Palbociclib Promotes Dephosphorylation of NPM/B23 at Threonine 199 and Inhibits Endometrial Cancer Cell Growth


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Exposure to Palbociclib Induced ERα Expression in Endometrial Cancer Cells
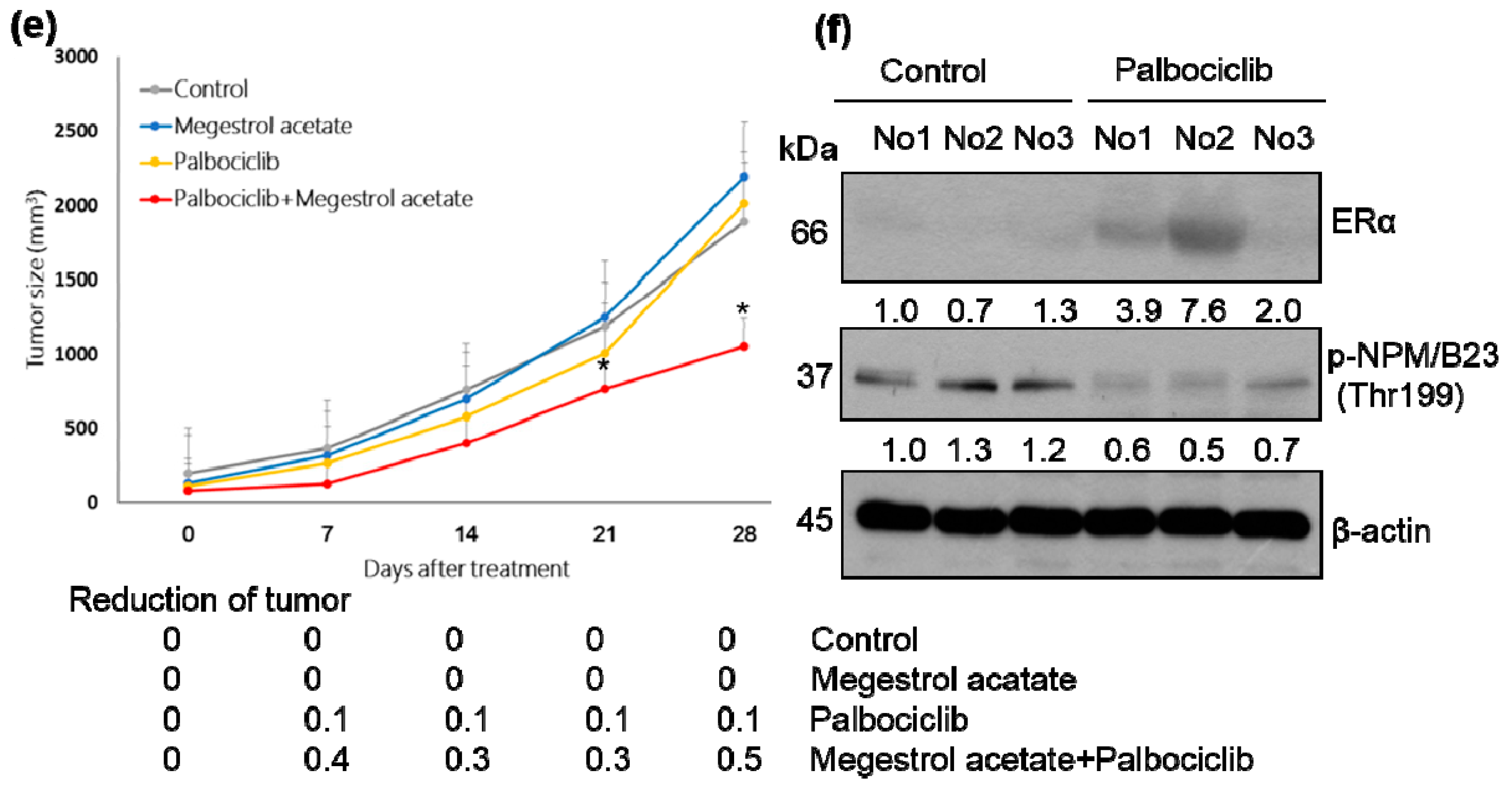
2.2. Palbociclib and Megestrol Acetate Synergistically Inhibit Survival, Increase Apoptosis, and Increase the Expression of ERα in Endometrial Cancer Cells
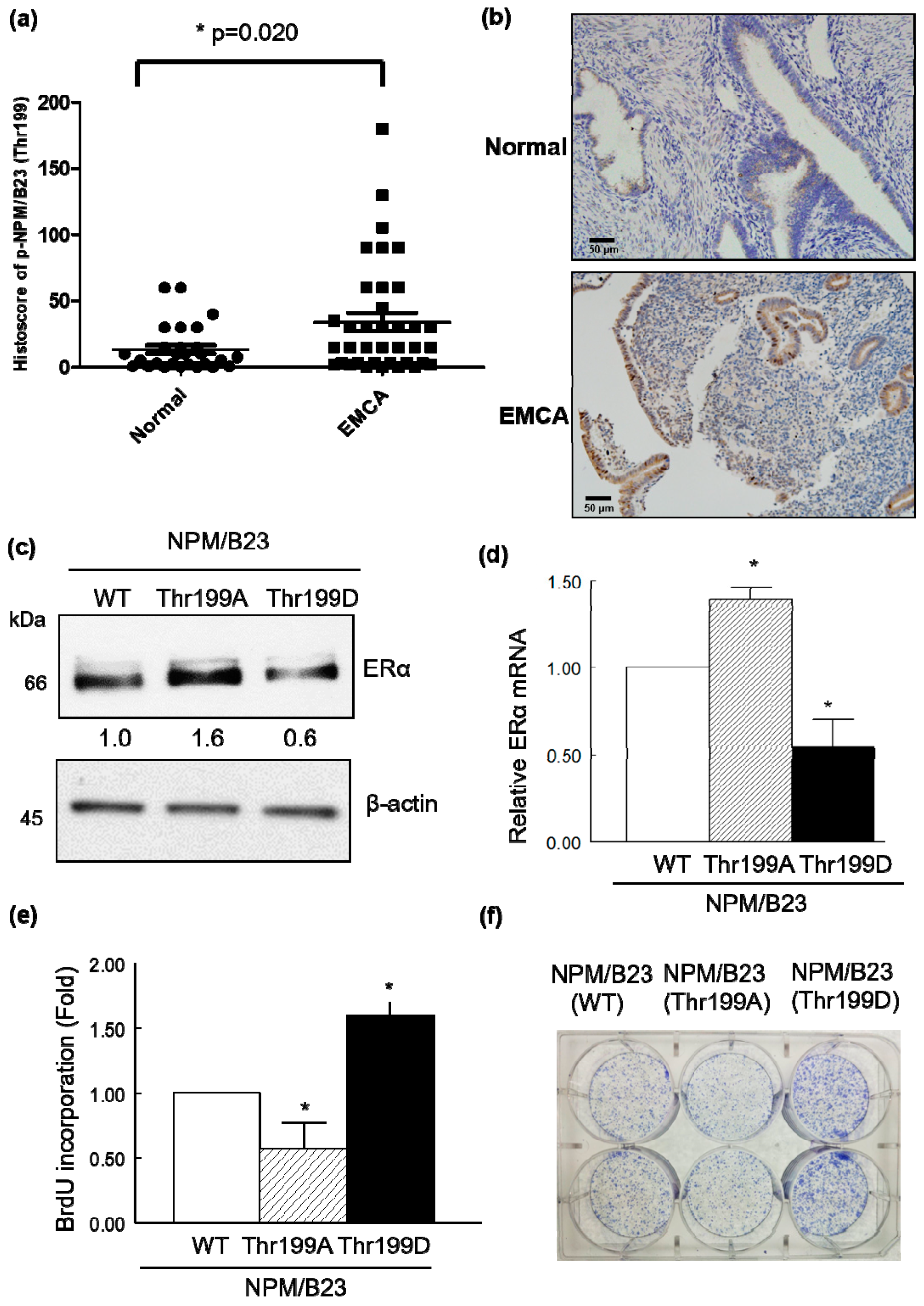
2.3. Phospho-NPM/B23 (Thr199) Is Involved in Endometrial Tumorigenesis
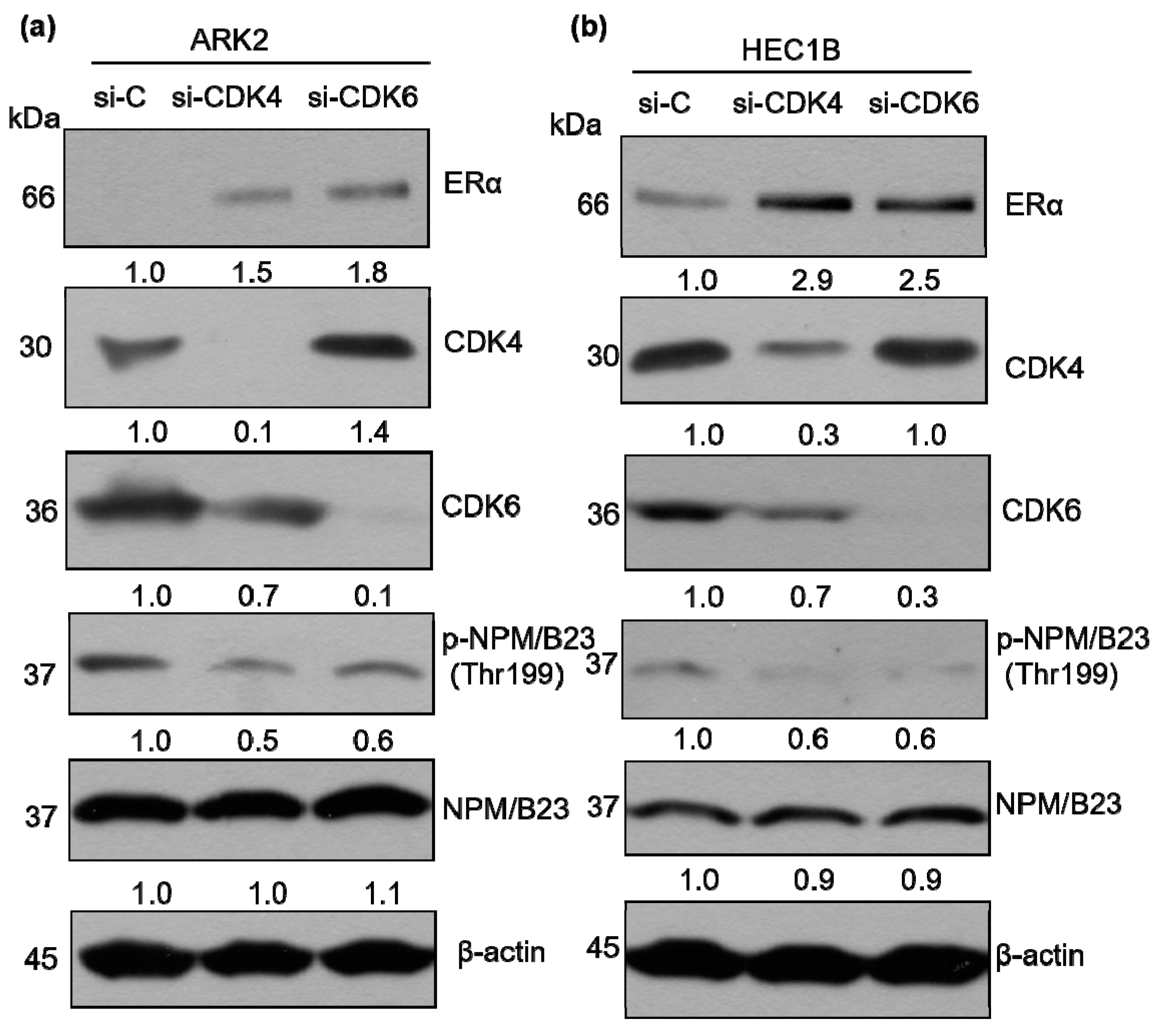
2.4. CDK6-Mediated Phosphorylation of NPM/B23 Promotes ERα Expression
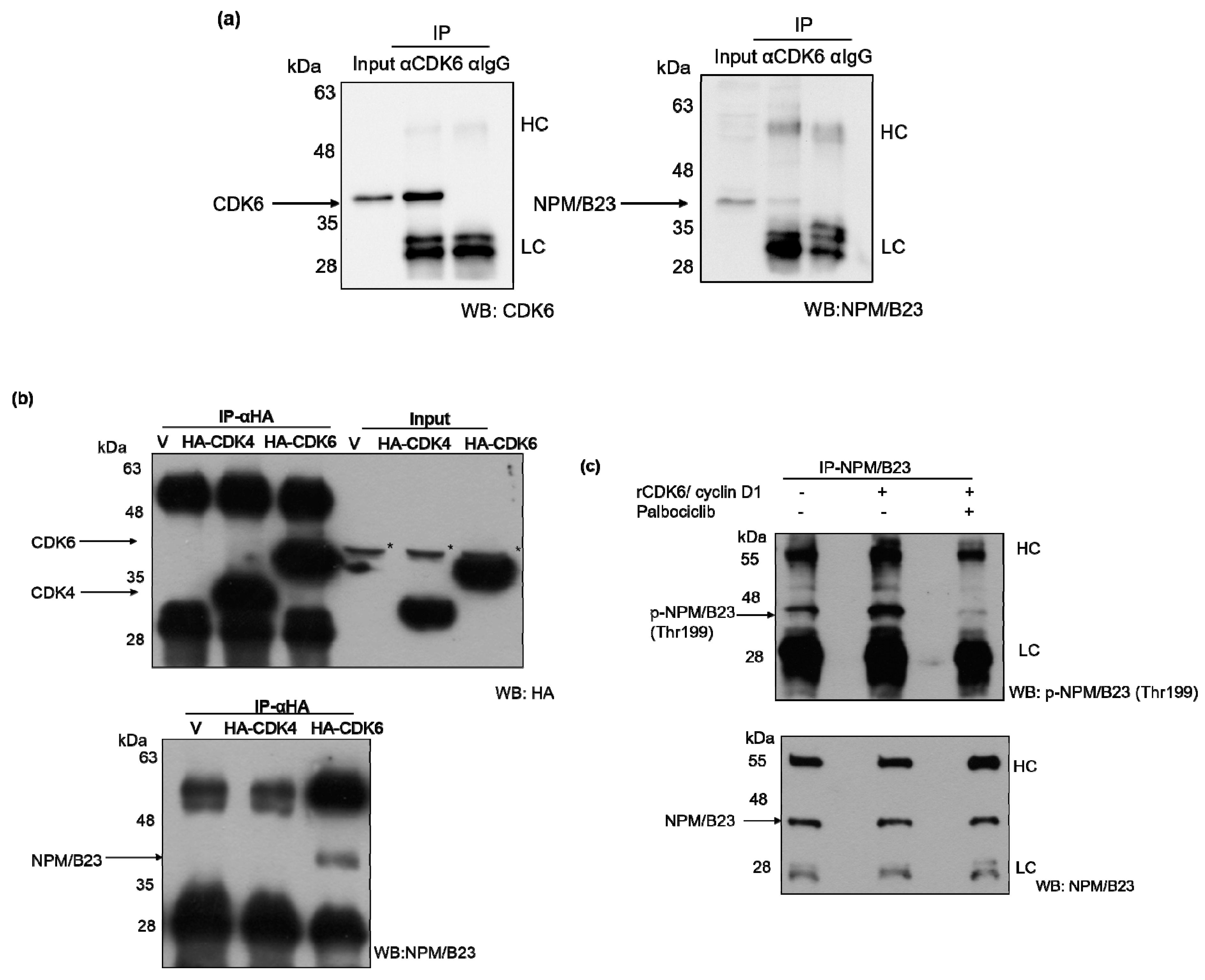
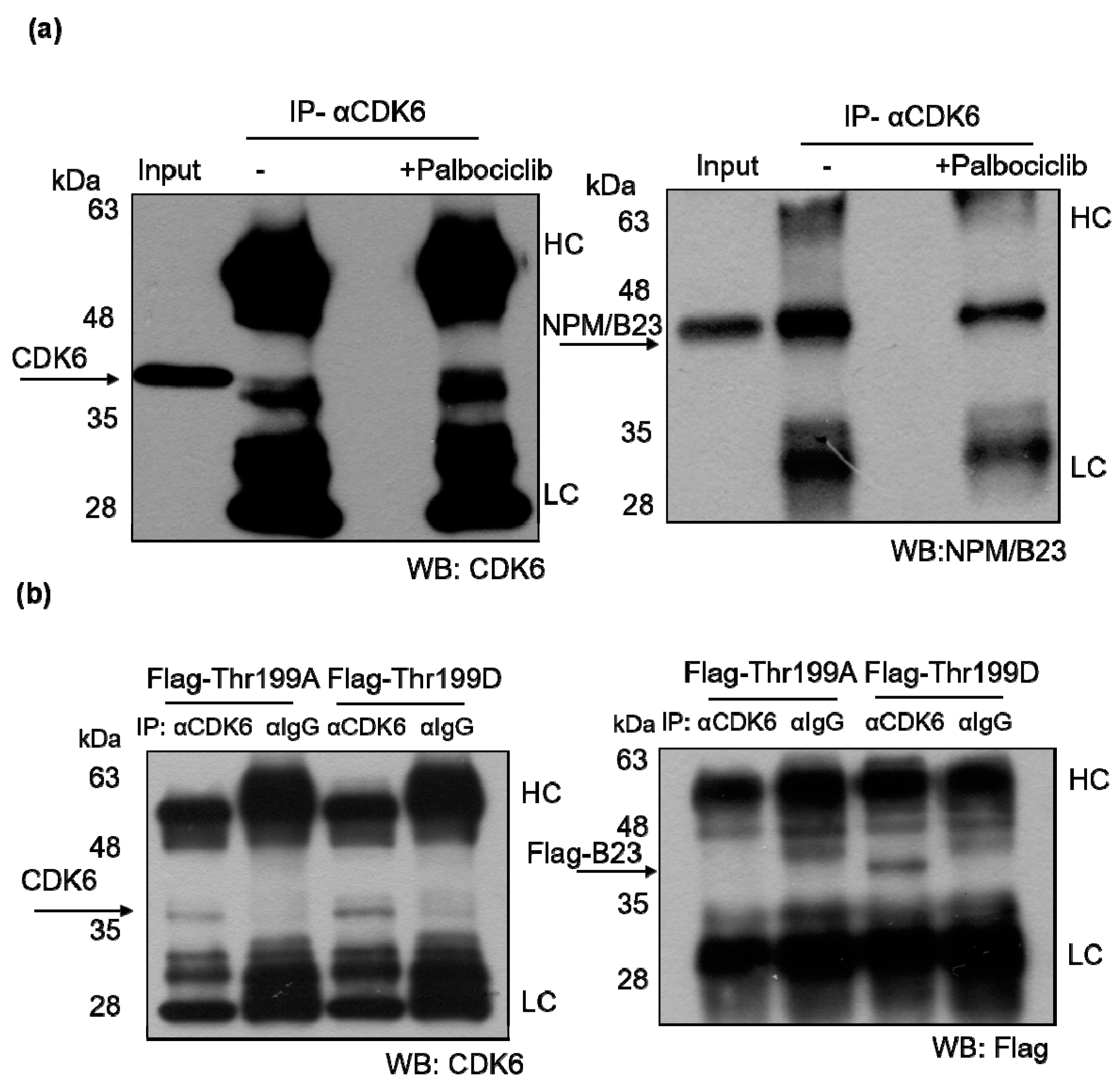
2.5. Phosphorylated NPM/B23 (Thr199) Forms a Preferential Complex with CDK6
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Antibodies, Reagents, and Plasmids
4.3. DNA Transfection Experiments
4.4. Western Blot Analysis
4.5. RNA Extraction and Real-time QPCR
4.6. Cell Viability Assay
4.7. Clonogenic Assays
4.8. Animals and Treatment
4.9. Synergistic Effect of the Combined Drug Treatment
4.10. RNA Interference Procedures
