Cancer Immune Therapy for Philadelphia Chromosome-Negative Chronic Myeloproliferative Neoplasms


Abstract
1. Introduction
The Philadelphia Chromosome-Negative MPN
2. The Immune System in MPN
2.1. Chronic Inflammation in MPN
2.2. The Immune System in MPN
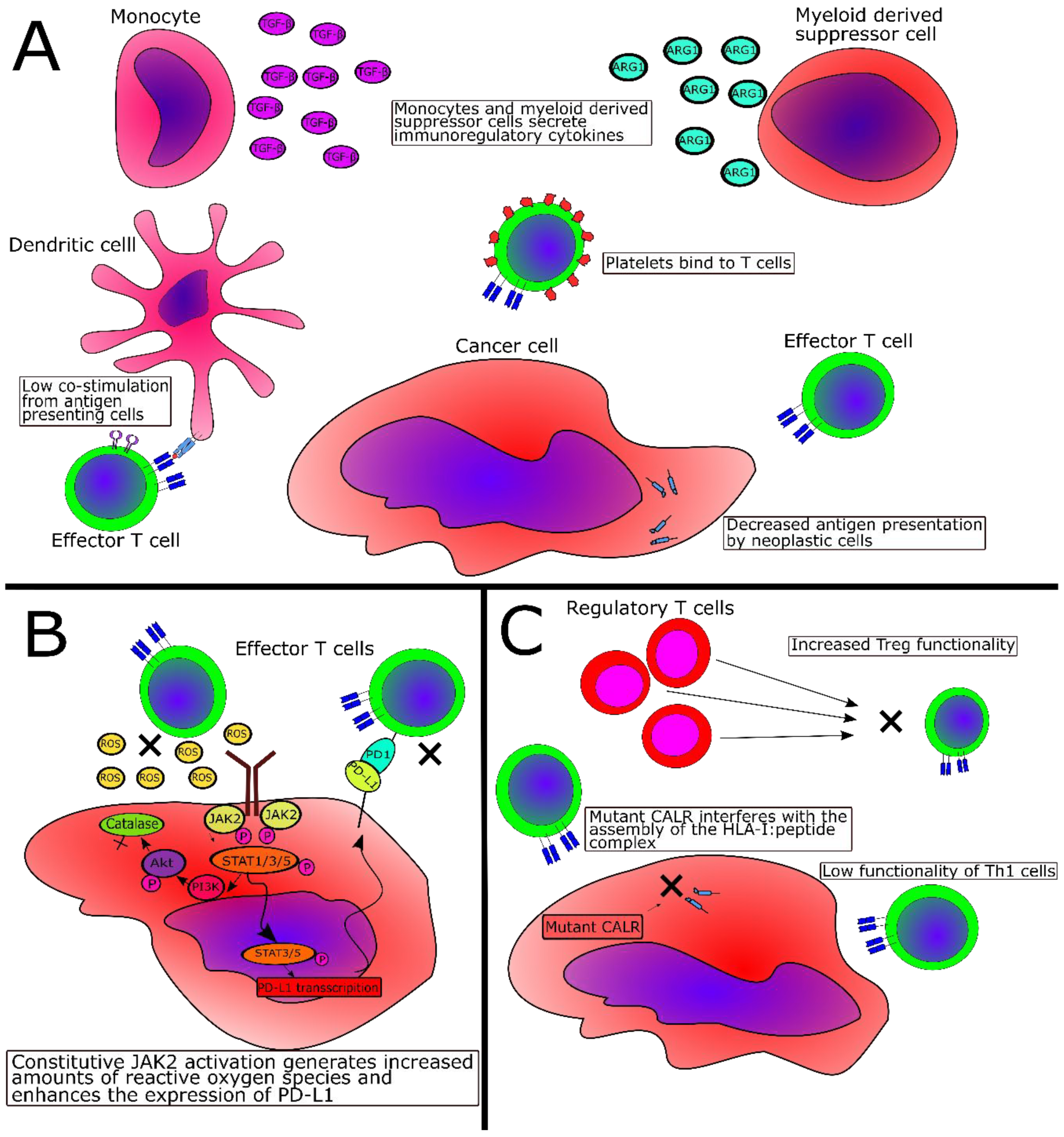
2.3. Immune-Subversive Mechanisms in MPN
2.4. Immunosuppressive Mechanisms Directly Mediated by JAK2V617F or CALR Exon 9 Mutations
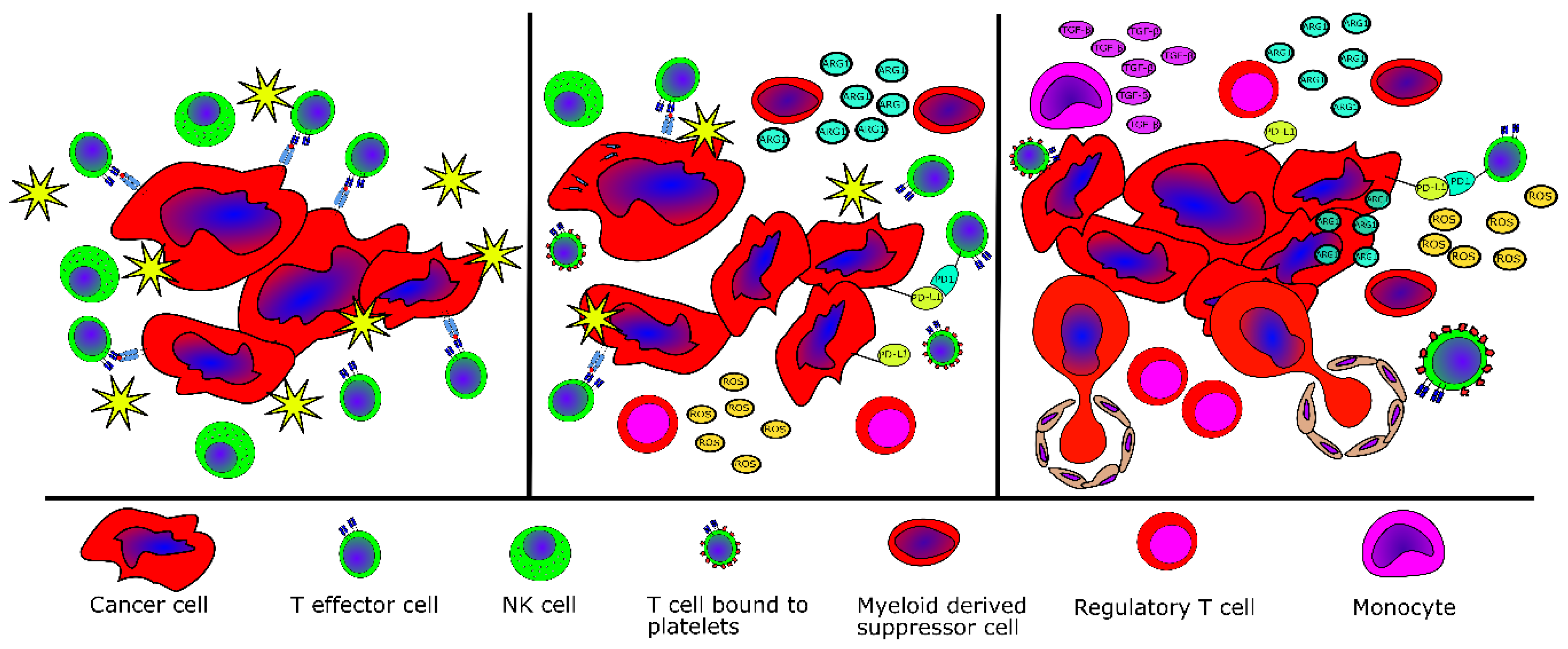
2.5. Evidence of Tumor Immne Escape in MPN
3. Potential Cancer Immune Therapeutic Strategies for MPN
3.1. The Role of IFN-α in MPN
3.2. Recognition of Neoplastic Cells Through Antigen Recognition by T cells
3.3. Cancer Immune Therapy for MPN—Targeting the Neoantigens
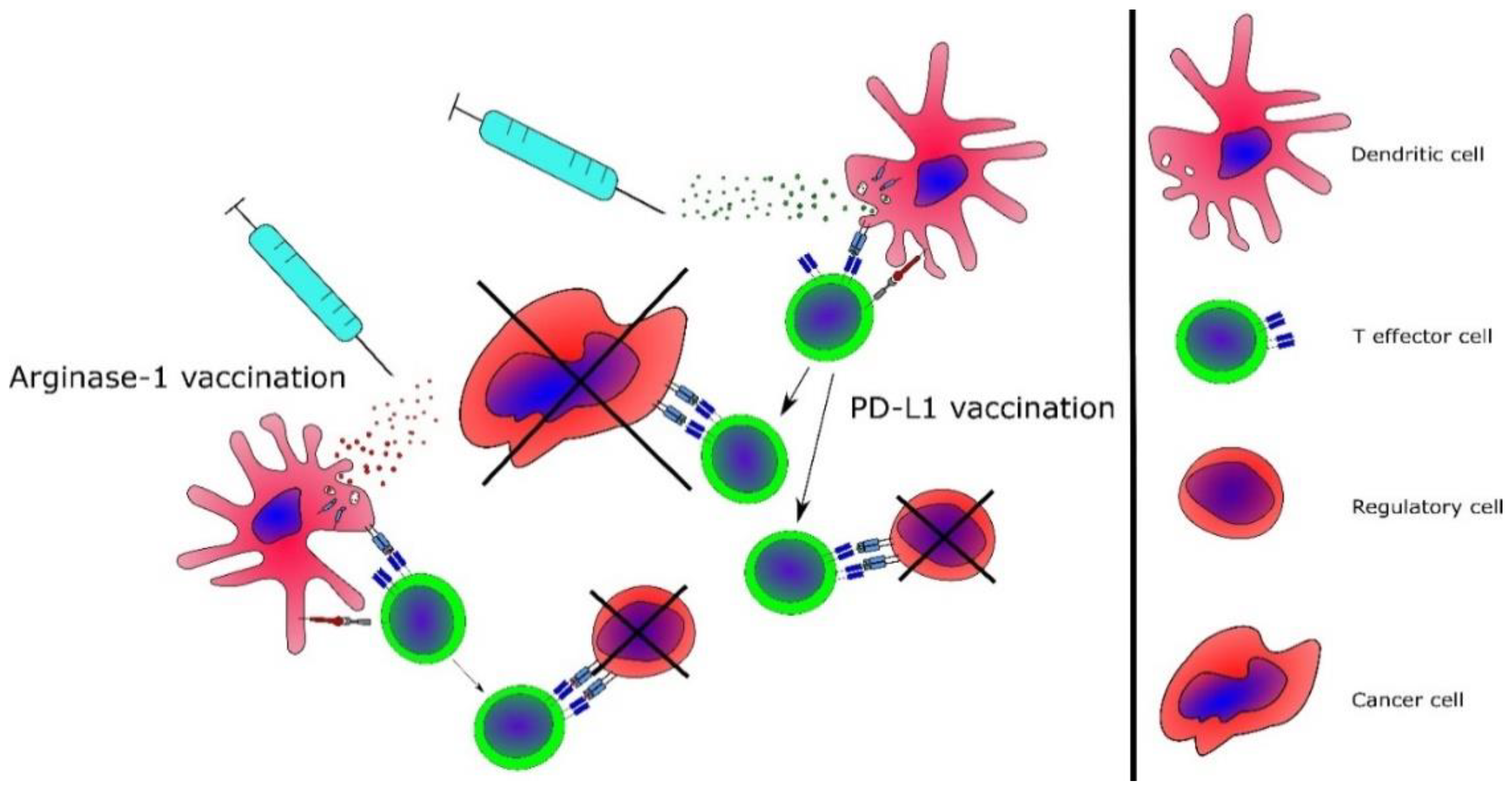
3.4. Targeting of Immunoregulatory Mechanisms in MPN
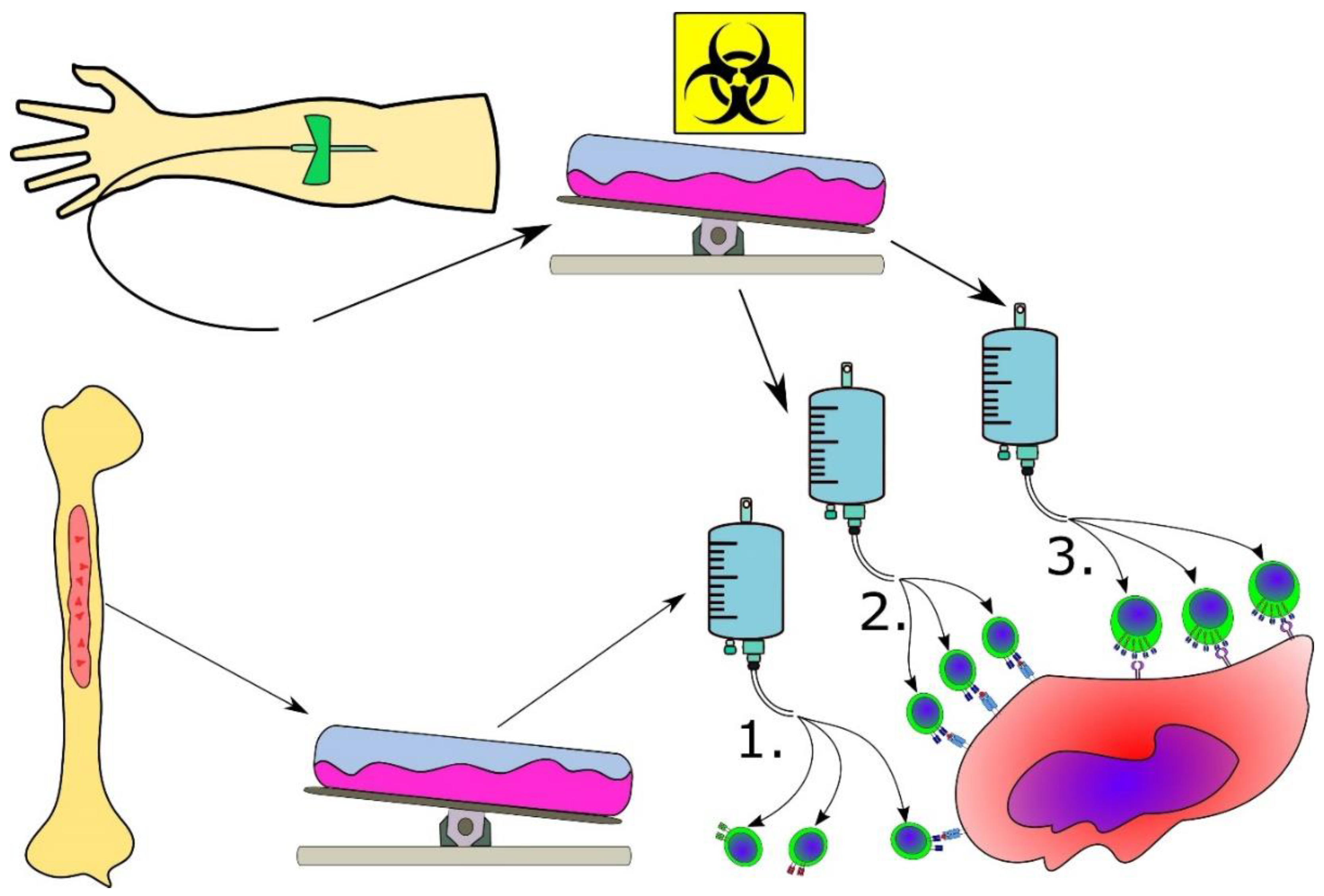
3.5. Advanced Cancer Immune Therapeutic Strategies for MPN
3.6. Combinatorial Strategies Involving Cancer Immune Therapy
4. Conclusions
Funding
Conflicts of Interest
References
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.P.J.; Green, A.A.R. The Myeloproliferative Disorders. N. Engl. J. Med. 2006, 355, 2452–2466. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Gilliland, D.G. Myeloproliferative disorders. Blood 2008, 112, 2190–2198. [Google Scholar] [CrossRef] [PubMed]
- Björkholm, M.; Derolf, Å.R.; Hultcrantz, M.; Kristinsson, S.Y.; Ekstrand, C.; Goldin, L.R.; Andreasson, B.; Birgegård, G.; Linder, O.; Malm, C.; et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J. Clin. Oncol. 2011, 29, 2410–2415. [Google Scholar] [CrossRef]
- Anderson, L.A.; McMullin, M.F. Epidemiology of MPN: What Do We Know? Curr. Hematol. Malig. Rep. 2014, 9, 340–349. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef]
- Geyer, H.L.; Scherber, R.M.; Dueck, A.C.; Kiladjian, J.J.; Xiao, Z.; Slot, S.; Zweegman, S.; Sackmann, F.; Fuentes, A.K.; Hernández-Maraver, D.; et al. Distinct clustering of symptomatic burden among myeloproliferative neoplasm patients: Retrospective assessment in 1470 patients. Blood 2014, 123, 3803–3810. [Google Scholar] [CrossRef]
- Emanuel, R.M.; Dueck, A.C.; Geyer, H.L.; Kiladjian, J.J.; Slot, S.; Zweegman, S.; Te Boekhorst, P.A.W.; Commandeur, S.; Schouten, H.C.; Sackmann, F.; et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: Prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J. Clin. Oncol. 2012, 30, 4098–4103. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.J.; Tichelli, A.; Cazzola, M.; Skoda, R.C.R. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.I.; Marty, C.; Gryshkova, V.; Defour, J.P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef]
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 2016, 127, 1307–1316. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Baral, A.J.; Beeson, D.; Rivera, J.F.; Ko, A.; Florescu, N.; Birrane, G.; Chen, E.; Mullally, A. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood 2018, 131, 782–786. [Google Scholar] [CrossRef]
- Hansen, I.O.; Sørensen, A.L.; Hasselbalch, H.C. Second malignancies in hydroxyurea and interferon-treated Philadelphia-negative myeloproliferative neoplasms. Eur. J. Haematol. 2017, 98, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.T.; Kiladjian, J.-J.; Hasselbalch, H.C. Interferon and the treatment of polycythaemia vera, essential thrombocytemia and myelofibrosis. Exp. Hematol. 2013, 6, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.S.; Møller, M.B.; de Stricker, K.; Nørgaard, P.; Samuelsson, J.; Marcher, C.; Andersen, M.T.; Bjerrum, O.W.; Hasselbalch, H.C. Minimal residual disease and normalization of the bone marrow after long-term treatment with alpha-interferon2b in polycythemia vera. A report on molecular response patterns in seven patients in sustained complete hematological remission. Hematology 2009, 14, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.