Tebentafusp: T Cell Redirection for the Treatment of Metastatic Uveal Melanoma


Abstract
1. Introduction
2. UM Biology
3. Metastatic Disease
4. Immunotherapy and UM
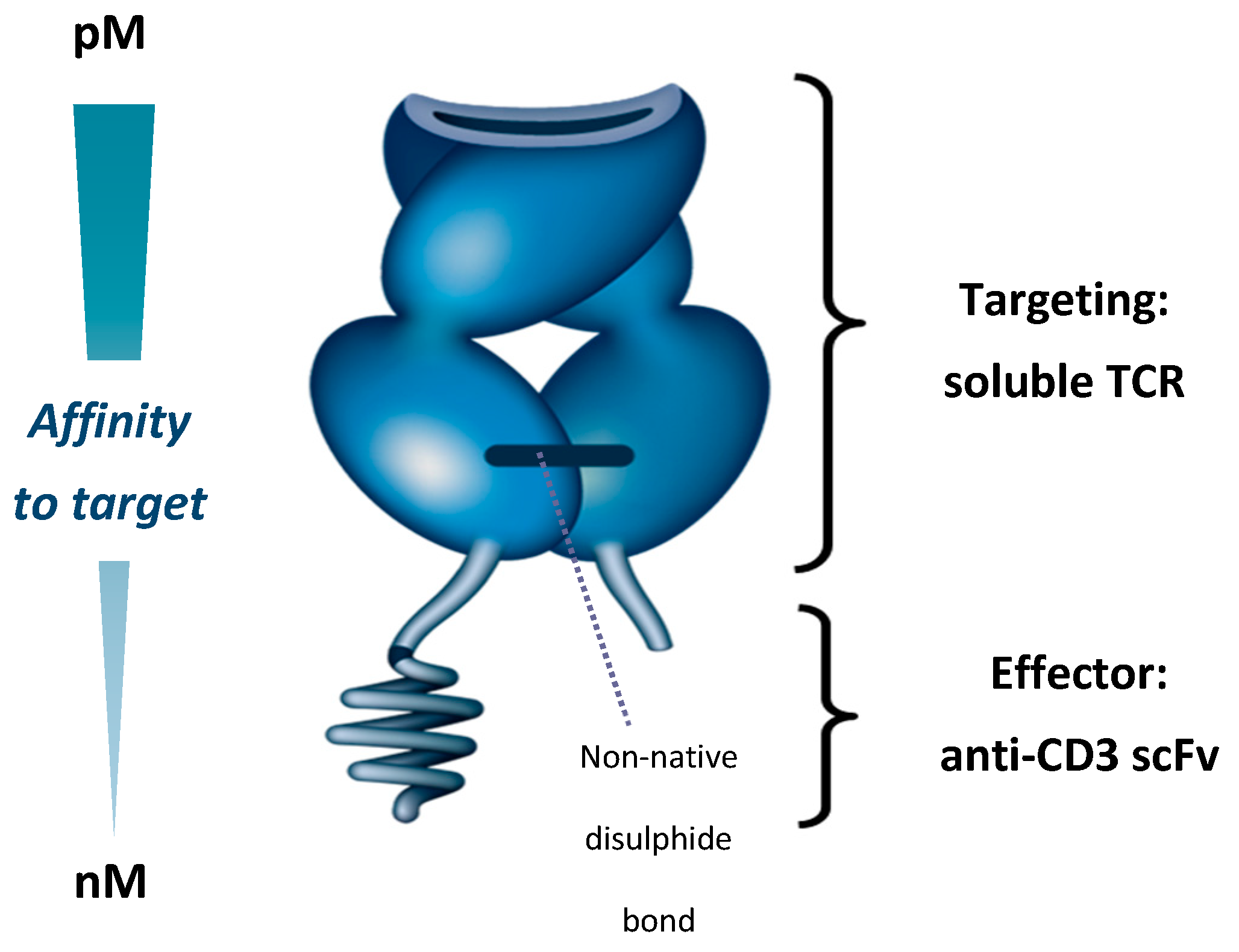
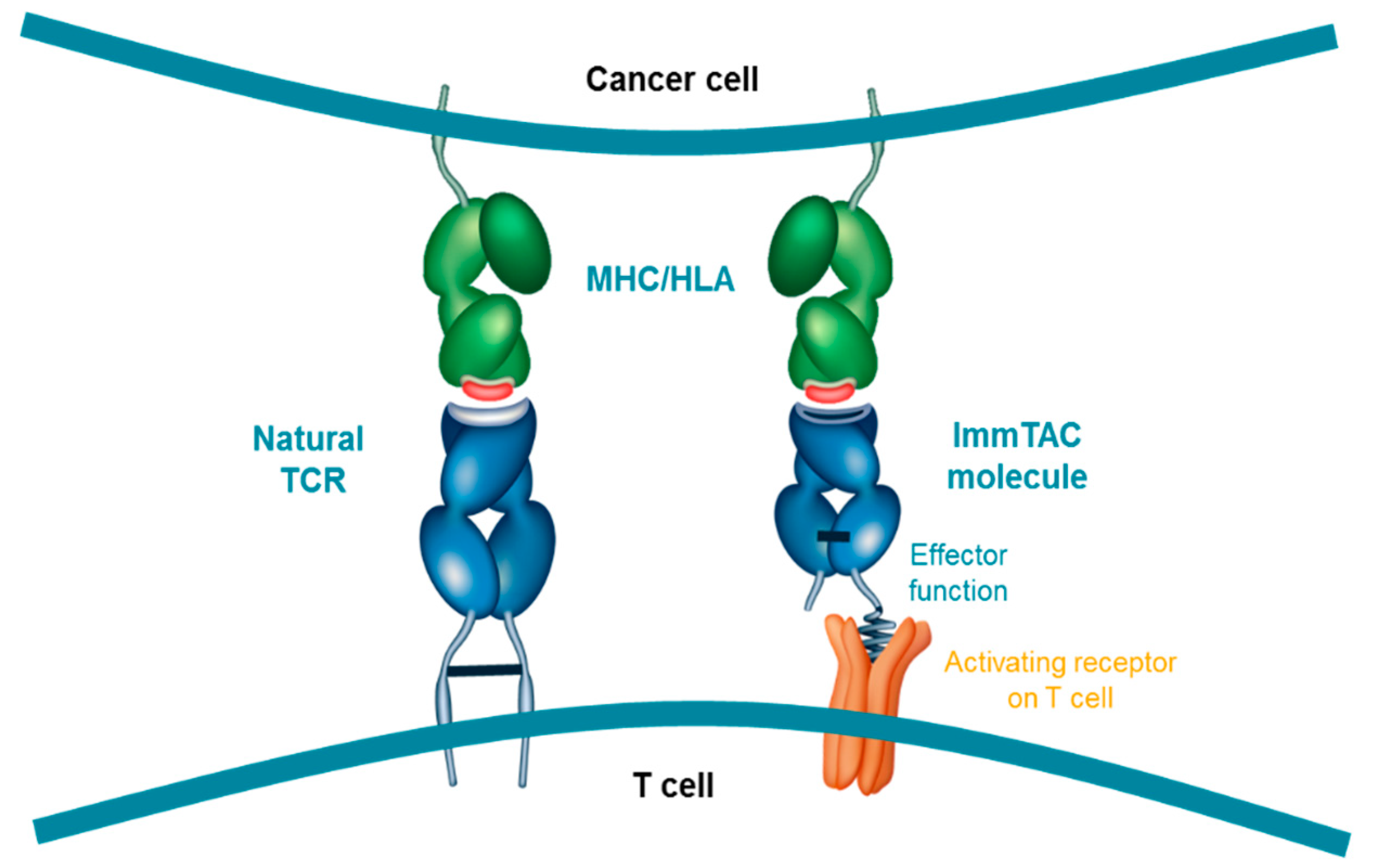
5. Development of ImmTAC Molecules
6. MoA of Tebentafusp
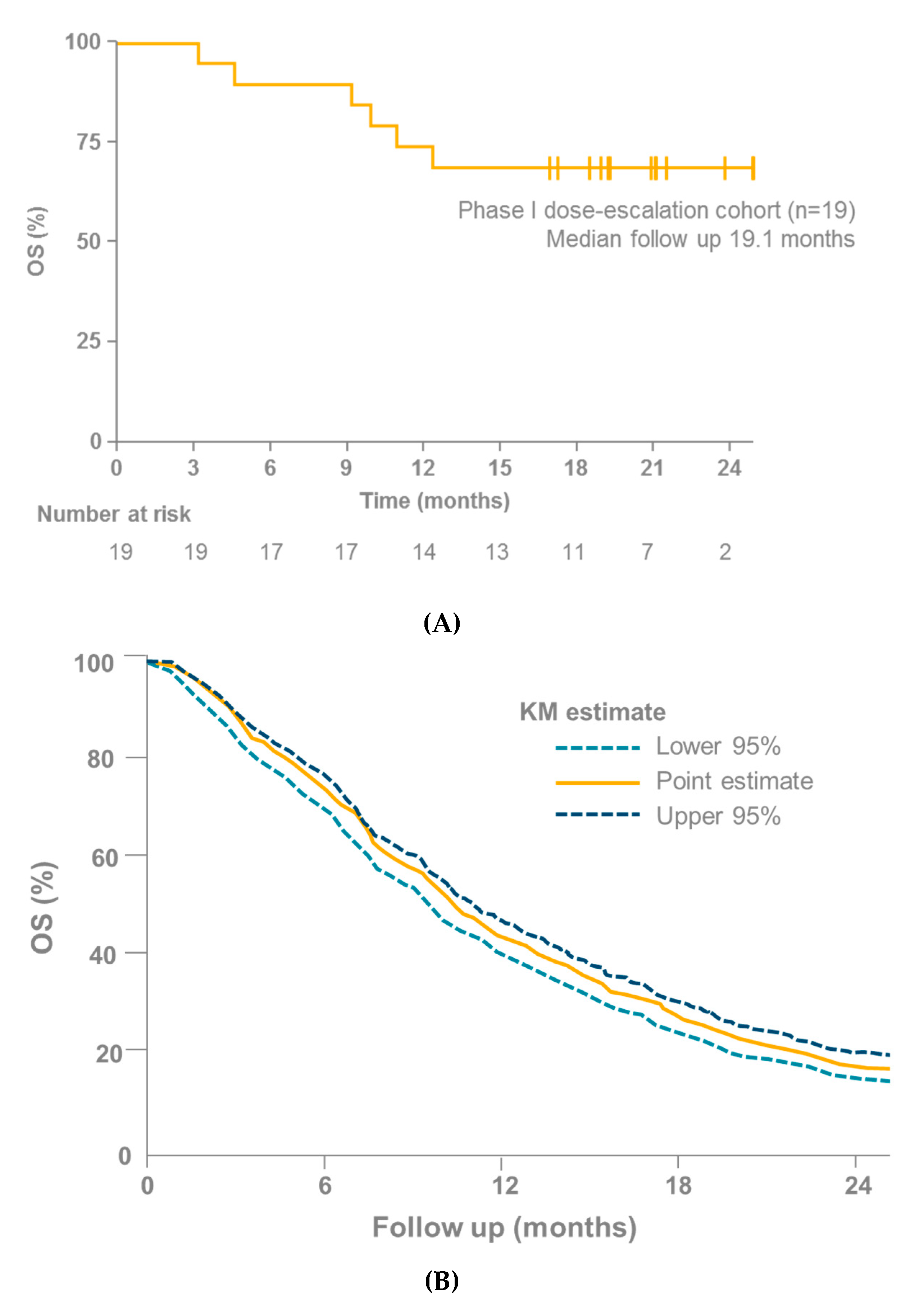
7. Tebentafusp in Clinical Studies
8. Future Prospects
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kujala, E.; Makitie, T.; Kivelä, T. Very long-term prognosis of patients with malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef]
- Lorenzo, D.; Piulats, J.M.; Ochoa, M.; Arias, L.; Gutierrez, C.; Catala, J.; Cobos, E.; Garcia-Bru, P.; Dias, B.; Padron-Perez, N.; et al. Clinical predictors of survival in metastatic uveal melanoma. Jpn. J. Ophthalmol. 2019, 63, 197–209. [Google Scholar] [CrossRef]
- Rantala, E.S.; Hernberg, M.; Kivelä, T.T. Overall survival after treatment for metastatic uveal melanoma: A systematic review and meta-analysis. Melanoma Res. 2019. [Google Scholar] [CrossRef]
- Sato, T.; Nathan, P.D.; Hernandez-Aya, L.; Sacco, J.; Orloff, M.; Engler, F.; Little, N.; Hulstine, A.; Coughlin, C.; Carvajal, R.D. Redirected T cell mediated lysis in patients with metastatic uveal melanoma with gp100-directed TCR IMCgp100: Overall survival findings. In Proceedings of the American Society of Clinical Oncology Annual Meeting, Chicago, IL, USA, 1–5 June 2018. Poster 9521. [Google Scholar]
- Middleton, M.; Steven, N.; Evans, J.; Infante, J.; Sznol, M. Safety, pharmacokinetics and efficacy of IMCgp100, a first-in-class soluble TCR anti-CD3 bispecific T cell redirector with solid tumour activity: Results from the first in human study in melanoma. In Proceedings of the American Society of Clinical Oncology Annual Meeting, Chicago, IL, USA, 3–7 June 2016. Poster 3016. [Google Scholar]
- Damato, B. Progress in the management of patients with uveal melanoma. The 2012 Ashton Lecture. Eye 2012, 26, 1157–1172. [Google Scholar] [CrossRef]
- Rodríguez, A.; Dueñas-Gonzalez, A.; Delgado-Pelayo, S. Clinical presentation and management of uveal melanoma. Mol. Clin. Oncol. 2016, 5, 675–677. [Google Scholar] [CrossRef]
- Krantz, B.A.; Dave, N.; Komatsubara, K.M.; Marr, B.P.; Carvajal, R.D. Uveal melanoma: Epidemiology, etiology, and treatment of primary disease. Clin. Ophthalmol. 2017, 11, 279–289. [Google Scholar] [CrossRef]
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef]
- Damato, B.E.; Coupland, S.E. Differences in uveal melanomas between men and women from the British Isles. Eye 2011, 26, 292. [Google Scholar] [CrossRef]
- Chua, V.; Lapadula, D.; Randolph, C.; Benovic, J.L.; Wedegaertner, P.B.; Aplin, A.E. Dysregulated GPCR Signaling and Therapeutic Options in Uveal Melanoma. Mol. Cancer Res. 2017, 15, 501–506. [Google Scholar] [CrossRef]
- Van den Bosch, T.; Kilic, E.; Paridaens, D.; de Klein, A. Genetics of uveal melanoma and cutaneous melanoma: Two of a kind? Derm. Res. Pract. 2010, 2010, 360136. [Google Scholar] [CrossRef]
