyomavirus large tumor antigen at serine 816 by ATM kinase induces apoptosis in host cells. J. Biol. Chem. 2015, 290, 1874–1884. [Google Scholar] [CrossRef]
- Diaz, J.; Wang, X.; Tsang, S.H.; Jiao, J.; You, J. Phosphorylation of large T antigen regulates merkel cell polyomavirus replication. Cancers 2014, 6, 1464–1486. [Google Scholar] [CrossRef] [PubMed]
- Baez, C.F.; Brandão Varella, R.; Villani, S.; Delbue, S. Human Polyomaviruses: The battle of large and small tumor antigens. Virology 2017, 8. [Google Scholar] [CrossRef]
- Kwun, H.J.; Shuda, M.; Camacho, C.J.; Gamper, A.M.; Thant, M.; Chang, Y.; Moore, P.S. Restricted protein phosphatase 2A targeting by Merkel cell polyomavirus small T antigen. J. Virol. 2015, 89, 4191–4200. [Google Scholar] [CrossRef]
- Liu, X.; Hein, J.; Richardson, S.C.; Basse, P.H.; Toptan, T.; Moore, P.S.; Gjoerup, O.V.; Chang, Y. Merkel cell polyomavirus large T antigen disrupts lysosome clustering by translocating human Vam6p from the cytoplasm to the nucleus. J. Biol. Chem. 2011, 286, 17079–17090. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Buck, C.B. The Merkel cell polyomavirus minor capsid protein. PLoS Pathog. 2013, 9, e1003558. [Google Scholar] [CrossRef]
- Pastrana, D.V.; Tolstov, Y.L.; Becker, J.C.; Moore, P.S.; Chang, Y.; Buck, C.B. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog. 2009, 5, e1000578. [Google Scholar] [CrossRef]
- Moens, U.; Krumbholz, A.; Ehlers, B.; Zell, R.; Johne, R.; Calvignac-Spencer, S.; Lauber, C. Biology, evolution, and medical importance of polyomaviruses: An update. Infect. Genet. Evol. 2017, 54, 18–38. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Rinaldo, C.H.; Funk, G.A.; Egli, A.; Ramos, E.; Drachenberg, C.B.; Hirsch, H.H. Polyomavirus BK with rearranged noncoding control region emerge in vivo in renal transplant patients and increase viral replication and cytopathology. J. Exp. Med. 2008, 205, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Kardas, P.; Major, E.O.; Hirsch, H.H. Rearranged JC virus noncoding control regions found in progressive multifocal leukoencephalopathy patient samples increase virus early gene expression and replication rate. J. Virol. 2010, 84, 10448–10456. [Google Scholar] [CrossRef]
- Delbue, S.; Elia, F.; Carloni, C.; Tavazzi, E.; Marchioni, E.; Carluccio, S.; Signorini, L.; Novati, S.; Maserati, R.; Ferrante, P. JC virus load in cerebrospinal fluid and transcriptional control region rearrangements may predict the clinical course of progressive multifocal leukoencephalopathy. J. Cell Physiol. 2012, 227, 3511–3517. [Google Scholar] [CrossRef] [PubMed]
- Abdulsalam, I.; Rasheed, K.; Sveinbjørnsson, B.; Ehlers, B.; Moens, U. Promoter activity of Merkel cell Polyomavirus variants in human dermal fibroblasts and a Merkel cell carcinoma cell line. Virol. J. 2020, 17, 54. [Google Scholar] [CrossRef]
- Prezioso, C.; Obregon, F.; Ambroselli, D.; Petrolo, S.; Checconi, P.; Rodio, D.M.; Coppola, L.; Nardi, A.; Vito, C.; Sarmati, L.; et al. Merkel cell Polyomavirus (MCPyV) in the context of immunosuppression: Genetic analysis of noncoding control region (NCCR) variability among a HIV-1-positive population. Viruses 2020, 12, E507. [Google Scholar] [CrossRef]
- Kitchen, A.; Miyamoto, M.M.; Mulligan, C.J. Utility of DNA viruses for studying human host history: Case study of JC virus. Mol. Phylogenet. Evol. 2008, 46, 673–682. [Google Scholar] [CrossRef]
- Krumbholz, A.; Bininda-Emonds, O.R.; Wutzler, P.; Zell, R. Evolution of four BK virus subtypes. Infect. Genet. Evol. 2008, 8, 632–643. [Google Scholar] [CrossRef]
- Mes, T.H.; van Doornum, G.J.; Schutten, M. Population genetic tests suggest that the epidemiologies of JCV and BKV are strikingly different. Infect. Genet. Evol. 2010, 10, 397–403. [Google Scholar] [CrossRef]
- Torres, C.; Barrios, M.E.; Cammarata, R.V.; Victoria, M.; Fernandez-Cassi, X.; Bofill-Mas, S.; Colina, R.; Blanco Fernández, M.D.; Mbayed, V.A. Phylodynamics of Merkel-cell polyomavirus and human polyomavirus 6: A long-term history with humans. Mol. Phylogenet. Evol. 2018, 126, 210–220. [Google Scholar] [CrossRef]
- Madinda, N.F.; Ehlers, B.; Wertheim, J.O.; Akoua-Koffi, C.; Bergl, R.A.; Boesch, C.; Akonkwa, D.B.; Eckardt, W.; Fruth, B.; Gillespie, T.R.; et al. Assessing host-virus codivergence for close relatives of Merkel cell polyomavirus infecting african great apes. J. Virol. 2016, 90, 8531–8541. [Google Scholar] [CrossRef] [PubMed]
- Leendertz, F.H.; Scuda, N.; Cameron, K.N.; Kidega, T.; Zuberbühler, K.; Leendertz, S.A.; Couacy-Hymann, E.; Boesch, C.; Calvignac, S.; Ehlers, B. African great apes are naturally infected with polyomaviruses closely related to Merkel cell polyomavirus. J. Virol. 2011, 85, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Martel-Jantin, C.; Pedergnana, V.; Nicol, J.T.; Leblond, V.; Trégouët, D.A.; Tortevoye, P.; Plancoulaine, S.; Coursaget, P.; Touzé, A.; Abel, L.; et al. Merkel cell polyomavirus infection occurs during early childhood and is transmitted between siblings. J. Clin. Virol. 2013, 58, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Van der Meijden, E.; Bialasiewicz, S.; Rockett, R.J.; Tozer, S.J.; Sloots, T.P.; Feltkamp, M.C. Different serologic behavior of MCPyV, TSPyV, HPyV6, HPyV7 and HPyV9 polyomaviruses found on the skin. PLoS ONE 2013, 8, e81078. [Google Scholar] [CrossRef]
- Gaudette, L.A.; Illing, E.M.; Hill, G.B. Canadian Cancer Statistics 1991. Health Rep. 1991, 3, 107–135. [Google Scholar]
- Tolstov, Y.L.; Pastrana, D.V.; Feng, H.; Becker, J.C.; Jenkins, F.J.; Moschos, S.; Chang, Y.; Buck, C.B.; Moore, P.S. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int. J. Cancer 2009, 125, 1250–1256. [Google Scholar] [CrossRef]
- Carter, J.J.; Paulson, K.G.; Wipf, G.C.; Miranda, D.; Madeleine, M.M.; Johnson, L.G.; Lemos, B.D.; Lee, S.; Warcola, A.H.; Iyer, J.G.; et al. Association of Merkel cell polyomavirus-specific antibodies with Merkel cell carcinoma. J. Natl. Cancer Inst. 2009, 101, 1510–1522. [Google Scholar] [CrossRef]
- Pastrana, D.V.; Brennan, D.C.; Cuburu, N.; Storch, G.A.; Viscidi, R.P.; Randhawa, P.S.; Buck, C.B. Neutralization serotyping of BK polyomavirus infection in kidney transplant recipients. PLoS Pathog. 2012, 8, e1002650. [Google Scholar] [CrossRef]
- Nicol, J.T.; Robinot, R.; Carpentier, A.; Carandina, G.; Mazzoni, E.; Tognon, M.; Touze, A.; Coursaget, P. Age-specific seroprevalences of merkel cell polyomavirus, human polyomaviruses 6, 7, and 9, and trichodysplasia spinulosa-associated polyomavirus. Clin. Vaccine Immunol. 2013, 20, 363–368. [Google Scholar] [CrossRef]
- Sroller, V.; Hamšíková, E.; Ludvíková, V.; Vochozková, P.; Kojzarová, M.; Fraiberk, M.; Saláková, M.; Morávková, A.; Forstová, J.; Němečková, S. Seroprevalence rates of BKV, JCV, and MCPyV polyomaviruses in the general Czech Republic population. J. Med. Virol. 2014, 86, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, F.; He, Z.; Deng, Q.; Pan, Y.; Liu, Y.; Zhang, C.; Ning, T.; Guo, C.; Liang, Y.; et al. Seroprevalence of Merkel cell polyomavirus in the general rural population of Anyang, China. PLoS ONE 2014, 9, e106430. [Google Scholar] [CrossRef]
- Paulson, K.G.; Lewis, C.W.; Redman, M.W.; Simonson, W.T.; Lisberg, A.; Ritter, D.; Morishima, C.; Hutchinson, K.; Mudgistratova, L.; Blom, A.; et al. Viral oncoprotein antibodies as a marker for recurrence of Merkel cell carcinoma: A prospective validation study. Cancer 2017, 123, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yang, R.; Payne, A.S.; Schowalter, R.M.; Spurgeon, M.E.; Lambert, P.F.; Xu, X.; Buck, C.B.; You, J. Identifying the target cells and mechanisms of Merkel Cell Polyomavirus infection. Cell Host Microbe 2016, 19, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Hashida, Y.; Nakajima, K.; Nakajima, H.; Shiga, T.; Tanaka, M.; Murakami, M.; Matsuzaki, S.; Naganuma, S.; Kuroda, N.; Seki, Y.; et al. High load of Merkel cell polyomavirus DNA detected in the normal skin of Japanese patients with Merkel cell carcinoma. J. Clin. Virol. 2016, 82, 101–107. [Google Scholar] [CrossRef]
- Liu, W.; Krump, N.A.; Buck, C.B.; You, J. Merkel cell Polyomavirus infection and detection. J. Vis. Exp. 2019, 144. [Google Scholar] [CrossRef]
- Peretti, A.; Borgogna, C.; Rossi, D.; De Paoli, L.; Bawadekar, M.; Zavattaro, E.; Boldorini, R.; De Andrea, M.; Gaidano, G.; Gariglio, M. Analysis of human β-papillomavirus and Merkel cell polyomavirus infection in skin lesions and eyebrow hair bulbs from a cohort of patients with chronic lymphocytic leukaemia. Br. J. Dermatol. 2014, 171, 1525–1528. [Google Scholar] [CrossRef]
- Toker, C. Trabecular carcinoma of the skin. Arch. Dermatol. 1972, 105, 107–110. [Google Scholar] [CrossRef]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbe, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis Primers 2017, 3, 17077. [Google Scholar] [CrossRef]