4.11. In Vitro Kinase Assay
4.12. Immunoprecipitation
4.13. Immunohistochemistry and Clinical Tissue Specimens
4.14. Proliferation Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lortet-Tieulent, J.; Ferlay, J.; Bray, F.; Jemal, A. International Patterns and Trends in Endometrial Cancer Incidence, 1978–2013. J. Natl. Cancer Inst. 2018, 110, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Health Promotion Administration Ministry of Health and Welfare Taiwan. Cancer Registry Annual Report, 2015 Taiwan; Health Promotion Administration Ministry of Health and Welfare Taiwan: Taipei, Taiwan, 2017.
- Creasman, W.T.; Miller, D.S. Adenocarcinoma of the uterine corpus. In Clinical Gynecologic Oncology; Di Saia, P.J., Creasman, W.T., Mannel, R.S., McMeekin, D.S., Mutch, D.G., Eds.; Elsevier: Philadelphia, PA, USA, 2012; pp. 121–154. [Google Scholar]
- Creasman, W.T.; Soper, J.T.; McCarty, K.S.; McCarty, K.S., Jr.; Hinshaw, W., Sr.; Clarke-Pearson, D.L. Influence of cytoplasmic steroid receptor content on prognosis of early stage endometrial carcinoma. Am. J. Obstet. Gynecol. 1985, 151, 922–932. [Google Scholar] [CrossRef]