-J.; Giraudier, S.; Cassinat, B. Interferon-alpha for the therapy of myeloproliferative neoplasms: Targeting the malignant clone. Leukemia 2015, 30, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Ostojic, A.; Vrhovac, R.; Verstovsek, S. Ruxolitinib: A new JAK1/2 inhibitor that offers promising options for treatment of myelofibrosis. Future Oncol. 2011, 7, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Leroy, E.; Gilles, L.; Marty, C.; Plo, I.; Constantinescu, S.N. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Research 2018, 7, 82. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.-J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef]
- Cervantes, F.; Pereira, A. Does ruxolitinib prolong the survival of patients with myelofibrosis? Blood 2017, 129, 832–838. [Google Scholar] [CrossRef]
- Deeg, H.J.; Gooley, T.A.; Flowers, M.E.D.; Sale, G.E.; Slattery, J.T.; Anasetti, C.; Chauncey, T.R.; Doney, K.; Georges, G.E.; Kiem, H.; et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Transplantation 2003, 102, 3912–3918. [Google Scholar] [CrossRef]
- Barosi, G. An Immune Dysregulation in MPN. Curr. Hematol. Malig. Rep. 2014, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: Clinical implications. Exp. Hematol. 2014, 42, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Bjørn, M.E.; Andersen, C.L.; Jensen, M.K.; Hasselbalch, H.C. Circulating YKL-40 in myelofibrosis a potential novel biomarker of disease activity and the inflammatory state. Eur. J. Haematol. 2014, 93, 224–228. [Google Scholar] [CrossRef]
- Nina, A.Q.; Øbro, F.; Grinfeld, J.; Belmonte, M.; Irvine, M.; Shepherd, M.S.; Rao, T.N.; Karow, A.; Riedel, L.M.; Harris, O.B.; et al. Longitudinal Cytokine Pro fi ling Identi fi es GRO- a and EGF as Potential Biomarkers of Disease Progression in Essential Thrombocythemia. HemaSphere 2020, 4, e371. [Google Scholar]
- Elli, E.M.; Baratè, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on the impact of JAK-inhibitor therapy upon inflammation-mediated comorbidities in myelofibrosis and related neoplasms. Expert Rev. Hematol. 2014, 7, 203–216. [Google Scholar] [CrossRef]
- Kristinsson, S.Y.; Landgren, O.; Samuelsson, J.; Björkholm, M.; Goldin, L.R. Autoimmunity and the risk of myeloproliferative neoplasms. Haematologica 2010, 95, 1216–1220. [Google Scholar] [CrossRef]
- Bak, M.; Sørensen, T.L.; Flachs, E.M.; Zwisler, A.D.; Juel, K.; Frederiksen, H.; Hasselbalch, H.C. Age-related macular degeneration in patients with chronic myeloproliferative neoplasms. JAMA Ophthalmol. 2017, 135, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z.; Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Hernández-Boluda, J.C.; Villamor, N.; Serra, A.; Montserrat, E. Assessment of peripheral blood lymphocyte subsets in idiopathic myelofibrosis. Eur. J. Haematol. 2000, 65, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.A.; Verstovsek, S.; Dingli, D.; Schwager, S.M.; Mesa, R.A.; Li, C.Y.; Tefferi, A. Monocytosis is an adverse prognostic factor for survival in younger patients with primary myelofibrosis. Leuk. Res. 2007, 31, 1503–1509. [Google Scholar] [CrossRef]
- Barraco, D.; Cerquozzi, S.; Gangat, N.; Patnaik, M.M.; Lasho, T.; Finke, C.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. Monocytosis in polycythemia vera: Clinical and molecular correlates. Am. J. Hematol. 2017, 92, 640–645. [Google Scholar] [CrossRef]
- Riley, C.H.; Hansen, M.; Brimnes, M.K.; Hasselbalch, H.C.; Bjerrum, O.W.; Straten, P.T.; Svane, I.M.; Jensen, M.K. Expansion of circulating CD56(bright) natural killer cells in patients with JAK2-positive chronic myeloproliferative neoplasms during treatment with interferon-α. Eur. J. Haematol. 2015, 94, 227–234. [Google Scholar] [CrossRef]