- Paulson, K.G.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463. [Google Scholar] [CrossRef]
- Harms, P.W.; Collie, A.M.; Hovelson, D.H.; Cani, A.K.; Verhaegen, M.E.; Patel, R.M.; Fullen, D.R.; Omata, K.; Dlugosz, A.A.; Tomlins, S.A.; et al. Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod. Pathol. 2016, 29, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Pulitzer, M.P.; Amin, B.D.; Busam, K.J. Merkel cell carcinoma: Review. Adv. Anat. Pathol. 2009, 16, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Agelli, M.; Clegg, L.X. Epidemiology of primary Merkel cell carcinoma in the United States. J. Am. Acad. Dermatol. 2003, 49, 832–841. [Google Scholar] [CrossRef]
- Howard, R.A.; Dores, G.M.; Curtis, R.E.; Anderson, W.F.; Travis, L.B. Merkel cell carcinoma and multiple primary cancers. Cancer Epidemiol. Biomarkers Prev. 2006, 15, 1545–1549. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2016, 7, 3403–3415. [Google Scholar] [CrossRef] [PubMed]
- Starrett, G.J.; Marcelus, C.; Cantalupo, P.G.; Katz, J.P.; Cheng, J.; Akagi, K.; Thakuria, M.; Rabinowits, G.; Wang, L.C.; Symer, D.E.; et al. Merkel cell Polyomavirus exhibits dominant control of the tumor genome and transcriptome in virus-associated Merkel cell carcinoma. mBio 2017, 8, e02079. [Google Scholar] [CrossRef]
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Peñas, P.F.; Nghiem, P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: The AEIOU features. J. Am. Acad. Dermatol. 2008, 58, 375–381. [Google Scholar] [CrossRef]
- Engels, E.A.; Frisch, M.; Goedert, J.J.; Biggar, R.J.; Miller, R.W. Merkel cell carcinoma and HIV infection. Lancet 2002, 359, 4974–4998. [Google Scholar] [CrossRef]
- Clarke, C.A.; Robbins, H.A.; Tatalovich, Z.; Lynch, C.F.; Pawlish, K.S.; Finch, J.L.; Hernandez, B.Y.; Fraumeni, J.F.J.; Madeleine, M.M.; Engels, E.A. Risk of merkel cell carcinoma after solid organ transplantation. J. Natl. Cancer Inst. 2015, 107, dju382. [Google Scholar] [CrossRef]
- Paulson, K.G.; Iyer, J.G.; Blom, A.; Warton, E.M.; Sokil, M.; Yelistratova, L.; Schuman, L.; Nagase, K.; Bhatia, S.; Asgari, M.M.; et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J. Investig. Dermatol. 2013, 133, 642–646. [Google Scholar] [CrossRef]
- Csoboz, B.; Rasheed, K.; Sveinbjørnsson, B.; Moens, U. Merkel cell polyomavirus and non-Merkel cell carcinomas: Guilty or circumstantial evidence? APMIS 2020, 128, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Harms, P.W. Update on Merkel cell carcinoma. Clin. Lab. Med. 2017, 3, 17077. [Google Scholar] [CrossRef]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K. Brownell I: The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef]
- Schrama, D.; Sarosi, E.M.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; König, E.M.; Utikal, J.; Becker, J.C.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019, 145, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.; Brandner, J.; Fuchs, F.; Moll, I.; Grundhoff, A. Detection of Merkel cell polyomavirus (MCPyV) in Merkel cell carcinoma cell lines: Cell morphology and growth phenotype do not reflect presence of the virus. Int. J. Cancer 2010, 126, 2133–2142. [Google Scholar] [CrossRef]
- Shuda, M.; Arora, R.; Kwun, H.J.; Feng, H.; Sarid, R.; Fernández-Figueras, M.T.; Tolstov, Y.; Gjoerup, O.; Mansukhani, M.M.; Swerdlow, S.H.; et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int. J. Cancer 2009, 125, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Cheng, J.; Wardzala, J.; DoRosario, A.; Scanlon, J.J.; Laga, A.C.; Martinez-Fernandez, A.; Barletta, J.A.; Bellizzi, A.M.; Sadasivam, S.; et al. Improved detection suggests all Merkel cell carcinomas harbor Merkel polyomavirus. J. Clin. Investig. 2012, 122, 4645–4653. [Google Scholar] [CrossRef]
- Velásquez, C.; Amako, Y.; Harold, A.; Toptan, T.; Chang, Y.; Shuda, M. Characterization of a Merkel Cell Polyomavirus-positive Merkel Cell carcinoma cell line CVG-1. Front. Microbiol. 2018, 9, 713. [Google Scholar] [CrossRef]
- Gould, V.E.; Moll, R.; Moll, I.; Lee, I.; Franke, W.W. Neuroendocrine (Merkel) cells of the skin: Hyperplasias, dysplasias, and neoplasms. Lab. Investig. 1985, 52, 334–353. [Google Scholar]
- Moll, I.; Kuhn, C.; Moll, R. Cytokeratin 20 is a general marker of cutaneous Merkel cells while certain neuronal proteins are absent. J. Investig. Dermatol. 1995, 104, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Ortonne, J.P.; Petchot-Bacque, J.P.; Verrando, P.; Pisani, A.; Pautrat, G.; Bernerd, F. Normal Merkel cells express a synaptophysin-like immunoreactivity. Dermatologica 1988, 177, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Nabeshima, K.; Akiyama, Y.; Maeda, S.; Nishida, T.; Nakayama, F.; Amano, M.; Ogata, K.; Setoyama, M. CD56: A useful marker for diagnosing Merkel cell carcinoma. J. Dermatol. Sci. 2003, 31, 219–224. [Google Scholar] [CrossRef]
- Foschini, M.P.; Eusebi, V. Divergent differentiation in endocrine and nonendocrine tumors of the skin. Semin. Diagn. Pathol. 2000, 17, 162–168. [Google Scholar]
- Spurgeon, M.E.; Lambert, P.F. Merkel cell polyomavirus: A newly discovered human virus with oncogenic potential. Virology 2013, 435, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.C.; Jahchan, N.S.; Sage, J.; Choi, J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene 2018, 37, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Cordoba, C.; Cheung, K.; Konya, J. Merkel cell carcinoma in situ: New insights into the cells of origin. Australasian J. Dermatol. 2019, 60, e311–e313. [Google Scholar] [CrossRef] [PubMed]
- Kervarrec, T.; Aljundi, M.; Appenzeller, S.; Samimi, M.; Maubec, E.; Cribier, B.; Deschamps, L.; Sarma, B.; Sarosi, E.M.; Berthon, P.; et al. Polyomavirus-positive Merkel cell carcinoma derived from a Trichoblastoma suggests an epithelial origin of this Merkel cell carcinoma. J. Investig. Dermatol. 2020, 140, 976–985. [Google Scholar] [CrossRef]
- Munde, P.B.; Khandekar, S.P.; Dive, A.M.; Sharma, A. Pathophysiology of merkel cell. J. Oral Maxillofac. Pathol. 2013, 17, 408–412. [Google Scholar] [CrossRef]
- Harms, P.W.; Patel, R.M.; Verhaegen, M.E.; Giordano, T.J.; Nash, K.T.; Johnson, C.N.; Daignault, S.; Thomas, D.G.; Gudjonsson, J.E.; Elder, J.T.; et al. Distinct gene expression profiles of viral- and nonviral-associated merkel cell carcinoma revealed by transcriptome analysis. J. Investig. Dermatol. 2013, 133, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Tilling, T.; Moll, I. Which are the cells of origin in merkel cell carcinoma? J. Skin Cancer 2012, 2012, 680410. [Google Scholar] [CrossRef] [PubMed]
- Moll, I.; Roessler, M.; Brandner, J.M.; Eispert, A.C.; Houdek, P.; Moll, R. Human Merkel cells--aspects of cell biology, distribution and functions. Eur. J. Cell Biol. 2005, 84, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Moll, I.; Zieger, W.; Schmelz, M. Proliferative Merkel cells were not detected in human skin. Arch. Dermatol. Res. 1996, 288, 1841–1887. [Google Scholar] [CrossRef] [PubMed]
- Sauer, C.M.; Haugg, A.M.; Chteinberg, E.; Rennspiess, D.; Winnepenninckx, V.; Speel, E.J.; Becker, J.C.; Kurz, A.K.; Zur Hausen, A. Reviewing the current evidence supporting early B-cells as the cellular origin of Merkel cell carcinoma. Crit. Rev. Oncol. Hematol. 2017, 116, 99–105. [Google Scholar] [CrossRef]
- Deichmann, M.; Kurzen, H.; Egner, U.; Altevogt, P.; Hartschuh, W. Adhesion molecules CD171 (L1CAM) and CD24 are expressed by primary neuroendocrine carcinomas of the skin (Merkel cell carcinomas). J. Cutan. Pathol. 2003, 30, 363–368. [Google Scholar] [CrossRef]
- Kervarrec, T.; Samimi, M.; Guyétant, S.; Sarma, B.; Chéret, J.; Blanchard, E.; Berthon, P.; Schrama, D.; Houben, R.; Touzé, A. Histogenesis of Merkel cell carcinoma: A Comprehensive review. Front. Oncol. 2019, 9, 451. [Google Scholar] [CrossRef]
- Scott, M.P.; Helm, K.F. Cytokeratin 20: A marker for diagnosing Merkel cell carcinoma. Am. J. Dermatopathol. 1999, 21, 16–20. [Google Scholar] [CrossRef]
- Shuda, M.; Guastafierro, A.; Geng, X.; Shuda, Y.; Ostrowski, S.M.; Lukianov, S.; Jenkins, F.J.; Honda, K.; Maricich, S.M.; Moore, P.S.; et al. Merkel Cell Polyomavirus Small T Antigen induces cancer and Embryonic Merkel Cell proliferation in a transgenic mouse model. PLoS ONE 2015, 10, e0142329. [Google Scholar] [CrossRef]
- Hwang, J.H.; Alanen, K.; Dabbs, K.D.; Danyluk, J.; Silverman, S. Merkel cell carcinoma with squamous and sarcomatous differentiation. J. Cutan. Pathol. 2008, 35, 955–959. [Google Scholar] [CrossRef]