- Thigpen, J.T.; Brady, M.F.; Alvarez, R.D.; Adelson, M.D.; Homesley, H.D.; Manetta, A.; Soper, J.T.; Given, F.T. Oral medroxyprogesterone acetate in the treatment of advanced or recurrent endometrial carcinoma: A dose-response study by the Gynecologic Oncology Group. J. Clin. Oncol. 1999, 17, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.J.; Thiel, K.W.; Leslie, K.K. Past, present, and future of hormonal therapy in recurrent endometrial cancer. Int. J. Women’s Health 2014, 6, 429–435. [Google Scholar]
- Jongen, V.; Briet, J.; de Jong, R.; ten Hoor, K.; Boezen, M.; van der Zee, A.; Nijman, H.; Hollema, H. Expression of estrogen receptor-alpha and -beta and progesterone receptor-A and -B in a large cohort of patients with endometrioid endometrial cancer. Gynecol. Oncol. 2009, 112, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Kuukasjarvi, T.; Kononen, J.; Helin, H.; Holli, K.; Isola, J. Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J. Clin. Oncol. 1996, 14, 2584–2589. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.; Nakagawa, J.; Doree, M.; Labbe, J.C.; Nigg, E.A. Identification of major nucleolar proteins as candidate mitotic substrates of cdc2 kinase. Cell 1990, 60, 791–801. [Google Scholar] [CrossRef]