- Shakil, S.; Chen, C.; Surahio, A.; Mirza, M.; Su, M.; Sun, Y.; Wang, J.-C. T Regulator Cells (Treg) In Patients with Myelofibrosis. ASH Annu. Meet. Abstr. 2010, 116, 5051. [Google Scholar] [CrossRef]
- Riley, C.H.; Jensen, M.K.; Brimnes, M.K.; Hasselbalch, H.C.; Bjerrum, O.W.; Straten, P.T.; Svane, I.M. Increase in circulating CD4+CD25+Foxp3+ T cells in patients with Philadelphia-negative chronic myeloproliferative neoplasms during treatment with IFN-α. Blood 2011, 118, 2170–2173. [Google Scholar] [CrossRef][Green Version]
- Keohane, C.; Kordasti, S.; Seidl, T.; Perez Abellan, P.; Thomas, N.S.B.; Harrison, C.N.; Mclornan, D.P.; Mufti, G.J. JAK inhibition induces silencing of T Helper cytokine secretion and a profound reduction in T regulatory cells. Br. J. Haematol. 2015, 171, 60–73. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-Derived Suppressor Cells: Linking Inflammation and Cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Kundra, A.; Andrei, M.; Baptiste, S.; Chen, C.; Wong, C. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leuk. Res. 2016, 43, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Baptiste, S.; Chen, C.; Sindhu, H.; Wang, J.-C. Programmed Cell Death Receptor (PD-1), PD-1 Ligand (PD-L1) Expression and Myeloid Derived Suppressor Cells (MDSC) In Myeloid Neoplasms Implicate The Mechanism Of IMiD Treatment Of Myelofibrosis. Blood 2013, 122, 2837. [Google Scholar]
- Larsen, T.S.; Christensen, J.H.; Hasselbalch, H.C.; Pallisgaard, N. The JAK2 V617F mutation involves B- and T-lymphocyte lineages in a subgroup of patients with Philadelphia-chromosome negative chronic myeloproliferative disorders. Br. J. Haematol. 2007, 136, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, L.; Holmström, M.O.; Cordua, S.; Thomassen, M.; Kruse, T.A.; Andersen, M.H.; Svane, I.M.; Pallisgaard, N.; Skov, V.; Hasselbalch, H.C. Sorted Peripheral Blood Cells Identify CALR Mutations in B- and T-lymphocytes. Leuk. Lymphoma 2017, 59, 973–977. [Google Scholar] [CrossRef]
- Raghavan, M.; Wijeyesakere, S.J.; Peters, L.R.; Del Cid, N. Calreticulin in the immune system: Ins and outs. Trends Immunol. 2013, 34, 13–21. [Google Scholar] [CrossRef]
- Ward, A.C.; Touw, I.; Yoshimura, A. Review article The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood J. Am. Soc. Hematol. 2016, 95, 19–30. [Google Scholar]
- Dupuis, B.M.; Schaerer, E.; Krause, K.; Tschopp, J. The Calcium-binding Protein Calreticulin Is a Major Constituent of Lytic Granules in Cytolytic T Lymphocytes. J. Exp. Med. 1993, 177, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sipione, S.; Ewen, C.; Shostak, I.; Bleackley, R.C.; Sipione, S.; Ewen, C.; Shostak, I.; Michalak, M.; Bleackley, R.C. Impaired cytolytic activity in calreticulin-deficient CTLs. J. Immunol. 2005, 174, 3212–3219. [Google Scholar] [CrossRef] [PubMed]
- Andrin, C.; Pinkoski, M.J.; Burns, K.; Atkinson, E.A.; Krahenbuhl, O.; Hudig, D.; Fraser, S.A.; Winkler, U.; Tschopp, J.; Opas, M.; et al. Interaction between a Ca2+-binding protein calreticulin and perforin, a component of the cytotoxic T-cell granules. Biochemistry 1998, 37, 10386–10394. [Google Scholar] [CrossRef] [PubMed]
- Porcellini, S.; Traggiai, E.; Schenk, U.; Ferrera, D.; Matteoli, M.; Lanzavecchia, A.; Michalak, M.; Grassi, F. Regulation of peripheral T cell activation by calreticulin. J. Exp. Med. 2006, 203, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Rameshwar, P.; Denny, T.N.; Stein, D.; Gascón, P.; Rameshwar, P.; Denny, T.N.; Stein, D.; Gascon, P. Monocyte adhesion in patients with bone marrow fibrosis is required for the production of fibrogenic cytokines. Potential role for interleukin-1 and TGF-beta. J. Immunol. 1994, 153, 2819–2830. [Google Scholar] [PubMed]