- Abbas, O.; Bhawan, J. Expression of stem cell markers nestin and cytokeratin 15 and 19 in cutaneous malignancies. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Lemasson, G.; Coquart, N.; Lebonvallet, N.; Boulais, N.; Galibert, M.D.; Marcorelles, P.; Misery, L. Presence of putative stem cells in Merkel cell carcinomas. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 789–795. [Google Scholar] [CrossRef]
- Laga, A.C.; Lai, C.Y.; Zhan, Q.; Huang, S.J.; Velazquez, E.F.; Yang, Q.; Hsu, M.Y.; Murphy, G.F. Expression of the embryonic stem cell transcription factor SOX2 in human skin: Relevance to melanocyte and merkel cell biology. Am. J. Pathol. 2010, 176, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Fradette, J.; Godbout, M.J.; Michel, M.; Germain, L. Localization of Merkel cells at hairless and hairy human skin sites using keratin 18. Biochem. Cell Biol. 1995, 73, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Murakami, I.; Takata, K.; Matsushita, M.; Nonaka, D.; Iwasaki, T.; Kuwamoto, S.; Kato, M.; Mohri, T.; Nagata, K.; Kitamura, Y.; et al. Immunoglobulin expressions are only associated with MCPyV-positive Merkel cell carcinomas but not with MCPyV-negative ones: Comparison of prognosis. Am. J. Surg. Pathol. 2014, 38, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, A.; Rennspiess, D.; Winnepenninckx, V.; Speel, E.J.; Kurz, A.K. Early B-cell differentiation in Merkel cell carcinomas: Clues to cellular ancestry. Cancer Res. 2013, 73, 4982–4987. [Google Scholar] [CrossRef]
- Shiver, M.B.; Mahmoud, F.; Gao, L. Response to Idelalisib in a patient with stage IV Merkel-cell carcinoma. N. Engl. J. Med. 2015, 373, 1580–1582. [Google Scholar] [CrossRef]
- Angermeyer, S.; Hesbacher, S.; Becker, J.C.; Schrama, D.; Houben, R. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J. Investig. Dermatol. 2013, 133, 2059–2064. [Google Scholar] [CrossRef]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef]
- Nwogu, N.; Ortiz, L.E.; Kwun, H.J. Surface charge of Merkel cell polyomavirus small T antigen determines cell transformation through allosteric FBW7 WD40 domain targeting. Oncogenesis 2020, 9, 53. [Google Scholar] [CrossRef]
- Woo, S.H.; Stumpfova, M.; Jensen, U.B.; Lumpkin, E.A.; Owens, D.M. Identification of epidermal progenitors for the Merkel cell lineage. Development 2010, 137, 3965–3971. [Google Scholar] [CrossRef] [PubMed]
- Larouche, D.; Tong, X.; Fradette, J.; Coulombe, P.A.; Germain, L. Vibrissa hair bulge houses two populations of skin epithelial stem cells distinct by their keratin profile. FASEB J. 2008, 22, 1404–1415. [Google Scholar] [CrossRef]
- Brownell, I.; Guevara, E.; Bai, C.B.; Loomis, C.A.; Joyner, A.L. Nerve-derived sonic hedgehog defines a niche for hair follicle stem cells capable of becoming epidermal stem cells. Cell Stem Cell 2011, 8, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.B.; Cohen, I.; Kumar, V.; Xu, Z.; Bar, C.; Dauber-Decker, K.L.; Tsai, P.C.; Marangoni, P.; Klein, O.D.; Hsu, Y.C.; et al. FGF signalling controls the specification of hair placode-derived SOX9 positive progenitors to Merkel cells. Nat. Commun. 2018, 9, 2333. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef]
- Houben, R.; Adam, C.; Baeurle, A.; Hesbacher, S.; Grimm, J.; Angermeyer, S.; Henzel, K.; Hauser, S.; Elling, R.; Bröcker, E.B.; et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int. J. Cancer 2012, 130, 847–856. [Google Scholar] [CrossRef]
- Houben, R.; Angermeyer, S.; Haferkamp, S.; Aue, A.; Goebeler, M.; Schrama, D.; Hesbacher, S. Characterization of functional domains in the Merkel cell polyoma virus Large T antigen. Int. J. Cancer 2015, 136, E290–E300. [Google Scholar] [CrossRef]
- Schrama, D.; Hesbacher, S.; Angermeyer, S.; Schlosser, A.; Haferkamp, S.; Aue, A.; Adam, C.; Weber, A.; Schmidt, M.; Houben, R. Serine 220 phosphorylation of the Merkel cell polyomavirus large T antigen crucially supports growth of Merkel cell carcinoma cells. Int. J. Cancer 2016, 138, 1153–1162. [Google Scholar] [CrossRef]
- Houben, R.; Grimm, J.; Willmes, C.; Weinkam, R.; Becker, J.C.; Schrama, D. Merkel cell carcinoma and Merkel cell polyomavirus: Evidence for hit-and-run oncogenesis. J. Investig. Dermatol. 2012, 132, 254–256. [Google Scholar] [CrossRef]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef]
- Temblador, A.; Topalis, D.; Andrei, G.; Snoeck, R. CRISPR/Cas9 Editing of the Polyomavirus Tumor antigens inhibits merkel cell carcinoma growth in vitro. Cancers 2019, 11, 1260. [Google Scholar] [CrossRef]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Macdonald, A. Effect of the large and small T-Antigens of human Polyomaviruses on signaling pathways. Int. J. Mol. Sci. 2019, 20, 3914. [Google Scholar] [CrossRef] [PubMed]
- Borchert, S.; Czech-Sioli, M.; Neumann, F.; Schmidt, C.; Wimmer, P.; Dobner, T.; Grundhoff, A.; Fischer, N. High-affinity Rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened Merkel cell polyomavirus large T antigens. J. Virol. 2014, 88, 3144–3160. [Google Scholar] [CrossRef]
- Martinez-Zapien, D.; Ruiz, F.X.; Poirson, J.; Mitschler, A.; Ramirez, J.; Forster, A.; Cousido-Siah, A.; Masson, M.; Vande Pol, S.; Podjarny, A.; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541–545. [Google Scholar] [CrossRef]
- Park, D.E.; Cheng, J.; Berrios, C.; Montero, J.; Cortés-Cros, M.; Ferretti, S.; Arora, R.; Tillgren, M.L.; Gokhale, P.C.; DeCaprio, J.A. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 2019, 116, 1027–1032. [Google Scholar] [CrossRef]
- Sherr, C.J. Divorcing ARF and p53: An unsettled case. Nat. Rev. Cancer 2006, 6, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Cantalupo, P.; Pipas, J.M. The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol. Cell Biol. 2000, 20, 6233–6243. [Google Scholar] [CrossRef]
- Moens, U.; Van Ghelue, M.; Johannessen, M. Oncogenic potentials of the human polyomavirus regulatory proteins. Cell Mol. Life Sci. 2007, 64, 1656–1678. [Google Scholar] [CrossRef]
- Martel-Jantin, C.; Filippone, C.; Cassar, O.; Peter, M.; Tomasic, G.; Vielh, P.; Brière, J.; Petrella, T.; Aubriot-Lorton, M.H.; Mortier, L.; et al. Genetic variability and integration of Merkel cell polyomavirus in Merkel cell carcinoma. Virology 2012, 426, 134–142. [Google Scholar] [CrossRef]
- Matsushita, M.; Iwasaki, T.; Kuwamoto, S.; Kato, M.; Nagata, K.; Murakami, I.; Kitamura, Y.; Hayashi, K. Merkel cell polyomavirus (MCPyV) strains in Japanese merkel cell carcinomas (MCC) are distinct from Caucasian type MCPyVs: Genetic variability and phylogeny of MCPyV genomes obtained from Japanese MCPyV-infected MCCs. Virus Genes 2014, 48, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Perdigoto, C.N.; Bardot, E.S.; Valdes, V.J.; Santoriello, F.J.; Ezhkova, E. Embryonic maturation of epidermal Merkel cells is controlled by a redundant transcription factor network. Development 2014, 141, 4690–4696. [Google Scholar] [CrossRef] [PubMed]
- Harold, A.; Amako, Y.; Hachisuka, J.; Bai, Y.; Li, M.Y.; Kubat, L.; Gravemeyer, J.; Franks, J.; Gibbs, J.R.; Park, H.J.; et al. Conversion of Sox2-dependent Merkel cell carcinoma to a differentiated neuron-like phenotype by T antigen inhibition. Proc. Natl. Acad. Sci. USA 2019, 116, 20104–20114. [Google Scholar] [CrossRef] [PubMed]
- Aguado-Llera, D.; Goormaghtigh, E.; de Geest, N.; Quan, X.J.; Prieto, A.; Hassan, B.A.; Gómez, J.; Neira, J.L. The basic helix-loop-helix region of human neurogenin 1 is a monomeric natively unfolded protein which forms a "fuzzy" complex upon DNA binding. Biochemistry 2010, 49, 1577–1589. [Google Scholar] [CrossRef]
- Fan, K.; Gravemeyer, J.; Ritter, C.; Rasheed, K.; Gambichler, T.; Moens, U.; Shuda, M.; Schrama, D.; Becker, J.C. MCPyV large T antigen-induced Atonal Homolog 1 Is a lineage-dependency Oncogene in Merkel cell carcinoma. J. Investig. Dermatol. 2020, 140, 56–65. [Google Scholar] [CrossRef]
- Czech-Sioli, M.; Siebels, S.; Radau, S.; Zahedi, R.P.; Schmidt, C.; Dobner, T.; Grundhoff, A.; Fischer, N. The Ubiquitin-specific Protease Usp7, a novel Merkel cell Polyomavirus large T-antigen interaction partner, modulates viral DNA replication. J. Virol. 2020, 94, e01638-19. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Chakraborty, D.; Basu, M.; Ghosh, M.K. Emerging insights into HAUSP (USP7) in physiology, cancer and other diseases. Signal. Transduct. Target. Ther. 2018, 3, 17. [Google Scholar] [CrossRef]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M.; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Schowalter, R.M.; Jiao, J.; Buck, C.B.; You, J. Bromodomain protein Brd4 plays a key role in Merkel cell polyomavirus DNA replication. PLoS Pathog. 2012, 8, e1003021. [Google Scholar] [CrossRef]