- Reddy, B.Y.; Miller, D.M.; Tsao, H. Somatic driver mutations in melanoma. Cancer 2017, 123, 2104–2117. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, H.; Höiom, V. The genetics of uveal melanoma: Current insights. Appl. Clin. Genet. 2016, 9, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Damato, B. Ocular treatment of choroidal melanoma in relation to the prevention of metastatic death—A personal view. Prog. Retin. Eye Res. 2018, 66, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Stannard, C.; Sauerwein, W.; Maree, G.; Lecuona, K. Radiotherapy for ocular tumours. Eye 2013, 27, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Damato, B.E.; Stewart, J.M.; Afshar, A.R.; Groenewald, C.; Foulds, W.S. Surgical resection of choroidal melanoma. In Ryan’s Retina, 6th ed.; Elsevier: Edinburgh, UK, 2018; Volume 3, pp. 2591–2600. [Google Scholar]
- Damato, B.; Lecuona, K. Conservation of eyes with choroidal melanoma by a multimodality approach to treatment: An audit of 1632 patients. Ophthalmology 2004, 111, 977–983. [Google Scholar] [CrossRef]
- Damato, B. Vasculopathy after treatment of choroidal melanoma. In Retinal Vascular Disease; Springer: Berlin, Germany, 2007. [Google Scholar]
- Damato, B.; Hope-Stone, L.; Cooper, B.; Brown, S.L.; Salmon, P.; Heimann, H.; Dunn, L.B. Patient-reported outcomes and quality of life after treatment of choroidal melanoma: A comparison of enucleation versus radiotherapy in 1596 patients. Am. J. Ophthalmol. 2018, 193, 230–251. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Ocular Melanoma Study Quality of Life Study Group. Quality of life after iodine 125 brachytherapy vs enucleation for choroidal melanoma: 5-year results from the collaborative ocular melanoma study: COMS QOLS Report No. 3. JAMA Ophthalmol. 2006, 124, 226–238. [Google Scholar] [CrossRef]
- Collaborative Ocular Melanoma Study Group. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: V. Twelve-year mortality rates and prognostic factors: COMS report No. 28. Arch. Ophthalmol. 2006, 124, 1684–1693. [Google Scholar] [CrossRef]
- Damato, B. Legacy of the collaborative ocular melanoma study. Arch. Ophthalmol. 2007, 125, 966–968. [Google Scholar] [CrossRef]
- Marshall, E.; Romaniuk, C.; Ghaneh, P.; Wong, H.; McKay, M.; Chopra, M.; Coupland, S.E.; Damato, B.E. MRI in the detection of hepatic metastases from high-risk uveal melanoma: A prospective study in 188 patients. Br. J. Ophthalmol. 2013, 97, 159–163. [Google Scholar] [CrossRef]
- Damato, B.; Eleuteri, A.; Taktak, A.F.; Coupland, S.E. Estimating prognosis for survival after treatment of choroidal melanoma. Prog. Retin. Eye Res. 2011, 30, 285–295. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2018, 33, 151. [Google Scholar] [CrossRef]
- Dogrusöz, M.; Jager, M.J. Genetic prognostication in uveal melanoma. Acta Ophthalmol. 2018, 96, 331–347. [Google Scholar] [CrossRef]
- Damato, B.; Duke, C.; Coupland, S.E.; Hiscott, P.; Smith, P.A.; Campbell, I.; Douglas, A.; Howard, P. Cytogenetics of uveal melanoma: A 7-year clinical experience. Ophthalmology 2007, 114, 1925–1931. [Google Scholar] [CrossRef]