- Cardoso, E.M.; Esgalhado, A.J.; Patrão, L.; Santos, M.; Neves, V.P.; Martinez, J. Distinctive CD8+ T cell and MHC class I signatures in polycythemia vera patients. Ann. Hematol. 2018, 97, 1563–1575. [Google Scholar] [CrossRef]
- Skov, V.; Riley, C.H.; Thomassen, M.; Larsen, T.S.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Whole blood transcriptional profiling reveals significant down-regulation of human leukocyte antigen class I and II genes in essential thrombocythemia, polycythemia vera and myelofibrosis. Leuk. Lymphoma 2013, 54, 2269–2273. [Google Scholar] [CrossRef]
- Skov, V.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C.; Larsen, T.S. Gene expression profiling with principal component analysis depicts the biological continuum from essential thrombocythemia over polycythemia vera to myelofibrosis. Exp. Hematol. 2012, 40, 771–780.e19. [Google Scholar] [CrossRef]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: Identification of deregulated genes of significance for inflammation and immune surveillance. Leuk. Res. 2012, 36, 1387–1392. [Google Scholar] [CrossRef]
- Romano, M.; Sollazzo, D.; Trabanelli, S.; Barone, M.; Polverelli, N.; Perricone, M.; Forte, D.; Luatti, S.; Cavo, M.; Vianelli, N.; et al. Mutations in JAK2 and Calreticulin genes are associated with specific alterations of the immune system in myelofibrosis. Oncoimmunology 2017, 6, e1345402. [Google Scholar] [CrossRef]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; Sullivan, D.O.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Boerries, M.; Busch, H.; et al. Oncogenic JAK2 V617F causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaam7729. [Google Scholar] [CrossRef]
- Wang, J.C.; Chen, C.; Kundra, A.; Kodali, S.; Pandey, A.; Wong, C.; Cheung, T.; Gotlieb, V.; Joseph, G.; Tribie, S. Programmed Cell Death Receptor (PD-1) Ligand (PD-L1) expression in Philadelphia chromosome-negative myeloproliferative neoplasms. Leuk. Res. 2019, 79, 52–59. [Google Scholar] [CrossRef]
- Bozkus, C.C.; Roudko, V.; Finnigan, J.P.; Mascarenhas, J.; Hoffman, R.; Iancu-rubin, C.; Bhardwaj, N. Immune Checkpoint Blockade Enhances Shared Neoantigen-Induced T-cell Immunity Directed against Mutated Calreticulin in Myeloproliferative Neoplasms. Cancer Discov. 2019, 9, 1192–1207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lv, Z.; Yu, H.; Zhu, J. The clinicopathological and prognostic role of thrombocytosis in patients with cancer: A meta-analysis. Oncol. Lett. 2017, 13, 5002–5008. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. The platelet-cancer loop in myeloproliferative cancer. Is thrombocythemia an enhancer of cancer invasiveness and metastasis in essential thrombocythemia, polycythemia vera and myelofibrosis? Leuk. Res. 2014, 38, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Rachidi, S.; Metelli, A.; Riesenberg, B.; Wu, B.X.; Nelson, M.H.; Wallace, C.; Paulos, C.M.; Rubinstein, M.P.; Garrett-Mayer, E.; Hennig, M.; et al. Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci. Immunol. 2017, 2, eaai7911. [Google Scholar] [CrossRef]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.-P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.-L.; Plo, I. A role for reactive oxygen species in JAK2V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef]
- Belikov, A.V.; Schraven, B.; Simeoni, L. T cells and reactive oxygen species. J. Biomed. Sci. 2015, 22, 85. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid. Med. Cell. Longev. 2016, 2016, 11–16. [Google Scholar] [CrossRef]
- Arshad, N.; Cresswell, P. Tumor-associated calreticulin variants functionally compromise the peptide loading complex and impair its recruitment of MHC-I. J. Biol. Chem. 2018, 293, 9555–9569. [Google Scholar] [CrossRef]