- Arora, R.; Vats, A.; Chimankar, V. MCV Truncated Large T antigen interacts with BRD4 in tumors. Matters 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Velásquez, C.; Cheng, E.; Cordek, D.G.; Kwun, H.J.; Chang, Y.; Moore, P.S. CDK1 substitutes for mTOR kinase to activate mitotic cap-dependent protein translation. Proc. Natl. Acad. Sci. USA 2015, 112, 5875–5882. [Google Scholar] [CrossRef]
- Velásquez, C.; Cheng, E.; Shuda, M.; Lee-Oesterreich, P.J.; Pogge von Strandmann, L.; Gritsenko, M.A.; Jacobs, J.M.; Moore, P.S.; Chang, Y. Mitotic protein kinase CDK1 phosphorylation of mRNA translation regulator 4E-BP1 Ser83 may contribute to cell transformation. Proc. Natl. Acad. Sci. USA 2016, 113, 8466–8471. [Google Scholar] [CrossRef]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Vozheiko, T.D.; Weick, J.W.; Wilbert, D.M.; Saunders, T.L.; Ermilov, A.N.; Bichakjian, C.K.; Johnson, T.M.; et al. Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J. Investig. Dermatol. 2015, 135, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Wendzicki, J.A.; Shuda, Y.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen induces genome instability by E3 ubiquitin ligase targeting. Oncogene 2017, 36, 6784–6792. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, D.A.; Abdul-Sada, H.; Knight, L.M.; Jackson, B.R.; Richards, K.; Prescott, E.L.; Peach, A.H.; Blair, G.E.; Macdonald, A.; Whitehouse, A. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J. Virol. 2013, 87, 13853–13867. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Sada, H.; Müller, M.; Mehta, R.; Toth, R.; Arthur, J.S.C.; Whitehouse, A.; Macdonald, A. The PP4R1 sub-unit of protein phosphatase PP4 is essential for inhibition of NF-κB by merkel polyomavirus small tumour antigen. Oncotarget 2017, 8, 25418–25432. [Google Scholar] [CrossRef]
- Cheng, J.; Park, D.E.; Berrios, C.; White, E.A.; Arora, R.; Yoon, R.; Branigan, T.; Xiao, T.; Westerling, T.; Federation, A.; et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 2017, 13, e1006668. [Google Scholar] [CrossRef]
- Knight, L.M.; Stakaityte, G.; Wood, J.J.; Abdul-Sada, H.; Griffiths, D.A.; Howell, G.J.; Wheat, R.; Blair, G.E.; Steven, N.M.; Macdonald, A.; et al. Merkel cell polyomavirus small T antigen mediates microtubule destabilization to promote cell motility and migration. J. Virol. 2015, 89, 35–47. [Google Scholar] [CrossRef]
- Stakaitytė, G.; Nwogu, N.; Dobson, S.J.; Knight, L.M.; Wasson, C.W.; Salguero, F.J.; Blackbourn, D.J.; Blair, G.E.; Mankouri, J.; Macdonald, A.; et al. Merkel cell Polyomavirus small T antigen drives cell motility via Rho-GTPase-induced Filopodium formation. J. Virol. 2018, 92, e00940-17. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Janji, B.; Viry, E.; Moussay, E.; Paggetti, J.; Arakelian, T.; Mgrditchian, T.; Messai, Y.; Noman, M.Z.; van Moer, K.; Hasmim, M.; et al. The multifaceted role of autophagy in tumor evasion from immune surveillance. Oncotarget 2016, 7, 17591–17607. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The role of autophagy in cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Xie, H.; Shi, H.; Gao, J.; Juhlin, C.C.; Björnhagen, V.; Höög, A.; Lee, L.; Larsson, C.; Lui, W.O. Merkel cell polyomavirus oncoproteins induce microRNAs that suppress multiple autophagy genes. Int. J. Cancer 2020, 146, 1652–1666. [Google Scholar] [CrossRef]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Eberl, M.; Wilbert, D.M.; Meireles, J.; Bichakjian, C.K.; Saunders, T.L.; Wong, S.Y.; Dlugosz, A.A. Merkel cell Polyomavirus small T antigen initiates Merkel cell carcinoma-like tumor development in mice. Cancer Res. 2017, 77, 3151–3157. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef]
- Bjornsti, M.A.; Houghton, P.J. Lost in translation: Dysregulation of cap-dependent translation and cancer. Cancer Cell 2004, 5, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Chang, Y.; Moore, P.S. Protein-mediated viral latency is a novel mechanism for Merkel cell polyomavirus persistence. Proc. Natl. Acad. Sci. USA 2017, 114, E4040–E4047. [Google Scholar] [CrossRef]
- Dye, K.N.; Welcker, M.; Clurman, B.E.; Roman, A.; Galloway, D.A. Merkel cell polyomavirus Tumor antigens expressed in Merkel cell carcinoma function independently of the ubiquitin ligases Fbw7 and β-TrCP. PLoS Pathog. 2019, 15, e1007543. [Google Scholar] [CrossRef]
- Zhan, S.; Wang, T.; Ge, W. Multiple functions of the E3 ubiquitin ligase CHIP in immunity. Int. Rev. Immunol. 2017, 36, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Shahzad, N.; Harold, A.; Shuda, M.; Venuti, A.; Romero-Medina, M.C.; Pacini, L.; Brault, L.; Robitaille, A.; Taverniti, V.; et al. Merkel Cell Polyomavirus Downregulates N-myc Downstream-Regulated Gene 1, Leading to Cellular Proliferation and Migration. J. Virol. 2020, 94, e00899-19. [Google Scholar] [CrossRef] [PubMed]
- Masterson, L.; Thibodeau, B.J.; Fortier, L.E.; Geddes, T.J.; Pruetz, B.L.; Malhotra, R.; Keidan, R.; Wilson, G.D. Gene expression differences predict treatment outcome of merkel cell carcinoma patients. J. Skin Cancer 2014, 2014, 596459. [Google Scholar] [CrossRef] [PubMed]
- Daily, K.; Coxon, A.; Williams, J.S.; Lee, C.R.; Coit, D.G.; Busam, K.J.; Brownell, I. Assessment of cancer cell line representativeness using microarrays for Merkel cell carcinoma. J. Investig. Dermatol. 2015, 135, 1138–1146. [Google Scholar] [CrossRef]
- Berrios, C.; Padi, M.; Keibler, M.A.; Park, D.E.; Molla, V.; Cheng, J.; Lee, S.M.; Stephanopoulos, G.; Quackenbush, J.; DeCaprio, J.A. Merkel cell Polyomavirus small T antigen promotes pro-Glycolytic metabolic perturbations required for transformation. PLoS Pathog. 2016, 12, e1006020. [Google Scholar] [CrossRef] [PubMed]
- Melotte, V.; Qu, X.; Ongenaert, M.; van Criekinge, W.; de Bruïne, A.P.; Baldwin, H.S.; van Engeland, M. The N-myc downstream regulated gene (NDRG) family: Diverse functions, multiple applications. FASEB J. 2010, 24, 4153–4166. [Google Scholar] [CrossRef]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Böhling, T.; Joensuu, H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin. Cancer Res. 2011, 17, 4806–4813. [Google Scholar] [CrossRef]
- Pei, D.; Zhang, Y.; Zheng, J. Regulation of p53: A collaboration between Mdm2 and Mdmx. Oncotarget 2012, 3, 228–235. [Google Scholar] [CrossRef]
- Arora, R.; Shuda, M.; Guastafierro, A.; Feng, H.; Toptan, T.; Tolstov, T.; Normolle, D.; Vollmer, L.L.; Vogt, A.; Dömling, A.; et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci. Transl. Med. 2012, 4, 133ra156. [Google Scholar] [CrossRef]
- Houben, R.; Dreher, C.; Angermeyer, S.; Borst, A.; Utikal, J.; Haferkamp, S.; Peitsch, W.K.; Schrama, D.; Hesbacher, S. Mechanisms of p53 restriction in Merkel cell carcinoma cells are independent of the Merkel cell polyoma virus T antigens. J. Investig. Dermatol. 2013, 133, 2453–2460. [Google Scholar] [CrossRef]
- Waltari, M.; Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Böhling, T.; Joensuu, H. Association of Merkel cell polyomavirus infection with tumor p53, KIT, stem cell factor, PDGFR-alpha and survival in Merkel cell carcinoma. Int. J. Cancer 2011, 129, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Peitsch, W.K.; Zapatka, M.; Kneitz, H.; Houben, R.; Eib, S.; Haferkamp, S.; Moore, P.S.; Shuda, M.; Thompson, J.F.; et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J. Investig. Dermatol. 2011, 131, 1631–1638. [Google Scholar] [CrossRef]
- Guergnon, J.; Godet, A.N.; Galioot, A.; Falanga, P.B.; Colle, J.H.; Cayla, X.; Garcia, A. PP2A targeting by viral proteins: A widespread biological strategy from DNA/RNA tumor viruses to HIV-1. Biochim. Biophys. Acta 2011, 1812, 1498–1507. [Google Scholar] [CrossRef]
- Ruvolo, P.P. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 2016, 6, 87–99. [Google Scholar] [CrossRef]
- Luo, W.; Xu, C.; Ayello, J.; Dela Cruz, F.; Rosenblum, J.M.; Lessnick, S.L.; Cairo, M.S. Protein phosphatase 1 regulatory subunit 1A in ewing sarcoma tumorigenesis and metastasis. Oncogene 2018, 37, 798–809. [Google Scholar] [CrossRef]
- Fowle, H.; Zhao, Z.; Graña, X. PP2A holoenzymes, substrate specificity driving cellular functions and deregulation in cancer. Adv. Cancer Res. 2019, 144, 55–93. [Google Scholar] [CrossRef]
- Park, J.; Lee, D.H. Functional roles of protein phosphatase 4 in multiple aspects of cellular physiology: A friend and a foe. BMB Rep. 2020, 53, 181–190. [Google Scholar] [CrossRef]
- Kolupaeva, V.; Janssens, V. PP1 and PP2A phosphatases--cooperating partners in modulating retinoblastoma protein activation. FEBS J. 2013, 280, 627–643. [Google Scholar] [CrossRef]