- Eleuteri, A.; Taktak, A.F.G.; Coupland, S.E.; Heimann, H.; Kalirai, H.; Damato, B. Prognostication of metastatic death in uveal melanoma patients: A Markov multi-state model. Comput. Biol. Med. 2018, 102, 151–156. [Google Scholar] [CrossRef]
- Christopher, A.B.; April, K.S. New NCCN Guidelines for uveal melanoma and treatment of recurrent or progressive distant metastatic melanoma. J. Natl. Compr. Cancer Netw. 2018, 16, 646–650. [Google Scholar] [CrossRef]
- Gomez, D.; Wetherill, C.; Cheong, J.; Jones, L.; Marshall, E.; Damato, B.; Coupland, S.E.; Ghaneh, P.; Poston, G.J.; Malik, H.Z.; et al. The Liverpool uveal melanoma liver metastases pathway: Outcome following liver resection. J. Surg. Oncol. 2014, 109, 542–547. [Google Scholar] [CrossRef]
- Leyvraz, S.; Piperno-Neumann, S.; Suciu, S.; Baurain, J.F.; Zdzienicki, M.; Testori, A.; Marshall, E.; Scheulen, M.; Jouary, T.; Negrier, S.; et al. Hepatic intra-arterial versus intravenous fotemustine in patients with liver metastases from uveal melanoma (EORTC 18021): A multicentric randomized trial. Ann. Oncol. 2014, 25, 742–746. [Google Scholar] [CrossRef]
- Pinqpank, J.; Hughes, M.; Alexander, H.; Faries, M.; Zager, J.; Siskin, G.; Agarwala, S.; Whitman, E.; Nutting, C.; Ozkan, O. Percutaneous hepatic perfusion (PHP) vs. best alternative care (BAC) for patients (pts) with melanoma liver metastases–efficacy update of the Phase 3 trial (NCT00324727). Eur. J. Cancer 2011, 47, S653. [Google Scholar] [CrossRef]
- Karydis, I.; Gangi, A.; Wheater, M.J.; Choi, J.; Wilson, I.; Thomas, K.; Pearce, N.; Takhar, A.; Gupta, S.; Hardman, D.; et al. Percutaneous hepatic perfusion with melphalan in uveal melanoma: A safe and effective treatment modality in an orphan disease. J. Surg. Oncol. 2018, 117, 1170–1178. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Schwartz, G.K.; Tezel, T.; Marr, B.; Francis, J.H.; Nathan, P.D. Metastatic disease from uveal melanoma: Treatment options and future prospects. Br. J. Ophthalmol. 2017, 101, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Scheulen, M.E.; Kaempgen, E.; Keilholz, U.; Heinzerling, L.; Ochsenreither, S.; Abendroth, A.; Hilger, R.A.; Grubert, M.; Wetter, A.; Guberina, N.; et al. STREAM: A randomized discontinuation, blinded, placebo-controlled phase II study of sorafenib (S) treatment of chemonaïve patients (pts) with metastatic uveal melanoma (MUM). J. Clin. Oncol. 2017, 35, 9511. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in combination with dacarbazine in patients with metastatic uveal melanoma: A Phase III, multicenter, randomized trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: A randomized clinical trial. JAMA 2014, 311, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Lewis, K.D.; Infante, J.R.; Gordon, M.S.; Vogelzang, N.J.; DeMarini, D.J.; Sun, P.; Moy, C.; Szabo, S.A.; Roadcap, L.T.; et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 782–789. [Google Scholar] [CrossRef]