- Sollazzo, D.; Forte, D.; Polverelli, N.; Perricone, M.; Romano, M.; Luatti, S.; Vianelli, N.; Cavo, M.; Palandri, F.; Catani, L. Circulating Calreticulin Is Increased in Myelofibrosis: Correlation with Interleukin-6 Plasma Levels, Bone Marrow Fibrosis, and Splenomegaly. Mediat. Inflamm. 2016, 2016, 5860657. [Google Scholar] [CrossRef]
- Han, L.; Schubert, C.; Köhler, J.; Schemionek, M.; Isfort, S.; Brümmendorf, T.H.; Koschmieder, S.; Chatain, N. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J. Hematol. Oncol. 2016, 9, 45. [Google Scholar] [CrossRef]
- Garbati, M.R.; Welgan, C.A.; Landefeld, S.H.; Newell, L.F.; Agarwal, A.; Dunlap, J.B.; Chourasia, T.K.; Lee, H.; Elferich, J.; Traer, E.; et al. Mutant calreticulin-expressing cells induce monocyte hyperreactivity through a paracrine mechanism. Am. J. Hematol. 2016, 91, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, L.; Loos, F.; Marty, C.; Xie, W.; Martins, I.; Lachkar, S.; Qu, B.; Waeckel-Énée, E.; Plo, I.; et al. Immunosuppression by Mutated Calreticulin Released from Malignant Cells Article Immunosuppression by Mutated Calreticulin Released from Malignant Cells. Mol. Cell 2020, 77, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Holmström, M.O.; Riley, C.H.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. The CALR exon 9 mutations are shared neoantigens in patients with CALR mutant chronic myeloproliferative neoplasms. Leukemia 2016, 30, 2413–2416. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, M.O.; Martinenaite, E.; Ahmad, S.M.; Met, O.; Friese, C.; Kjaer, L.; Riley, C.H.; thor Straten, P.; Svane, I.M.; Hasselbalch, H.C.; et al. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia 2018, 32, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Holmström, M.O.; Ahmad, S.M.; Klausen, U.; Bendtsen, S.K.; Martinenaite, E.; Riley, C.H.; Svane, I.M.; Kjær, L.; Skov, V.; Ellervik, C.; et al. High frequencies of circulating memory T cells specific for calreticulin exon 9 mutations in healthy individuals. Blood Cancer J. 2019, 9, 8. [Google Scholar] [CrossRef]
- Tubb, V.M.; Schrikkema, D.S.; Croft, N.P.; Purcell, A.W.; Linnemann, C.; Freriks, M.R.; Chen, F.; Long, H.M.; Lee, S.P.; Bendle, G.M. Isolation of T cell receptors targeting recurrent neoantigens in hematological malignancies. J. Immunother. Cancer 2018, 6, 70. [Google Scholar] [CrossRef]
- Cordua, S.; Kjaer, L.; Skov, V.; Pallisgaard, N.; Hasselbalch, H.C.; Ellervik, C. Prevalence and phenotypes of JAK2 V617F and Calreticulin mutations in a Danish general population. Blood 2019, 134, 469–479. [Google Scholar] [CrossRef]
- Holmström, M.O.; Cordua, S.; Skov, V.; Kjær, L.; Pallisgaard, N.; Ellervik, C.; Hasselbalch, H.C.; Hald Andersen, M. Evidence of immune elimination, immuno-editing and immune escape in patients with hematological cancer. Cancer Immunol. Immunother. 2020, 69, 315–324. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immuno- surveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, H.; Knutsen, H.; Holmberg, E.; Andréasson, B. Increased incidence of another cancer in myeloproliferative neoplasms patients at the time of diagnosis. Eur. J. Haematol. 2014, 94, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Landtblom, A.R.; Bower, H.; Andersson, T.M.-L.; Dickman, P.W.; Samuelsson, J.; Björkholm, M.; Kristinsson, S.Y.; Hultcrantz, M. Second malignancies in patients with myeloproliferative neoplasms: A population-based cohort study of 9379 patients. Leukemia 2018, 32, 2203–2210. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.; Farkas, D.K.; Christiansen, C.F.; Hasselbalch, H.C.; Sørensen, H.T.; Sorensen, H.T. Chronic myeloproliferative neoplasms and subsequent cancer risk: A Danish population-based cohort study. Blood 2011, 118, 6515–6520. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef]
- Minn, A.J. Interferons and the Immunogenic Effects of Cancer Therapy. Trends Immunol. 2015, 36, 725–737. [Google Scholar] [CrossRef]