- Janssens, V.; Longin, S.; Goris, J. PP2A holoenzyme assembly: In cauda venenum (the sting is in the tail). Trends Biochem. Sci. 2008, 33, 113–121. [Google Scholar] [CrossRef]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Henkel, T. Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Iwasaki, T.; Nonaka, D.; Kuwamoto, S.; Nagata, K.; Kato, M.; Kitamura, Y.; Hayashi, K. Higher expression of activation-induced Cytidine Deaminase is significantly associated with Merkel cell Polyomavirus-negative Merkel cell carcinomas. Yonago Acta Med. 2017, 60, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Nwogu, N.; Boyne, J.R.; Dobson, S.J.; Poterlowicz, K.; Blair, G.E.; Macdonald, A.; Mankouri, J.; Whitehouse, A. Cellular sheddases are induced by Merkel cell polyomavirus small tumour antigen to mediate cell dissociation and invasiveness. PLoS Pathog. 2018, 14, e1007276. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The TORrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 266–275. [Google Scholar] [CrossRef]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef]
- Hafner, C.; Houben, R.; Baeurle, A.; Ritter, C.; Schrama, D.; Landthaler, M.; Becker, J.C. Activation of the PI3K/AKT pathway in Merkel cell carcinoma. PLoS ONE 2012, 7, e31255. [Google Scholar] [CrossRef]
- Iwasaki, T.; Matsushita, M.; Nonaka, D.; Kuwamoto, S.; Kato, M.; Murakami, I.; Nagata, K.; Nakajima, H.; Sano, S.; Hayashi, K. Comparison of Akt/mTOR/4E-BP1 pathway signal activation and mutations of PIK3CA in Merkel cell polyomavirus-positive and Merkel cell polyomavirus-negative carcinomas. Hum. Pathol. 2015, 46, 210–216. [Google Scholar] [CrossRef]
- Nardi, V.; Song, Y.; Santamaria-Barria, J.A.; Cosper, A.K.; Lam, Q.; Faber, A.C.; Boland, G.M.; Yeap, B.Y.; Bergethon, K.; Scialabba, V.L.; et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin. Cancer Res. 2012, 18, 1227–1236. [Google Scholar] [CrossRef]
- Fang, B.; Kannan, A.; Zhao, S.; Nguyen, Q.H.; Ejadi, S.; Yamamoto, M.; Camilo Barreto, J.; Zhao, H.; Gao, L. Inhibition of PI3K by copanlisib exerts potent antitumor effects on Merkel cell carcinoma cell lines and mouse xenografts. Sci. Rep. 2020, 10, 8867. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C: Perfectly balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230. [Google Scholar] [CrossRef]
- Jain, K.; Basu, A. The Multifunctional Protein Kinase C-ε in Cancer Development and Progression. Cancers 2014, 6, 860–878. [Google Scholar] [CrossRef]
- Costa, A.; Mackelfresh, J.; Gilbert, L.; Bonner, M.Y.; Arbiser, J.L. Activation of Protein Kinase C ε in Merkel Cell Polyomavirus-Induced Merkel Cell Carcinoma. JAMA Dermatol. 2017, 153, 931–932. [Google Scholar] [CrossRef]
- Kovall, R.A.; Gebelein, B.; Sprinzak, D.; Kopan, R. The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Dev. Cell 2017, 41, 228–241. [Google Scholar] [CrossRef]
- Wardhani, L.O.; Matsushita, M.; Kuwamoto, S.; Nonaka, D.; Nagata, K.; Kato, M.; Kitamura, Y.; Hayashi, K. Expression of Notch 3 and Jagged 1 Is Associated With Merkel Cell Polyomavirus Status and Prognosis in Merkel Cell Carcinoma. Anticancer Res. 2019, 39, 319–329. [Google Scholar] [CrossRef]
- Hooper, J.E.; Scott, M.P. Communicating with Hedgehogs. Nat. Rev. Mol. Cell Biol. 2005, 6, 306–317. [Google Scholar] [CrossRef]
- Kuromi, T.; Matsushita, M.; Iwasaki, T.; Nonaka, D.; Kuwamoto, S.; Nagata, K.; Kato, M.; Akizuki, G.; Kitamura, Y.; Hayashi, K. Association of expression of the hedgehog signal with Merkel cell polyomavirus infection and prognosis of Merkel cell carcinoma. Hum. Pathol. 2017, 69, 8–14. [Google Scholar] [CrossRef]
- Kennedy, M.M.; Blessing, K.; King, G.; Kerr, K.M. Expression of bcl-2 and p53 in Merkel cell carcinoma. An immunohistochemical study. Am. J. Dermatopathol 1996, 18, 273–277. [Google Scholar] [CrossRef]
- Feinmesser, M.; Halpern, M.; Fenig, E.; Tsabari, C.; Hodak, E.; Sulkes, J.; Brenner, B.; Okon, E. Expression of the apoptosis-related oncogenes bcl-2, bax, and p53 in Merkel cell carcinoma: Can they predict treatment response and clinical outcome? Hum. Pathol. 1999, 30, 1367–1372. [Google Scholar] [CrossRef]
- Brunner, M.; Thurnher, D.; Pammer, J.; Geleff, S.; Heiduschka, G.; Reinisch, C.M.; Petzelbauer, P.; Erovic, B.M. Expression of VEGF-A/C, VEGF-R2, PDGF-alpha/beta, c-kit, EGFR, Her-2/Neu, Mcl-1 and Bmi-1 in Merkel cell carcinoma. Mod. Pathol. 2008, 21, 876–884. [Google Scholar] [CrossRef]
- Sahi, H.; Koljonen, V.; Kavola, H.; Haglund, C.; Tukiainen, E.; Sihto, H.; Böhling, T. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012, 461, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, M.E.; Mangelberger, D.; Weick, J.W.; Vozheiko, T.D.; Harms, P.W.; Nash, K.T.; Quintana, E.; Baciu, P.; Johnson, T.M.; Bichakjian, C.K.; et al. Merkel cell carcinoma dependence on bcl-2 family members for survival. J. Investig. Dermatol. 2014, 134, 2241–2250. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Varker, K.A.; Collamore, M.; Zwiebel, J.A.; Coit, D.; Kelsen, D.; Chung, K.Y. G3139 (Genasense) in patients with advanced merkel cell carcinoma. Am. J. Clin. Oncol 2009, 32, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Church, C.D.; Nghiem, P. How does the Merkel polyomavirus lead to a lethal cancer? Many answers, many questions, and a new mouse model. J. Investig. Dermatol. 2015, 135, 1221–1224. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Tegeder, A.; Willmes, C.; Iyer, J.G.; Afanasiev, O.K.; Schrama, D.; Koba, S.; Thibodeau, R.; Nagase, K.; Simonson, W.T.; et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol. Res. 2014, 2, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Afanasiev, O.K.; Yelistratova, L.; Miller, N.; Nagase, K.; Paulson, K.; Iyer, J.G.; Ibrani, D.; Koelle, D.M.; Nghiem, P. Merkel polyomavirus-specific T cells fluctuate with merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clin. Cancer Res. 2013, 19, 5351–5360. [Google Scholar] [CrossRef]
- Afanasiev, O.K.; Nagase, K.; Simonson, W.; Vandeven, N.; Blom, A.; Koelle, D.M.; Clark, R.; Nghiem, P. Vascular E-selectin expression correlates with CD8 lymphocyte infiltration and improved outcome in Merkel cell carcinoma. J. Investig. Dermatol. 2013, 133, 2065–2073. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Lipson, E.J.; Vincent, J.G.; Loyo, M.; Kagohara, L.T.; Luber, B.S.; Wang, H.; Xu, H.; Nayar, S.K.; Wang, T.S.; Sidransky, D.; et al. PD-L1 expression in the Merkel cell carcinoma microenvironment: Association with inflammation, Merkel cell polyomavirus and overall survival. Cancer Immunol. Res. 2013, 1, 54–63. [Google Scholar] [CrossRef]
- Mitteldorf, C.; Berisha, A.; Tronnier, M.; Pfaltz, M.C.; Kempf, W. PD-1 and PD-L1 in neoplastic cells and the tumor microenvironment of Merkel cell carcinoma. J. Cutan. Pathol. 2017, 44, 740–746. [Google Scholar] [CrossRef]
- Schönrich, G.; Raftery, M.J. The PD-1/PD-L1 Axis and virus infections: A delicate balance. Front. Cell Infect. Microbiol. 2019, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, N.; Shuda, M.; Gheit, T.; Kwun, H.J.; Cornet, I.; Saidj, D.; Zannetti, C.; Hasan, U.; Chang, Y.; Moore, P.S.; et al. The T antigen locus of Merkel cell polyomavirus downregulates human Toll-like receptor 9 expression. J. Virol. 2013, 87, 13009–13019. [Google Scholar] [CrossRef] [PubMed]
- Jouhi, L.; Koljonen, V.; Böhling, T.; Haglund, C.; Hagström, J. The expression of Toll-like receptors 2, 4, 5, 7 and 9 in Merkel cell carcinoma. Anticancer Res. 2015, 35, 1843–1849. [Google Scholar]
- Wu, S.Y.; Chiang, C.M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef] [PubMed]
- Hajmirza, A.; Emadali, A.; Gauthier, A.; Casasnovas, O.; Gressin, R.; Callanan, M.B. BET family protein BRD4: An emerging actor in NFκB signaling in inflammation and cancer. Biomedicines 2018, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Malmgaard, L. Induction and regulation of IFNs during viral infections. J. Interferon Cytokine Res. 2004, 24, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Ichinohe, T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015, 23, 55–63. [Google Scholar] [CrossRef]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef]
- Germano, G.; Allavena, P.; Mantovani, A. Cytokines as a key component of cancer-related inflammation. Cytokine 2008, 43, 374–379. [Google Scholar] [CrossRef]
- Richards, K.F.; Guastafierro, A.; Shuda, M.; Toptan, T.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus T antigens promote cell proliferation and inflammatory cytokine gene expression. J. Gen. Virol. 2015, 96, 3532–3544. [Google Scholar] [CrossRef]
- Monnier, J.; Samson, M. Prokineticins in angiogenesis and cancer. Cancer Lett. 2010, 296, 144–149. [Google Scholar] [CrossRef]
- Lauttia, S.; Sihto, H.; Kavola, H.; Koljonen, V.; Böhling, T.; Joensuu, H. Prokineticins and Merkel cell polyomavirus infection in Merkel cell carcinoma. Br. J. Cancer 2014, 110, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, K.; Abdulsalam, I.; Fismen, S.; Grimstad, O.; Sveinbjornsson, B.; Moens, U. CCL17/TARC and CCR4 expression in Merkel cell carcinoma. Oncotarget 2018, 9, 31432–31447. [Google Scholar] [CrossRef][Green Version]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef]