- Sacco, J.J.; Nathan, P.D.; Danson, S.; Lorigan, P.; Nicholson, S.; Ottensmeier, C.; Corrie, P.; Steven, N.; Goodman, A.; Larkin, J.M.G.; et al. Sunitinib versus dacarbazine as first-line treatment in patients with metastatic uveal melanoma. J. Clin. Oncol. 2013, 31, 9031. [Google Scholar] [CrossRef]
- Algazi, A.P.; Tsai, K.K.; Shoushtari, A.N.; Munhoz, R.R.; Eroglu, Z.; Piulats, J.M.; Ott, P.A.; Johnson, D.B.; Hwang, J.; Daud, A.I.; et al. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer 2016, 122, 3344–3353. [Google Scholar] [CrossRef] [PubMed]
- Khoja, L.; Atenafu, E.G.; Suciu, S.; Leyvraz, S.; Sato, T.; Marshall, E.; Keilholz, U.; Zimmer, L.; Patel, S.P.; Piperno-Neumann, S.; et al. Meta-analysis in metastatic uveal melanoma to determine progression-free and overall survival benchmarks: An International Rare Cancers Initiative (IRCI) ocular melanoma study. Ann. Oncol. 2019. [Google Scholar] [CrossRef]
- Coley, W.B. The Treatment of Malignant Tumors by Repeated Innoculations of Erysipelas: With a Report of Ten Original Cases. Am. J. Med Sci. 1893, 10, 487–511. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Xing, Y.; Hogquist, K.A. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Domogalla, M.P.; Rostan, P.V.; Raker, V.K.; Steinbrink, K. Tolerance through education: How tolerogenic dendritic cells shape immunity. Front. Immunol. 2017, 8, 1764. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Thompson, C.B. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Davoodzadeh Gholami, M.; Kardar, G.A.; Saeedi, Y.; Heydari, S.; Garssen, J.; Falak, R. Exhaustion of T lymphocytes in the tumor microenvironment: Significance and effective mechanisms. Cell. Immunol. 2017, 322, 1–14. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef]
- Margolin, K.A. Interleukin-2 in the treatment of renal cancer. Semin. Oncol. 2000, 27, 194–203. [Google Scholar]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 165. [Google Scholar] [CrossRef]
- Yun, S.; Vincelette, N.D.; Green, M.R.; Wahner Hendrickson, A.E.; Abraham, I. Targeting immune checkpoints in unresectable metastatic cutaneous melanoma: A systematic review and meta-analysis of anti-CTLA-4 and anti-PD-1 agents trials. Cancer Med. 2016, 5, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Guillerey, C.; Harjunpaa, H.; Carrie, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT immune checkpoint blockade restores CD8(+) T-cell immunity against multiple myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [PubMed]
- Lichtenegger, F.S.; Rothe, M.; Schnorfeil, F.M.; Deiser, K.; Krupka, C.; Augsberger, C.; Schlüter, M.; Neitz, J.; Subklewe, M. Targeting LAG-3 and PD-1 to enhance T cell activation by antigen-presenting cells. Front. Immunol. 2018, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.S.; Somerville, R.P.T.; Yang, J.C.; Sherry, R.M.; Klebanoff, C.A.; Goff, S.L.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.; Paria, B.C.; et al. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: A single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 792–802. [Google Scholar] [CrossRef]