- Schiavoni, G.; Mattei, F.; Gabriele, L. Type I interferons as stimulators of DC-mediated cross-priming: Impact on anti-tumor response. Front. Immunol. 2013, 4, 483. [Google Scholar] [CrossRef]
- Bracci, L.; Proietti, E.; Belardelli, F. IFN-alpha and novel strategies of combination therapy for cancer. Ann. N. Y. Acad. Sci. 2007, 1112, 256–268. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Larsen, T.S.; Riley, C.H.; Jensen, M.K.; Kiladjian, J.-J. Interferon-alpha in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Status and perspectives. Curr. Drug Targets 2011, 12, 392–419. [Google Scholar] [CrossRef]
- Cassinat, B.; Verger, E.; Kiladjian, J.-J. Interferon Alfa Therapy in CALR-Mutated Essential Thrombocythemia. N. Engl. J. Med. 2014, 371, 188–189. [Google Scholar] [CrossRef] [PubMed]
- Kjær, L.; Cordua, S.; Holmström, M.O.; Thomassen, M.; Kruse, T.A.; Pallisgaard, N.; Larsen, T.S.; de Stricker, K.; Skov, V.; Hasselbalch, H.C. Differential Dynamics of CALR Mutant Allele Burden in Myeloproliferative Neoplasms during Interferon Alfa Treatment. PLoS ONE 2016, 11, e0165336. [Google Scholar] [CrossRef] [PubMed]
- Riley, C.H.; Brimnes, M.K.; Hansen, M.; Jensen, M.K.; Hasselbalch, H.C.; Kjaer, L.; Straten, P.T.; Svane, I.M. Interferon-α induces marked alterations in circulating regulatory T cells, NK cell subsets, and dendritic cells in patients with JAK2V617F-positive essential thrombocythemia and polycythemia vera. Eur. J. Haematol. 2016, 97, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Skov, V.; Riley, C.H.; Thomassen, M.; Kjær, L.; Larsen, T.S.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. The impact of interferon-alpha2 on HLA genes in patients with polycythemia vera and related neoplasms. Leuk. Lymphoma 2016, 58, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Kovacsovics-Bankowski, M.; Kelley, T.W.; Efimova, O.; Kim, S.J.; Wilson, A.; Swierczek, S.; Prchal, J. Changes in peripheral blood lymphocytes in polycythemia vera and essential thrombocythemia patients treated with pegylated-interferon alpha and correlation with JAK2 V617F allelic burden. Exp. Hematol. Oncol. 2015, 5, 28. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Holmström, M.O.; Hjortsø, M.D.; Ahmad, S.M.; Met, Ö.; Martinenaite, E.; Riley, C.; Straten, P.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. The JAK2V617F mutation is a target for specific T cells in the JAK2V617F-positive myeloproliferative neoplasms. Leukemia 2017, 31, 495–498. [Google Scholar] [CrossRef]
- Schischlik, F.; Jäger, R.; Rosebrock, F.; Hug, E.; Schuster, M.K.; Holly, R.; Fuchs, E.; Milosevic Feenstra, J.D.; Bogner, E.; Gisslinger, B.; et al. Mutational Landscape of the Transcriptome Offers Putative Targets for Immunotherapy of Myeloproliferative Neoplasms. Blood 2019, 134, 199–210. [Google Scholar] [CrossRef]
- Posthuma, E.F.; Falkenburg, J.H.; Apperley, J.F.; Gratwohl, A.; Roosnek, E.; Hertenstein, B.; Schipper, R.F.; Schreuder, G.M.; D’Amaro, J.; Oudshoorn, M.; et al. HLA-B8 and HLA-A3 coexpressed with HLA-B8 are associated with a reduced risk of the development of chronic myeloid leukemia. The Chronic Leukemia Working Party of the EBMT. Blood 1999, 93, 3863–3865. [Google Scholar]
- Kuželová, K.; Brodská, B.; Fuchs, O.; Dobrovolná, M.; Soukup, P.; Cetkovský, P. Altered HLA class I profile associated with type A/D nucleophosmin mutation points to possible anti-nucleophosmin immune response in acute myeloid leukemia. PLoS ONE 2015, 10, e0127637. [Google Scholar] [CrossRef] [PubMed]