- Negi, S.S.; Olson, M.O. Effects of interphase and mitotic phosphorylation on the mobility and location of nucleolar protein B23. J. Cell Sci. 2006, 119, 3676–3685. [Google Scholar] [CrossRef]
- Chan, P.K.; Aldrich, M.; Cook, R.G.; Busch, H. Amino acid sequence of protein B23 phosphorylation site. J. Biol. Chem. 1986, 261, 1868–1872. [Google Scholar]
- Okuwaki, M.; Tsujimoto, M.; Nagata, K. The RNA binding activity of a ribosome biogenesis factor, nucleophosmin/B23, is modulated by phosphorylation with a cell cycle-dependent kinase and by association with its subtype. Mol. Biol. Cell 2002, 13, 2016–2030. [Google Scholar] [CrossRef]
- Okuda, M. The role of nucleophosmin in centrosome duplication. Oncogene 2002, 21, 6170–6174. [Google Scholar] [CrossRef]
- Adon, A.M.; Zeng, X.; Harrison, M.K.; Sannem, S.; Kiyokawa, H.; Kaldis, P.; Saavedra, H.I. Cdk2 and Cdk4 regulate the centrosome cycle and are critical mediators of centrosome amplification in p53-null cells. Mol. Cell. Biol. 2010, 30, 694–710. [Google Scholar] [CrossRef]