- Van Loenen, M.M.; de Boer, R.; Hagedoorn, R.S.; Jankipersadsing, V.; Amir, A.L.; Falkenburg, J.H.; Heemskerk, M.H. Multi-cistronic vector encoding optimized safety switch for adoptive therapy with T-cell receptor-modified T cells. Gene Ther. 2013, 20, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, E.M.V.; Lindberg, M.F.; Jespersen, H.; Alsén, S.; Olofsson Bagge, R.; Donia, M.; Svane, I.M.; Nilsson, O.; Ny, L.; Nilsson, L.M.; et al. HER2 CAR-T cells eradicate uveal melanoma and T cell therapy-resistant human melanoma in interleukin-2 (IL-2) transgenic NOD/SCID IL-2 receptor knockout mice. Cancer Res. 2019, 79, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Novartis. KYMRIAH Prescribing Information. Available online: https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kymriah.pdf (accessed on 9 July 2019).
- Kite Pharma. YESCARTA Prescribing Information. Available online: https://www.yescarta.com/files/yescarta-pi.pdf (accessed on 9 July 2019).
- Retèl, V.P.; Steuten, L.M.G.; Geukes Foppen, M.H.; Mewes, J.C.; Lindenberg, M.A.; Haanen, J.B.A.G.; van Harten, W.H. Early cost-effectiveness of tumor infiltrating lymphocytes (TIL) for second line treatment in advanced melanoma: A model-based economic evaluation. BMC Cancer 2018, 18, 895. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.; Mount, N. Genetically modified T cells in cancer therapy: Opportunities and challenges. Dis. Model. Mech. 2015, 8, 337–350. [Google Scholar] [CrossRef]
- Jung, I.-Y.; Lee, J. Unleashing the therapeutic potential of CAR-T cell therapy using gene-editing technologies. Mol. Cells 2018, 41, 717–723. [Google Scholar] [CrossRef]
- Kalos, M.; June, C.H. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity 2013, 39, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; Bhoj, V.G. The pharmacology of T cell therapies. Mol. Ther. Methods Clin. Dev. 2018, 8, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Bernatchez, C.; Haymaker, C.; Wong, M.K.K.; Hwu, P.; Bentebibel, S.E.; Cho, D.; Tykodi, S.S.; Puzanov, I.; Kluger, H.; et al. 1212TiPPIVOT-02: A phase 1/2, open-label, multicenter, dose escalation and dose expansion study of NKTR-214 and nivolumab in patients with select, locally advanced or metastatic solid tumor malignancies. Ann. Oncol. 2017, 28. [Google Scholar] [CrossRef]
- Weinberg, A.D.; Morris, N.P.; Kovacsovics-Bankowski, M.; Urba, W.J.; Curti, B.D. Science gone translational: The OX40 agonist story. Immunol. Rev. 2011, 244, 218–231. [Google Scholar] [CrossRef]
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Johnson, D.B.; Bao, R.; Ancell, K.K.; Daniels, A.B.; Wallace, D.; Sosman, J.A.; Luke, J.J. Response to anti-PD-1 in uveal melanoma without high-volume liver metastasis. J. Natl. Compr. Cancer Netw. 2019, 17, 114–117. [Google Scholar] [CrossRef]
- Karlsson, A.K.; Saleh, S.N. Checkpoint inhibitors for malignant melanoma: A systematic review and meta-analysis. Clin. Cosmet. Investig. Dermatol. 2017, 10, 325–339. [Google Scholar] [CrossRef]