- Rusakiewicz, S.; Madrigal, A.; Travers, P.; Dodi, A.I. BCR/ABL-specific CD8+ T cells can be detected from CML patients, but are only expanded from healthy donors. Cancer Immunol. Immunother. 2009, 58, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Greiner, J.; Ono, Y.; Hofmann, S.; Schmitt, A.; Mehring, E.; Götz, M.; Guillaume, P.; Döhner, K.; Mytilineos, J.; Döhner, H.; et al. Mutated regions of nucleophosmin 1 elicit both CD4+ and CD8+ T-cell responses in patients with acute myeloid leukemia. Blood 2016, 97, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Ribas, A. Adaptive immune resistance: How cancer protects from immune attack. Cancer Discov. 2015, 5, 915–919. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2014, 372, 311–319. [Google Scholar] [CrossRef]
- Goodman, A.; Patel, S.P.; Kurzrock, R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat. Rev. Clin. Oncol. 2017, 14, 203–220. [Google Scholar] [CrossRef]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Rie Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef]
- Schnorfeil, F.M.; Lichtenegger, F.S.; Emmerig, K.; Schlueter, M.; Neitz, J.S.; Draenert, R.; Hiddemann, W.; Subklewe, M. T cells are functionally not impaired in AML: Increased PD-1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment. J. Hematol. Oncol. 2015, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.-R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Ørskov, A.D.; Treppendahl, M.B.; Skovbo, A.; Holm, M.S.; Friis, L.S.; Hokland, M.; Grønbæk, K. Hypomethylation and up-regulation of PD-1in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget 2015, 6, 9612–9626. [Google Scholar] [CrossRef] [PubMed]
- Ravandi-Kashani, F.; Garcia-Manero, G.; Ning, J.; Alhamal, Z.; DiNardo, C.D.; Pierce, S.A.; Pemmaraju, N.; Patel, K.P.; Blando, J.; Alfayez, M.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Non-randomized, Open-label, Phase 2 Study. Cancer Discov. 2018, 9, 370–383. [Google Scholar]
- Spiers, L.; Coupe, N.; Payne, M. Toxicities associated with checkpoint inhibitors-An overview. Rheumatology (UK) 2019, 58, vii7–vii16. [Google Scholar] [CrossRef]
- Munir, S.; Larsen, S.K.; Iversen, T.Z.; Donia, M.; Klausen, T.W.; Svane, I.M.; thor Straten, P.; Andersen, M.H. Natural CD4+ T-cell responses against indoleamine 2,3-dioxygenase. PLoS ONE 2012, 7, e34568. [Google Scholar] [CrossRef]
- Sørensen, R.B.; Køllgaard, T.; Andersen, R.S.; Van Den Berg, J.H.; Svane, I.M.; Straten, P.T.; Andersen, M.H. Spontaneous cytotoxic T-cell reactivity against indoleamine 2,3-dioxygenase-2. Cancer Res. 2011, 71, 2038–2044. [Google Scholar] [CrossRef]
- Munir, S.; Andersen, G.H.; Met, Ö.; Donia, M.; Frøsig, T.M.; Larsen, S.K.; Klausen, T.W.; Svane, I.M.; Andersen, M.H. HLA-restricted CTL that are specific for the immune checkpoint ligand PD-L1 occur with high frequency in cancer patients. Cancer Res. 2013, 73, 1764–1776. [Google Scholar] [CrossRef]
- Munir, S.; Andersen, G.H.; Svane, I.M.; Andersen, M.H. The immune checkpoint regulator PD-L1 is a specific target for naturally occurring CD4+ T cells. Oncoimmunology 2013, 2, e23991. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Svane, I.M.; Andersen, M.H. The stimulation of PD-L1-specific cytotoxic T lymphocytes can both directly and indirectly enhance antileukemic immunity. Blood Cancer J. 2014, 4, e230. [Google Scholar] [CrossRef]
- Munir Ahmad, S.; Martinenaite, E.; Hansen, M.; Junker, N.; Borch, T.H.; Met, Ö.; Donia, M.; Svane, I.M.; Andersen, M.H. PD-L1 peptide co-stimulation increases immunogenicity of a dendritic cell-based cancer vaccine. Oncoimmunology 2016, 5, e1202391. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H. Anti-regulatory T cells. Semin. Immunopathol. 2017, 39, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Holmström, M.O.; Riley, C.H.; Skov, V.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. Spontaneous T-cell responses against the immune check point programmed-death-ligand 1 (PD-L1) in patients with chronic myeloproliferative neoplasms correlate with disease stage and clinical response. Oncoimmunology 2018, 7, e1433521. [Google Scholar] [CrossRef] [PubMed]