- Liu, W.; Kim, G.B.; Krump, N.A.; Zhou, Y.; Riley, J.L.; You, J. Selective reactivation of STING signaling to target Merkel cell carcinoma. Proc. Natl. Acad. Sci. USA 2020, 117, 13730–13739. [Google Scholar] [CrossRef] [PubMed]
- Jouary, T.; Leyral, C.; Dreno, B.; Doussau, A.; Sassolas, B.; Beylot-Barry, M.; Renaud-Vilmer, C.; Guillot, B.; Bernard, P.; Lok, C.; et al. Groupe de Cancérologie Cutanée of the Société Francaise de Dermatologie. Adjuvant prophylactic regional radiotherapy versus observation in stage I Merkel cell carcinoma: A multicentric prospective randomized study. Ann. Oncol. 2012, 23, 1074–1080. [Google Scholar] [CrossRef]
- Hasan, S.; Liu, L.; Triplet, J.; Li, Z.; Mansur, D. The role of postoperative radiation and chemoradiation in merkel cell carcinoma: A systematic review of the literature. Front. Oncol. 2013, 3, 276. [Google Scholar] [CrossRef]
- Rush, Z.; Fields, R.C.; Lee, N.; Brownell, I. Radiation therapy in the management of Merkel cell carcinoma: Current perspectives. Expert Rev. Dermatol. 2011, 6, 395–404. [Google Scholar] [CrossRef]
- Miles, B.A.; Goldenberg, D. Merkel cell carcinoma: Do you know your guidelines? Head Neck 2016, 38, 647–652. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-dependent Cytosolic DNA sensing promotes radiation-induced type I Interferon-Dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Paulson, K.G.; Carter, J.J.; Johnson, L.G.; Cahill, K.W.; Iyer, J.G.; Schrama, D.; Becker, J.C.; Madeleine, M.M.; Nghiem, P.; Galloway, D.A. Antibodies to merkel cell polyomavirus T antigen oncoproteins reflect tumor burden in merkel cell carcinoma patients. Cancer Res. 2010, 70, 8388–8397. [Google Scholar] [CrossRef] [PubMed]
- Asioli, S.; Righi, A.; Volante, M.; Eusebi, V.; Bussolati, G. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer 2007, 110, 640–647. [Google Scholar] [CrossRef]
- Hall, B.J.; Pincus, L.B.; Yu, S.S.; Oh, D.H.; Wilson, A.R.; McCalmont, T.H. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J. Cutan. Pathol. 2012, 39, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Stetsenko, G.Y.; Malekirad, J.; Paulson, K.G.; Iyer, J.G.; Thibodeau, R.M.; Nagase, K.; Schmidt, M.; Storer, B.E.; Argenyi, Z.B.; Nghiem, P. p63 expression in Merkel cell carcinoma predicts poorer survival yet may have limited clinical utility. Am. J. Clin. Pathol. 2013, 140, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Fleming, K.E.; Ly, T.Y.; Pasternak, S.; Godlewski, M.; Doucette, S.; Walsh, N.M. Support for p63 expression as an adverse prognostic marker in Merkel cell carcinoma: Report on a Canadian cohort. Hum. Pathol. 2014, 45, 952–960. [Google Scholar] [CrossRef]
- Asioli, S.; Righi, A.; de Biase, D.; Morandi, L.; Caliendo, V.; Picciotto, F.; Macripò, G.; Maletta, F.; di Cantogno, L.V.; Chiusa, L.; et al. Expression of p63 is the sole independent marker of aggressiveness in localised (stage I-II) Merkel cell carcinomas. Mod. Pathol. 2011, 24, 1451–1461. [Google Scholar] [CrossRef]
- Patel, R.M.; Walters, L.L.; Kappes, F.; Mehra, R.; Fullen, D.R.; Markovitz, D.M.; Ma, L. DEK expression in Merkel cell carcinoma and small cell carcinoma. J. Cutan. Pathol. 2012, 39, 753–757. [Google Scholar] [CrossRef]
- Sandén, C.; Gullberg, U. The DEK oncoprotein and its emerging roles in gene regulation. Leukemia 2015, 29, 1632–1636. [Google Scholar] [CrossRef]
- Pryor, J.G.; Simon, R.A.; Bourne, P.A.; Spaulding, B.O.; Scott, G.A.; Xu, H. Merkel cell carcinoma expresses K homology domain-containing protein overexpressed in cancer similar to other high-grade neuroendocrine carcinomas. Hum. Pathol. 2009, 40, 238–243. [Google Scholar] [CrossRef]
- Pryor, J.G.; Bourne, P.A.; Yang, Q.; Spaulding, B.O.; Scott, G.A.; Xu, H. IMP-3 is a novel progression marker in malignant melanoma. Mod. Pathol. 2008, 21, 431–437. [Google Scholar] [CrossRef]
- Johnson, B.; Khalil, M.; Blansfield, J.; Lin, F.; Zhu, S.; Kirchner, H.L.; Weir, A.B. 3rd. Investigating the prognostic value of KOC (K homology domain containing protein overexpressed in cancer) overexpression after curative intent resection of pancreatic ductal adenocarcinoma. J. Gastrointest. Oncol. 2016, 7, E113–E117. [Google Scholar] [CrossRef] [PubMed]
- Kase, S.; Yoshida, K.; Osaki, M.; Adachi, H.; Ito, H.; Ohno, S. Expression of erythropoietin receptor in human Merkel cell carcinoma of the eyelid. Anticancer Res. 2006, 26, 4535–4537. [Google Scholar] [PubMed]
- Werchau, S.; Toberer, F.; Enk, A.; Dammann, R.; Helmbold, P. Merkel cell carcinoma induces lymphatic microvessel formation. J. Am. Acad. Dermatol. 2012, 67, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Vlahova, L.; Doerflinger, Y.; Houben, R.; Becker, J.C.; Schrama, D.; Weiss, C.; Goebeler, M.; Helmbold, P.; Goerdt, S.; Peitsch, W.K. P-cadherin expression in Merkel cell carcinomas is associated with prolonged recurrence-free survival. Br. J. Dermatol. 2012, 166, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Samimi, M.; Touzé, A.; Laude, H.; Le Bidre, E.; Arnold, F.; Carpentier, A.; Gardair, C.; Carlotti, A.; Maubec, E.; Dupin, N.; et al. Vitamin D deficiency is associated with greater tumor size and poorer outcome in Merkel cell carcinoma patients. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 298–308. [Google Scholar] [CrossRef]
- Rand, J.; Balzer, B.L.; Frishberg, D.P.; Essner, R.; Shon, W. Prevalence of Delta-Like Protein 3 expression in Merkel cell carcinoma. J. Am. Acad. Dermatol. 2019. [Google Scholar] [CrossRef]
- Xie, H.; Kaye, F.J.; Isse, K.; Sun, Y.; Ramoth, J.; French, D.M.; Flotte, T.J.; Luo, Y.; Saunders, L.R.; Mansfield, A.S. Delta-like Protein 3 Expression and Targeting in Merkel Cell Carcinoma. The Oncol. 2020. [CrossRef]
- Toberer, F.; Haenssle, H.A.; Heinzel-Gutenbrunner, M.; Enk, A.; Hartschuh, W.; Helmbold, P.; Kutzner, H. Metabolic reprogramming and angiogenesis in primary cutaneous Merkel cell carcinoma: Expression of hypoxia inducible factor-1α and its central downstream factors. J. Eur. Acad. Dermatol. Venereol. 2020. [Google Scholar] [CrossRef]
- Konstatinell, A.; Coucheron, D.H.; Sveinbjørnsson, B.; Moens, U. MicroRNAs as Potential Biomarkers in Merkel Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, 1873. [Google Scholar] [CrossRef]
- Xie, H.; Lee, L.; Caramuta, S.; Höög, A.; Browaldh, N.; Björnhagen, V.; Larsson, C.; Lui, W.O. MicroRNA expression patterns related to merkel cell polyomavirus infection in human merkel cell carcinoma. J. Investig. Dermatol. 2014, 134, 507–517. [Google Scholar] [CrossRef]
- Renwick, N.; Cekan, P.; Masry, P.A.; McGeary, S.E.; Miller, J.B.; Hafner, M.; Li, Z.; Mihailovic, A.; Morozov, P.; Brown, M.; et al. Multicolor microRNA FISH effectively differentiates tumor types. J. Clin. Investig. 2013, 123, 2694–2702. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Ritter, C.; Nghiem, P.; Blom, A.; Verhaegen, M.E.; Dlugosz, A.; Ødum, N.; Woetmann, A.; Tothill, R.W.; Hicks, R.J.; et al. Circulating Cell-Free miR-375 as surrogate marker of tumor burden in merkel cell carcinoma. Clin. Cancer Res. 2018, 24, 5873–5882. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Paulson, K.G.; Murchison, E.P.; Afanasiev, O.K.; Alkan, C.; Leonard, J.H.; Byrd, D.R.; Hannon, G.J.; Nghiem, P. Identification and validation of a novel mature microRNA encoded by the Merkel cell polyomavirus in human Merkel cell carcinomas. J. Clin. Virol. 2011, 52, 272–275. [Google Scholar] [CrossRef]
- Becker, J.C.; Stang, A.; Hausen, A.Z.; Fischer, N.; DeCaprio, J.A.; Tothill, R.W.; Lyngaa, R.; Hansen, U.K.; Ritter, C.; Nghiem, P.; et al. Epidemiology, biology and therapy of Merkel cell carcinoma: Conclusions from the EU project IMMOMEC. Cancer Immunol. Immunother. 2018, 67, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-approved immune checkpoint inhibitors per NCCN Guidelines with the level of evidence. Cancers 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in advanced Merkel-cell carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A. Durable tumor regression and overall survival in patients with advanced Merkel cell carcinoma receiving Pembrolizumab as first-line therapy. J. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef]
- Topalian, S.L.; Bhatia, S.; Amin, A.; Kudchadkar, R.R.; Sharfman, W.H.; Lebbé, C.; Delord, J.P.; Dunn, L.A.; Shinohara, M.M.; Kulikauskas, R.; et al. Neoadjuvant Nivolumab for Patients With Resectable Merkel Cell Carcinoma in the CheckMate 358 Trial. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Bhatia, S.; Brohl, A.S.; Hamid, O.; Mehnert, J.M.; Terheyden, P.; Shih, K.C.; Brownell, I.; Lebbé, C.; Lewis, K.D.; et al. Avelumab in patients with previously treated metastatic Merkel cell carcinoma: Long-term data and biomarker analyses from the single-arm phase 2 JAVELIN Merkel 200 trial. J. Immunother. Cancer 2020, 8, e000674. [Google Scholar] [CrossRef]