- Shen, X.; Zhao, B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: Meta-analysis. BMJ 2018, 362, k3529. [Google Scholar] [CrossRef]
- Javed, A.; Arguello, D.; Johnston, C.; Gatalica, Z.; Terai, M.; Weight, R.M.; Orloff, M.; Mastrangelo, M.J.; Sato, T. PD-L1 expression in tumor metastasis is different between uveal melanoma and cutaneous melanoma. Immunotherapy 2017, 9, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Terai, M.; Mastrangleo, M.J.; Sato, T. Immunological aspect of the liver and metastatic uveal melanoma. J. Cancer Metastasis Treat. 2017, 3, 231–243. [Google Scholar] [CrossRef]
- Oates, J.; Hassan, N.J.; Jakobsen, B.K. ImmTACs for targeted cancer therapy: Why, what, how, and which. Mol. Immunol. 2015, 67, 67–74. [Google Scholar] [CrossRef] [PubMed]
- De Souza, J.E.; Galante, P.A.; de Almeida, R.V.; da Cunha, J.P.; Ohara, D.T.; Ohno-Machado, L.; Old, L.J.; de Souza, S.J. SurfaceomeDB: A cancer-orientated database for genes encoding cell surface proteins. Cancer Immun. 2012, 12, 15. [Google Scholar] [PubMed]
- Stone, J.D.; Kranz, D.M. Role of T cell receptor affinity in the efficacy and specificity of adoptive T cell therapies. Front. Immunol. 2013, 4, 244. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M.; Boniface, J.J.; Reich, Z.; Lyons, D.; Hampl, J.; Arden, B.; Chien, Y. Ligand recognition by alpha beta T cell receptors. Annu. Rev. Immunol. 1998, 16, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Pecorari, F.; Tissot, A.C.; Plückthun, A. Folding, heterodimeric association and specific peptide recognition of a murine αβ T-cell receptor expressed in Escherichia coli. J. Mol. Biol. 1999, 285, 1831–1843. [Google Scholar] [CrossRef]
- Bossi, G.; Gerry, A.B.; Paston, S.J.; Sutton, D.H.; Hassan, N.J.; Jakobsen, B.K. Examining the presentation of tumor-associated antigens on peptide-pulsed T2 cells. Oncoimmunology 2013, 2, e26840. [Google Scholar] [CrossRef]
- Li, Y.; Moysey, R.; Molloy, P.E.; Vuidepot, A.L.; Mahon, T.; Baston, E.; Dunn, S.; Liddy, N.; Jacob, J.; Jakobsen, B.K.; et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005, 23, 349–354. [Google Scholar] [CrossRef]
- Dunn, S.M.; Rizkallah, P.J.; Baston, E.; Mahon, T.; Cameron, B.; Moysey, R.; Gao, F.; Sami, M.; Boulter, J.; Li, Y.; et al. Directed evolution of human T cell receptor CDR2 residues by phage display dramatically enhances affinity for cognate peptide-MHC without increasing apparent cross-reactivity. Protein Sci. 2006, 15, 710–721. [Google Scholar] [CrossRef]
- Boudousquie, C.; Bossi, G.; Hurst, J.M.; Rygiel, K.A.; Jakobsen, B.K.; Hassan, N.J. Polyfunctional response by ImmTAC (IMCgp100) redirected CD8(+) and CD4(+) T cells. Immunology 2017, 152, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.B.; Schreurs, M.W.; de Boer, A.J.; Kawakami, Y.; Rosenberg, S.A.; Adema, G.J.; Figdor, C.G. Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J. Exp. Med. 1994, 179, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Crabb, W.J.; Hu, B.; Crabb, J.S.; Triozzi, P.; Sauntharajah, Y.; Tubbs, R.; Singh, A.D. iTRAQ Quantitative Proteomic Comparison of Metastatic and Non-Metastic Uveal Melanoma Tumors. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.N.; Wagner, C.; Schultewolter, T.; Goos, M. Analysis of Pmel17/gp100 expression in primary human tissue specimens: Implications for melanoma immuno- and gene-therapy. Cancer Immunol. Immunother. 1997, 44, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Skipper, J.C.; Gulden, P.H.; Hendrickson, R.C.; Harthun, N.; Caldwell, J.A.; Shabanowitz, J.; Engelhard, V.H.; Hunt, D.F.; Slingluff, C.L., Jr. Mass-spectrometric evaluation of HLA-A*0201-associated peptides identifies dominant naturally processed forms of CTL epitopes from MART-1 and gp100. Int. J. Cancer 1999, 82, 669–677. [Google Scholar] [CrossRef]
- Marincola, F.M.; Venzon, D.; White, D.; Rubin, J.T.; Lotze, M.T.; Simonis, T.B.; Balkissoon, J.; Rosenberg, S.A.; Parkinson, D.R. HLA association with response and toxicity in melanoma patients treated with interleukin 2-based immunotherapy. Cancer Res. 1992, 52, 6561–6566. [Google Scholar] [PubMed]
- Harper, J.; Adams, K.J.; Bossi, G.; Wright, D.E.; Stacey, A.R.; Bedke, N.; Martinez-Hague, R.; Blat, D.; Humbert, L.; Buchanan, H.; et al. An approved in vitro approach to preclinical safety and efficacy evaluation of engineered T cell receptor anti-CD3 bispecific (ImmTAC) molecules. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 2012, 18, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Bossi, G.; Buisson, S.; Oates, J.; Jakobsen, B.K.; Hassan, N.J. ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol. Immunother. 2014, 63, 437–448. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Sato, T.; Shoushtari, A.N.; Sacco, J.; Nathan, P.; Orloff, M.; Corrie, P.; Steven, N.; Evans, J.; Infante, J.; et al. Safety, efficacy and biology of the gp100 TCR-based bispecific T cell redirector, IMCgp100 in advanced uveal melanoma in two Phase 1 trials. In Proceedings of the Society for Immunotherapy of Cancer Annual Meeting, National Harbor, MD, USA, 8–12 November 2017. Poster P208. [Google Scholar]
- de Vries, T.J.; Trancikova, D.; Ruiter, D.J.; van Muijen, G.N. High expression of immunotherapy candidate proteins gp100, MART-1, tyrosinase and TRP-1 in uveal melanoma. Br. J. Cancer 1998, 78, 1156–1161. [Google Scholar] [CrossRef]
- Luyten, G.P.; van der Spek, C.W.; Brand, I.; Sintnicolaas, K.; de Waard-Siebinga, I.; Jager, M.J.; de Jong, P.T.; Schrier, P.I.; Luider, T.M. Expression of MAGE, gp100 and tyrosinase genes in uveal melanoma cell lines. Melanoma Res. 1998, 8, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T cell-inflamed versus non-T cell-inflamed tumors: A conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy Class | Therapy Examples | Efficacy in mUM |
|---|---|---|
| Liver-directed therapies | Surgical resection | Surgical resection of isolated liver deposits appears to result in more favourable survival than no resection [31]. |