- Sarek, G.; Jarviluoma, A.; Moore, H.M.; Tojkander, S.; Vartia, S.; Biberfeld, P.; Laiho, M.; Ojala, P.M. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathog. 2010, 6, e1000818. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; Andre, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Beaver, J.A.; Amiri-Kordestani, L.; Charlab, R.; Chen, W.; Palmby, T.; Tilley, A.; Zirkelbach, J.F.; Yu, J.; Liu, Q.; Zhao, L.; et al. FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2015, 21, 4760–4766. [Google Scholar] [CrossRef]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef]
- Tanaka, T.; Terai, Y.; Ashihara, K.; Fujiwara, S.; Tanaka, Y.; Sasaki, H.; Tsunetoh, S.; Ohmichi, M. The efficacy of the cyclin-dependent kinase 4/6 inhibitor in endometrial cancer. PLoS ONE 2017, 12, e0177019. [Google Scholar] [CrossRef]
- Dosil, M.A.; Mirantes, C.; Eritja, N.; Felip, I.; Navaridas, R.; Gatius, S.; Santacana, M.; Colas, E.; Moiola, C.; Schoenenberger, J.A.; et al. Palbociclib has antitumour effects on Pten-deficient endometrial neoplasias. J. Pathol. 2017, 242, 152–164. [Google Scholar] [CrossRef]
- Chao, A.; Lin, C.Y.; Tsai, C.L.; Hsueh, S.; Lin, Y.Y.; Lin, C.T.; Chou, H.H.; Wang, T.H.; Lai, C.H.; Wang, H.S. Estrogen stimulates the proliferation of human endometrial cancer cells by stabilizing nucleophosmin/B23 (NPM/B23). J. Mol. Med. 2013, 91, 249–259. [Google Scholar] [CrossRef]