- Winkler, J.K.; Dimitrakopoulou-Strauss, A.; Sachpekidis, C.; Enk, A.; Hassel, J.C. Ipilimumab has efficacy in metastatic Merkel cell carcinoma: A case series of five patients. J. Eur. Acad. Dermatol. Venereol. 2017, 31, e389–e391. [Google Scholar] [CrossRef]
- Tabachnick-Cherny, S.; Pulliam, T.; Church, C.; Koelle, D.M.; Nghiem, P. Polyomavirus-driven Merkel cell carcinoma: Prospects for therapeutic vaccine development. Mol. Carcinogs. 2020, 59, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Gomez, B.P.; Viscidi, R.P.; Peng, S.; He, L.; Ma, B.; Wu, T.C.; Hung, C.F. Development of a DNA vaccine targeting Merkel cell polyomavirus. Vaccine 2012, 30, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Gomez, B.; He, L.; Tsai, Y.C.; Wu, T.C.; Viscidi, R.P.; Hung, C.F. Creation of a Merkel cell polyomavirus small T antigen-expressing murine tumor model and a DNA vaccine targeting small T antigen. Cell Biosci. 2013, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Gomez, B.P.; Wang, C.; Viscidi, R.P.; Peng, S.; He, L.; Wu, T.C.; Hung, C.F. Strategy for eliciting antigen-specific CD8+ T cell-mediated immune response against a cryptic CTL epitope of merkel cell polyomavirus large T antigen. Cell Biosci. 2012, 2, 36. [Google Scholar] [CrossRef]
- Togtema, M.; Jackson, R.; Grochowski, J.; Villa, P.L.; Mellerup, M.; Chattopadhyaya, J.; Zehbe, I. Synthetic siRNA targeting human papillomavirus 16 E6: A perspective on in vitro nanotherapeutic approaches. Nanomedicine 2018, 13, 455–474. [Google Scholar] [CrossRef]
- Rosa, J.; Suzuki, I.; Kravicz, M.; Caron, A.; Pupo, A.V.; Praça, F.G.; Bentley, M. Current non-viral siRNA delivery systems as a promising treatment of skin diseases. Curr. Pharm. Des. 2018, 24, 2644–2663. [Google Scholar] [CrossRef]
- Seguin, S.P.; Ireland, A.W.; Gupta, T.; Wright, C.M.; Miyata, Y.; Wipf, P.; Pipas, J.M.; Gestwicki, J.E.; Brodsky, J.L. A screen for modulators of large T antigen’s ATPase activity uncovers novel inhibitors of Simian Virus 40 and BK virus replication. Antiviral Res. 2012, 96, 70–81. [Google Scholar] [CrossRef][Green Version]
- Randhawa, P.; Zeng, G.; Bueno, M.; Salgarkar, A.; Lesniak, A.; Isse, K.; Seyb, K.; Perry, A.; Charles, I.; Hustus, C.; et al. Inhibition of large T antigen ATPase activity as a potential strategy to develop anti-polyomavirus JC drugs. Antiviral Res. 2014, 112, 113–119. [Google Scholar] [CrossRef]
- Sarma, B.; Willmes, C.; Angerer, L.; Adam, C.; Becker, J.C.; Kervarrec, T.; Schrama, D.; Houben, R. Artesunate Affects T Antigen expression and survival of virus-positive Merkel cell carcinoma. Cancers 2020, 12, 919. [Google Scholar] [CrossRef]
- Deeken, J.F.; Wang, H.; Hartley, M.; Cheema, A.K.; Smaglo, B.; Hwang, J.J.; He, A.R.; Weiner, L.M.; Marshall, J.L.; Giaccone, G.; et al. A phase I study of intravenous artesunate in patients with advanced solid tumor malignancies. Cancer Chemother. Pharmacol. 2018, 81, 587–596. [Google Scholar] [CrossRef]
- Kim, J.; McNiff, J.M. Nuclear expression of survivin portends a poor prognosis in Merkel cell carcinoma. Mod. Pathol. 2008, 21, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Dresang, L.R.; Guastafierro, A.; Arora, R.; Normolle, D.; Chang, Y.; Moore, P.S. Response of Merkel cell polyomavirus-positive merkel cell carcinoma xenografts to a survivin inhibitor. PLoS ONE 2013, 8, e80543. [Google Scholar] [CrossRef] [PubMed]
- Fewell, S.W.; Smith, C.M.; Lyon, M.A.; Dumitrescu, T.P.; Wipf, P.; Day, B.W.; Brodsky, J.L. Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J. Biol. Chem. 2004, 279, 51131–51140. [Google Scholar] [CrossRef]
- Adam, C.; Baeurle, A.; Brodsky, J.L.; Wipf, P.; Schrama, D.; Becker, J.C.; Houben, R. The HSP70 modulator MAL3-101 inhibits Merkel cell carcinoma. PLoS ONE 2014, 9, e92041. [Google Scholar] [CrossRef]
- Ocaña-Guzman, R.; Torre-Bouscoulet, L.; Sada-Ovalle, I. TIM-3 Regulates Distinct Functions in Macrophages. Front. Immunol. 2016, 7, 229. [Google Scholar] [CrossRef] [PubMed]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Carey, B.W.; Kim, D.Y.; Kovacs, D.M. Presenilin/gamma-secretase and alpha-secretase-like peptidases cleave human MHC Class I proteins. Biochem. J. 2007, 401, 121–127. [Google Scholar] [CrossRef]
- Anzivino, E.; Rodio, D.M.; Mischitelli, M.; Bellizzi, A.; Sciarra, A.; Salciccia, S.; Gentile, V.; Pietropaolo, V. High Frequency of JCV DNA Detection in prostate cancer tissues. Cancer Genom. Proteom. 2015, 12, 189–200. [Google Scholar]
- Tognon, M.; Provenzano, M. New insights on the association between the prostate cancer and the small DNA tumour virus, BK polyomavirus. J. Transl. Med. 2015, 13, 387. [Google Scholar] [CrossRef]
- Delbue, S.; Comar, M.; Ferrante, P. Review on the role of the human Polyomavirus JC in the development of tumors. Infect. Agent Cancer 2017, 12, 10. [Google Scholar] [CrossRef]
- Levican, J.; Acevedo, M.; León, O.; Gaggero, A.; Aguayo, F. Role of BK human polyomavirus in cancer. Infect. Agent Cancer 2018, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Dela Cruz, F.N., Jr.; Giannitti, F.; Li, L.; Woods, L.W.; Del Valle, L.; Delwart, E.; Pesavento, P.A. Novel polyomavirus associated with brain tumors in free-ranging raccoons, western United States. Emerg. Infect. Dis. 2013, 19, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Konno, A.; Nagata, M.; Nanko, H. Immunohistochemical diagnosis of a Merkel cell tumor in a dog. Vet. Pathol. 1998, 35, 538–540. [Google Scholar] [CrossRef] [PubMed]
- Bagnasco, G.; Properzi, R.; Porto, R.; Nardini, V.; Poli, A.; Abramo, F. Feline cutaneous neuroendocrine carcinoma (Merkel cell tumour): Clinical and pathological findings. Vet. Dermatol. 2003, 14, 111–115. [Google Scholar] [CrossRef]
- Ozaki, K.; Narama, I. Merkel cell carcinoma in a cat. J. Vet. Med. Sci. 2009, 71, 1093–1096. [Google Scholar] [CrossRef]
- Joiner, K.S.; Smith, A.N.; Henderson, R.A.; Brawner, W.R.; Spangler, E.A.; Sartin, E.A. Multicentric cutaneous neuroendocrine (Merkel cell) carcinoma in a dog. Vet. Pathol. 2010, 47, 1090–1094. [Google Scholar] [CrossRef]
- Teh, A.P.P.; Izzati, U.Z.; Diep, N.V.; Hirai, T.; Yamaguchi, R. Merkel Cell Carcinoma in a Steer. J. Comp. Pathol. 2018, 158, 17–21. [Google Scholar] [CrossRef]
- Sumi, A.; Chambers, J.K.; Doi, M.; Kudo, T.; Omachi, T.; Uchida, K. Clinical features and outcomes of Merkel cell carcinoma in 20 cats. Vet. Comp. Oncol. 2018, 16, 554–561. [Google Scholar] [CrossRef]
- Schuurman, R.; Sol, C.; van der Noordaa, J. The complete nucleotide sequence of bovine polyomavirus. J. Gen. Virol. 1990, 71, 1723–1735. [Google Scholar] [CrossRef]
- Delwart, E.; Kapusinszky, B.; Pesavento, P.A.; Estrada, M.; Seguin, M.A.; Leutenegger, C.M. Genome sequence of canine Polyomavirus in respiratory secretions of dogs with pneumonia of unknown etiology. Genome Announc. 2017, 5, e00615-17. [Google Scholar] [CrossRef]
- Gheit, T.; Dutta, S.; Oliver, J.; Robitaille, A.; Hampras, S.; Combes, J.D.; McKay-Chopin, S.; Le Calvez-Kelm, F.; Fenske, N.; Cherpelis, B.; et al. Isolation and characterization of a novel putative human polyomavirus. Virology 2017, 506, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Fahsbender, E.; Altan, E.; Estrada, M.; Seguin, M.A.; Young, P.; Leutenegger, C.M.; Delwart, E. Lyon-IARC Polyomavirus DNA in feces of Diarrheic cats. Microbiol. Resour. Announc. 2019, 8, e00550-19. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cell of Origin | Supporting Cell of Origin | Arguing Against Cell of Origin |
|---|---|---|
| Merkel cell (VN-MCC) |
| |
| (Epi)dermal stem cell (VN-MCC) | ||
| Pro/pre-B cell (VP-MCC) | ||
| Skin-derived precursors (VN-MCC) |
|
|
| Dermal fibroblasts (VP-MCC) | ||
| Keratinocytes (VN- and VP-MCC) |
| T Antigen | Protein | Functional Class | Biological Role | Reference |
|---|---|---|---|---|
| sT | abhydrolyse domain containing 12 (ABHD12) | metabolism | unknown | [129] |
| sT | ankyrin repeat domain 13A (ANKRD13Aa) | protein stability | unknown | [129] |
| sT | ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 | metabolism | unknown | [129] |
| LT | ATP binding cassette subfamily A Member 13 (ABCA13) | signaling | unknown | [129] |
| sT | ATP binding cassette subfamily D member 3 (ABCD3) | signaling | unknown | [129] |
| LT | ATP binding cassette subfamily D member 13 (ABCD13) | signaling | unknown | [129] |
| sT | aryl hydrocarbon receptor interacting protein (AIP) | transcription | unknown | [129] |
| LT | adaptor related protein complex 2 subunit A and M (AP2A1 and M1) | intracellular transport | unknown | [129] |