| Bland embolisation Chemoembolisation Radioembolisation Immunoembolisation | Limited prospective data for non-surgical therapies. Response rates have been superior to systemic chemotherapy [32,33], but only liver sites are targeted. Results with liver-directed melphalan have been particularly impressive in patients with liver metastases from mUM (ORR 47%) [34]. | |
| Chemotherapy | Dacarbazine Temozolomide Cisplatin Treosulfan Fotemustine Various combinations | Results to date have been disappointing [35]. |
| Kinase inhibitors | Sorafenib (multikinase inhibitor) | A randomised phase II trial of sorafenib showed PFS to be superior to placebo (median 5.5 vs 1.9 months) [36]. |
| Selmutinib (MEK inhibitor) | A randomised phase III trial of selmutinib + dacarbazine failed to show a benefit, compared to placebo + dacarbazine (ORR 3% vs 0%, respectively), in contrast to promising phase II results [37,38]. | |
| Trametinib (MEK inhibitor) | A phase I trial of trametinib demonstrated limited clinical activity (0% ORR, 50% achieved stable disease) [39]. | |
| Sunitinib (multikinase inhibitor) | A phase II trial of sunitinib showed no benefit vs dacarbazine (ORR 0% vs 8%, respectively) [40]. | |
| Immunotherapy | Pembrolizumab (PD-1 inhibitor) Nivolumab (PD-1 inhibitor) Ipilimumab (CTLA-4 inhibitor) | A meta-analysis of mUM trials and a real-world study both concluded limited benefit of checkpoint inhibitors in mUM (median PFS 2.6–2.8 months) [41,42]. |
| Study | N Total | Number of Studies | Therapy | Median OS (months) | OS Rate at 1 Year (%) |
|---|---|---|---|---|---|
| Algazi, 2016 [41] | 56 | 9 | Anti-PD-1 or Anti-PD-L1 antibodies | 7.7 | ~45 |
| Khoja, 2016 [42] | 915 | 29 | Immunotherapy, kinase inhibitors, anti-angiogenic agent, intra-hepatic chemotherapy or immunotherapy, LDTs | 10.2 | 43 |
| Rantala, 2019 [3] | 2494 | 78 | Immunotherapy, chemotherapy, LDTs, surgery | 12.8 | 52 |
| IMCgp100-01 [96] | 15 (evaluable in UM cohort) | 1 | Tebentafusp | Not reached after 16 months follow-up | 73 |
| IMCgp100-102 [4] | 19 | 1 | Tebentafusp | Not reached after 16 months follow-up | 74 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damato, B.E.; Dukes, J.; Goodall, H.; Carvajal, R.D. Tebentafusp: T Cell Redirection for the Treatment of Metastatic Uveal Melanoma. Cancers 2019, 11, 971. https://doi.org/10.3390/cancers11070971
Damato BE, Dukes J, Goodall H, Carvajal RD. Tebentafusp: T Cell Redirection for the Treatment of Metastatic Uveal Melanoma. Cancers. 2019; 11(7):971. https://doi.org/10.3390/cancers11070971
Chicago/Turabian StyleDamato, Bertil E., Joseph Dukes, Howard Goodall, and Richard D. Carvajal. 2019. "Tebentafusp: T Cell Redirection for the Treatment of Metastatic Uveal Melanoma" Cancers 11, no. 7: 971. https://doi.org/10.3390/cancers11070971
APA StyleDamato, B. E., Dukes, J., Goodall, H., & Carvajal, R. D. (2019). Tebentafusp: T Cell Redirection for the Treatment of Metastatic Uveal Melanoma. Cancers, 11(7), 971. https://doi.org/10.3390/cancers11070971