- Lin, C.Y.; Chao, A.; Wang, T.H.; Lee, L.Y.; Yang, L.Y.; Tsai, C.L.; Wang, H.S.; Lai, C.H. Nucleophosmin/B23 is a negative regulator of estrogen receptor alpha expression via AP2gamma in endometrial cancer cells. Oncotarget 2016, 7, 60038–60052. [Google Scholar]
- Grisendi, S.; Mecucci, C.; Falini, B.; Pandolfi, P.P. Nucleophosmin and cancer. Nat. Rev. Cancer 2006, 6, 493–505. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef]
- Lin, C.Y.; Tan, B.C.; Liu, H.; Shih, C.J.; Chien, K.Y.; Lin, C.L.; Yung, B.Y. Dephosphorylation of nucleophosmin by PP1beta facilitates pRB binding and consequent E2F1-dependent DNA repair. Mol. Biol. Cell 2010, 21, 4409–4417. [Google Scholar] [CrossRef]
- Ching, R.H.; Lau, E.Y.; Ling, P.M.; Lee, J.M.; Ma, M.K.; Cheng, B.Y.; Lo, R.C.; Ng, I.O.; Lee, T.K. Phosphorylation of nucleophosmin at threonine 234/237 is associated with HCC metastasis. Oncotarget 2015, 6, 43483–43495. [Google Scholar] [CrossRef]