| sT | ADAM metallopeptidase domain 9 (ADAM9) | cytoskeleton/extracellular matrix | unknown | [129] |
| LT | ataxia telangiectasia mutated (ATM kinase) | DNA replication and repair | LT phosphorylation | [14] |
| LT, sT | BCL2 associated anthanogene 2, 3 and 5 (BAG2, 3 and 5) | protein stability/apoptosis | unknown | [129] |
| LT | bromodomain protein 4 (Brd4) | cell cycle/DNA replication | viral genome replication | [130,131] |
| sT | cadherin 1 (CDH1) | cytoskeleton/extracellular matrix | unknown | [129] |
| LT | casein kinase 2 beta (CK2β) | Signaling | unknown | [129] |
| sT | cathepsin B (CTSB) | protein stability/modification | unknown | [129] |
| LT | caveolae associated protein 2 (CAVIN2) | intracellular transport | unknown | [129] |
| sT | CCHC-type Zinc finger nucleic acid binding protein (CNBP) | transcription | unknown | [129] |
| sT | cell surface glycoprotein 44 (CD44) | cell-cell interaction, cell adhesion, migration | unknown | [129] |
| sT | cell division cycle 20 (CDC20) | cell cycle | sT-mediated phosphorylation of 4E-BP1 | [129,132,133] |
| sT | coatomer protein complex subunit γ2 | intracellular transport | unknown | [129] |
| sT | 2′, 3′-cyclic nucleotide 3′ phosphodiesterase (CNP) | nucleotide metabolism | unknown | [129] |
| LT | DEAD-box helicase (DDX24) | post-transcription/translation | unknown | [129] |
| sT | heat shock protein 40 members A1 and B4 (DnaJA1 and B4) | chaperone | unknown | [129] |
| LT, sT | heat shock protein 40 member C7 (DnaJC7) | chaperone | unknown | [129] |
| LT | transcription factors E2F3 and 4 (E2F3 and 4) | transcription | unknown | [129] |
| sT | EGF containing fibulin extracellular matrix protein 2 (EFEM2) | cytoskeleton/extracellular matrix | unknown | [129] |
| sT | eukaryotic translation initiation factor 4E binding protein 1 (eIF-4EBP1) | translation | disregulated cap-dependent translation which promotes tumorigenesis | [99,133] |
| LT, sT | emerin (EMD) | cytoskeleton | unknown | [129] |
| LT | family with sequence similarity 71 member E2 (FAM71E2) | unknown | unknown | [129] |
| sT | F-box and WD repeat domain containing 7 (Fbxw7) | protein stability | tumorigenic properties of the virus (stabilization of LT and cellular proteins) | [134,135] |
| LT | general transcription factor IIIC subunit 1 (GTF3C1) | transcription | unknown | [129] |
| LT | high density lipoprotein binding protein (HDLBP) | metabolism | unknown | [129] |
| LT, sT | heat shock protein 70 (HSPA1 and A4) | chaperone | cell cycle progression | [129] |
| sT | insulin like growth factor 2 receptor (IGF2R) | signaling | unknown | [129] |
| LT, sT | inhibitor of nuclear factor kappa-B kinase-interacting protein (IκBIP) | signaling | unknown | [129] |
| LT | karyopherin subunit α2, 3 and 4 (KPNA2, 3 and 4) | intracellular transport | unknown | [129] |
| sT | lysyl oxidase (LOX) | metabolism | unknown | [129] |
| LT | microtubulin-associated protein 4 (MAP4) | cytoskeleton | unknown | [129] |
| sT | membrane bound O-acetyltransferase domain containing 7 | metabolism/plasma membrane lipid organization | unknown | [129] |
| LT | mediator complex subunit 14 (MED14) | transcription | unknown | [129] |
| sT | matrix metalloproteinase 14 | extracellular matrix | unknown | [129] |
| sT | myelin protein zero like 1 (MPZL1) | signaling | unknown | [129] |
| sT | mitochondrial carrier 2 (MTCH2) | metabolism | unknown | [129] |
| sT | myoferlin (MYOF) | membrane morphology | unknown | [129] |
| sT | NF-kappa-B essential modulator (NEMO=IKBKG) | signaling | inhibition NFκB signaling; immune evasion | [136] |
| sT | Notch 2 receptor (NOTCH2) | signaling | unknown | [129] |
| sT | nuclear receptor binding SET domain protein1 (NSD1) | transcription | unknown | [129] |
| LT | prolyl 4-hydroxylase subunit alpha 3 (P4HA3) | metabolism | unknown | [129] |
| sT | prolyl 4-hydroxylase subunit β (P4HB) | metabolism | unknown | [129] |
| sT | platelet-derived growth factor receptor subunit β (PDGFRβ) | signaling | unknown | [129] |
| LT | PGAM family member 5, mitochondrial Ser/Thr protein phosphatase (PGAM5) | signaling | unknown | [129] |
| sT | progesterone receptor membrane component 2 (PGRMC2) | signaling | unknown | [129] |
| LT | phosphatidylinositol-5-phosphate 4-kinase type 2 beta (PIP4K2β) | signaling | unknown | [129] |
| LT | protein phosphatase 2 scaffold subunit α (PP2AR1α) | signaling | unknown | [129] |
| sT | protein phosphatase 2 catalytic subunit α and β (PPP2CA and CB) | Signaling | mutation in PP2A binding site had no effect on the known activities of sT | [17,136,137] |
| sT | PRA1 domain family member 2 (PRAF2) | intracellular transport | unknown | [129] |
| sT | protein phosphatase 2 regulatory subunit Aα and Aβ (PP2R1A and B) | signaling | unknown | [138] |
| sT | protein phosphatase regulatory subunit 1 (PP4R1) | signaling | microtubule destabilization and cell motility (metastasis?); inhibition NFκB signaling (immune evasion?) | [17,136,139,140] |
| sT | protein phosphatase Mg2+/Mn2+ dependent 1A, 1B and 1G (PPM1A, B and G) | signaling | unknown | [129] |
| sT | proteasome 26S ATPase 2,3 and 4 (PSMC2, 3 and 4) | protein stability | unknown | [129] |
| LT | caveolae associated protein 1 (PTRF) | transcription | unknown | [129] |
| sT | pituitary tumor-transforming gene 1 protein-interacting protein (PTTPG1P) | intracellular transport | unknown | [129] |
| sT | Rab18 (RAB18) | signaling | unknown | [129] |
| LT | Retinoblastoma protein 1 (RB1) | cell cycle | cell cycle progression | [111,115,117] |
| sT | ribonuclease/angiogenin inhibitor 1 (RNH1) | transcription/translation | unknown | [129] |
| sT | ribosomal protein L21 | translation | unknown | [129] |
| sT | ribosomal protein S27 like | translation | unknown | [129] |
| LT | recitulon 4 (RTN4) | intracellular transport | unknown | [129] |
| LT | sphingosine-1-phosphate lyase 1 (SGPL1) | metabolism | unknown | [129] |
| sT | secreted protein acidic and cysteine rich (SPARC) | extracellular matrix | unknown | [129] |
| sT | sulfide quinone oxidoreductase (SQRDL) | metabolism | unknown | [129] |
| LT | signal recognition particle 14 (SRP14) | intracellular transport | unknown | [129] |
| LT, sT | signal recognition particle receptor subunit b (SRPRB) | intracellular transport | unknown | [129] |
| sT | ser/thr kinase 38 (STK38) | signaling | unknown | [129] |
| LT, sT | STIP1 homology and U-box containing protein 1 (STUB1) | protein stability | unknown | [129] |
| sT | surfeit 4 (SURF4) | intracellular transport | unknown | [129] |
| LT | Ubiquitin-specific protease (USP7) | protein stability | inhibition viral DNA replication | [127] |
| LT | transcription elongation factor B subunit 1 (TCEB1) | transcription | unknown | [129] |
| LT | transcription factor DP1 (TFDP1) | transcription | unknown | [129] |
| sT | translocase of inner mitochondrial membrane 8A (TIMM8A) | intracellular transport | unknown | [129] |
| sT | transmembrane protein 165 (TMEM165) | protein glycosylation | unknown | [129] |
| sT | thioredoxin related transmembrane protein 3 (TMX3) | protein folding | unknown | [129] |
| sT | toll interacting protein (TOLLIP) | signaling | unknown | [129] |
| LT | tripartite motif containing 38 (TRIM38) | protein stability | unknown | [129] |
| LT | testis-specific Y-encoded-like protein 1 (TSPYL1) | transcription | unknown | [129] |
| sT | tubulin α1 (TUBA1B) | protein folding and gap junctions | unknown | [17] |
| sT | tubulin β2α (TUBB2A) | mitosis and intracellular transport | unknown | [17] |
| LT, sT | upregulated during skeletal muscle growth 5 (USMG5) | nucleotide synthesis | unknown | [129] |
| LT | VPS39 subunit Of HOPS complex (Vam6p) | intracellular transport | role in DNA replication (?) | [18,129] |
| LT | VAMP associated proteins A and B (VAPA and VAPB) | intracellular transport | unknown | [129] |
| sT | vitamin K epoxide reductase complex subunit 1 (VKORC1) | metabolism | unknown | [129] |
| LT | vacuolar protein sorting-associated protein 11 homolog (VSP11) | intracellular transport | unknown | [129] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietropaolo, V.; Prezioso, C.; Moens, U. Merkel Cell Polyomavirus and Merkel Cell Carcinoma. Cancers 2020, 12, 1774. https://doi.org/10.3390/cancers12071774
Pietropaolo V, Prezioso C, Moens U. Merkel Cell Polyomavirus and Merkel Cell Carcinoma. Cancers. 2020; 12(7):1774. https://doi.org/10.3390/cancers12071774
Chicago/Turabian StylePietropaolo, Valeria, Carla Prezioso, and Ugo Moens. 2020. "Merkel Cell Polyomavirus and Merkel Cell Carcinoma" Cancers 12, no. 7: 1774. https://doi.org/10.3390/cancers12071774
APA StylePietropaolo, V., Prezioso, C., & Moens, U. (2020). Merkel Cell Polyomavirus and Merkel Cell Carcinoma. Cancers, 12(7), 1774. https://doi.org/10.3390/cancers12071774