- Aaboe-Jørgensen, M.; Holmström, M.O.; Martinenaite, E.; Riley, C.H.; Hasselbalch, H.C.; Hald Andersen, M. Spontaneous T-cell responses against Arginase-1 in chronic myeloproliferative neoplasms relative to disease stage and type of driver mutation. Oncoimmunology 2018, 7, e1468957. [Google Scholar] [CrossRef]
- Qazilbash, M.H.; Wieder, E.; Thall, P.F.; Wang, X.; Rios, R.; Lu, S.; Kanodia, S.; Ruisaard, K.E.; Giralt, S.A.; Estey, E.H.; et al. PR1 peptide vaccine induces specific immunity with clinical responses in myeloid malignancies. Leukemia 2017, 31, 697–704. [Google Scholar] [CrossRef]
- Melief, C.J.M.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S. a Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients With Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results From a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Blanc, C.; Granier, C.; Saldmann, A.; Tanchot, C.; Tartour, E.; Tran, T. Therapeutic cancer vaccine: Building the future from lessons of the past. Semin. Immunopathol. 2018, 41, 69–85. [Google Scholar]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; Van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 2007, 220, 22–34. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Escape Mechanism | MPN Type | Reference |
|---|---|---|
| Fewer effector T cells in peripheral blood | PMF | [40] |
| Fewer NK cells in peripheral blood | All MPN | [43] |
| Elevated numbers of MDSC in peripheral blood | All MPN | [47,48] |
| Higher inhibitory potential of MDSC in peripheral blood | All MPN | [47] |
| Elevated TGFβ production by bone-marrow-derived monocytes | PMF | [57] |
| Lower levels of naïve T-cells and higher levels of terminally differentiated effector T-cells | PV | [58] |
| Lower levels HLA-I on monocytes | PV | [58] |
| Lower levels of HLA-I on mutant cells | CALR-mutant MPN | [72] |
| Deregulation of genes related to antigen processing and activation as well as immune cell activation and inflammation | All MPN | [59,60,61] |
| Lower number of myeloid dendritic cells and lower priming potential of myeloid dendritic cells | PMF (lower number of myeloid dendritic cells), all MPN | [62] |
| Reduced Th1 compartment and more suppressive regulatory T-cells | CALR-mutant MPN | [62] |
| Increased expression of PD-L1 on JAK2V617F+ cells | JAK2V617F+ MPN | [63] |
| Increased expression of PD-1 and PD-L1 on CD4+ and CD8+ T-cells, monocytes and CD34+ hematopoietic stem cells | All MPN | [64] |
| Increased expression of PD-1 and CTLA-4 on T cells | All MPN | [65] |
| Overproduction of reactive oxygen species | JAK2V617F+ MPN | [69] |
| Overproduction of IL-10 and additional immunosuppressive cytokines by monocytes | CALR-mutant MPN | [75] |
| Abrogation of immunogenic cell death | CALR-mutant MPN | [76] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holmström, M.O.; Hasselbalch, H.C.; Andersen, M.H. Cancer Immune Therapy for Philadelphia Chromosome-Negative Chronic Myeloproliferative Neoplasms. Cancers 2020, 12, 1763. https://doi.org/10.3390/cancers12071763
Holmström MO, Hasselbalch HC, Andersen MH. Cancer Immune Therapy for Philadelphia Chromosome-Negative Chronic Myeloproliferative Neoplasms. Cancers. 2020; 12(7):1763. https://doi.org/10.3390/cancers12071763
Chicago/Turabian StyleHolmström, Morten Orebo, Hans Carl Hasselbalch, and Mads Hald Andersen. 2020. "Cancer Immune Therapy for Philadelphia Chromosome-Negative Chronic Myeloproliferative Neoplasms" Cancers 12, no. 7: 1763. https://doi.org/10.3390/cancers12071763
APA StyleHolmström, M. O., Hasselbalch, H. C., & Andersen, M. H. (2020). Cancer Immune Therapy for Philadelphia Chromosome-Negative Chronic Myeloproliferative Neoplasms. Cancers, 12(7), 1763. https://doi.org/10.3390/cancers12071763