- Tarapore, P.; Shinmura, K.; Suzuki, H.; Tokuyama, Y.; Kim, S.H.; Mayeda, A.; Fukasawa, K. Thr199 phosphorylation targets nucleophosmin to nuclear speckles and represses pre-mRNA processing. FEBS Lett. 2006, 580, 399–409. [Google Scholar] [CrossRef]
- Konecny, G.E.; Winterhoff, B.; Kolarova, T.; Qi, J.; Manivong, K.; Dering, J.; Yang, G.; Chalukya, M.; Wang, H.J.; Anderson, L.; et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin. Cancer Res. 2011, 17, 1591–1602. [Google Scholar] [CrossRef]
- Di Matteo, A.; Franceschini, M.; Chiarella, S.; Rocchio, S.; Travaglini-Allocatelli, C.; Federici, L. Molecules that target nucleophosmin for cancer treatment: An update. Oncotarget 2016, 7, 44821–44840. [Google Scholar] [CrossRef]
- Zhao, S.; Choi, M.; Overton, J.D.; Bellone, S.; Roque, D.M.; Cocco, E.; Guzzo, F.; English, D.P.; Varughese, J.; Gasparrini, S.; et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 2916–2921. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Chao, A.; Lin, C.Y.; Wu, R.C.; Lee, Y.S.; Lee, L.Y.; Tsai, C.L.; Yang, L.Y.; Liu, H.; Chen, S.J.; Wang, T.H.; et al. The combination of everolimus and terameprocol exerts synergistic antiproliferative effects in endometrial cancer: Molecular role of insulin-like growth factor binding protein 2. J. Mol. Med. 2018, 96, 1251–1266. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-Y.; Lee, L.-Y.; Wang, T.-H.; Hsu, C.-L.; Tsai, C.-L.; Chao, A.; Lai, C.-H. Palbociclib Promotes Dephosphorylation of NPM/B23 at Threonine 199 and Inhibits Endometrial Cancer Cell Growth. Cancers 2019, 11, 1025. https://doi.org/10.3390/cancers11071025
Lin C-Y, Lee L-Y, Wang T-H, Hsu C-L, Tsai C-L, Chao A, Lai C-H. Palbociclib Promotes Dephosphorylation of NPM/B23 at Threonine 199 and Inhibits Endometrial Cancer Cell Growth. Cancers. 2019; 11(7):1025. https://doi.org/10.3390/cancers11071025
Chicago/Turabian StyleLin, Chiao-Yun, Li-Yu Lee, Tzu-Hao Wang, Cheng-Lung Hsu, Chia-Lung Tsai, Angel Chao, and Chyong-Huey Lai. 2019. "Palbociclib Promotes Dephosphorylation of NPM/B23 at Threonine 199 and Inhibits Endometrial Cancer Cell Growth" Cancers 11, no. 7: 1025. https://doi.org/10.3390/cancers11071025
APA StyleLin, C.-Y., Lee, L.-Y., Wang, T.-H., Hsu, C.-L., Tsai, C.-L., Chao, A., & Lai, C.-H. (2019). Palbociclib Promotes Dephosphorylation of NPM/B23 at Threonine 199 and Inhibits Endometrial Cancer Cell Growth. Cancers, 11(7), 1025. https://doi.org/10.3390/cancers11071025