Compound C Inhibits B16-F1 Tumor Growth in a Syngeneic Mouse Model via the Blockage of Cell Cycle Progression and Angiogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
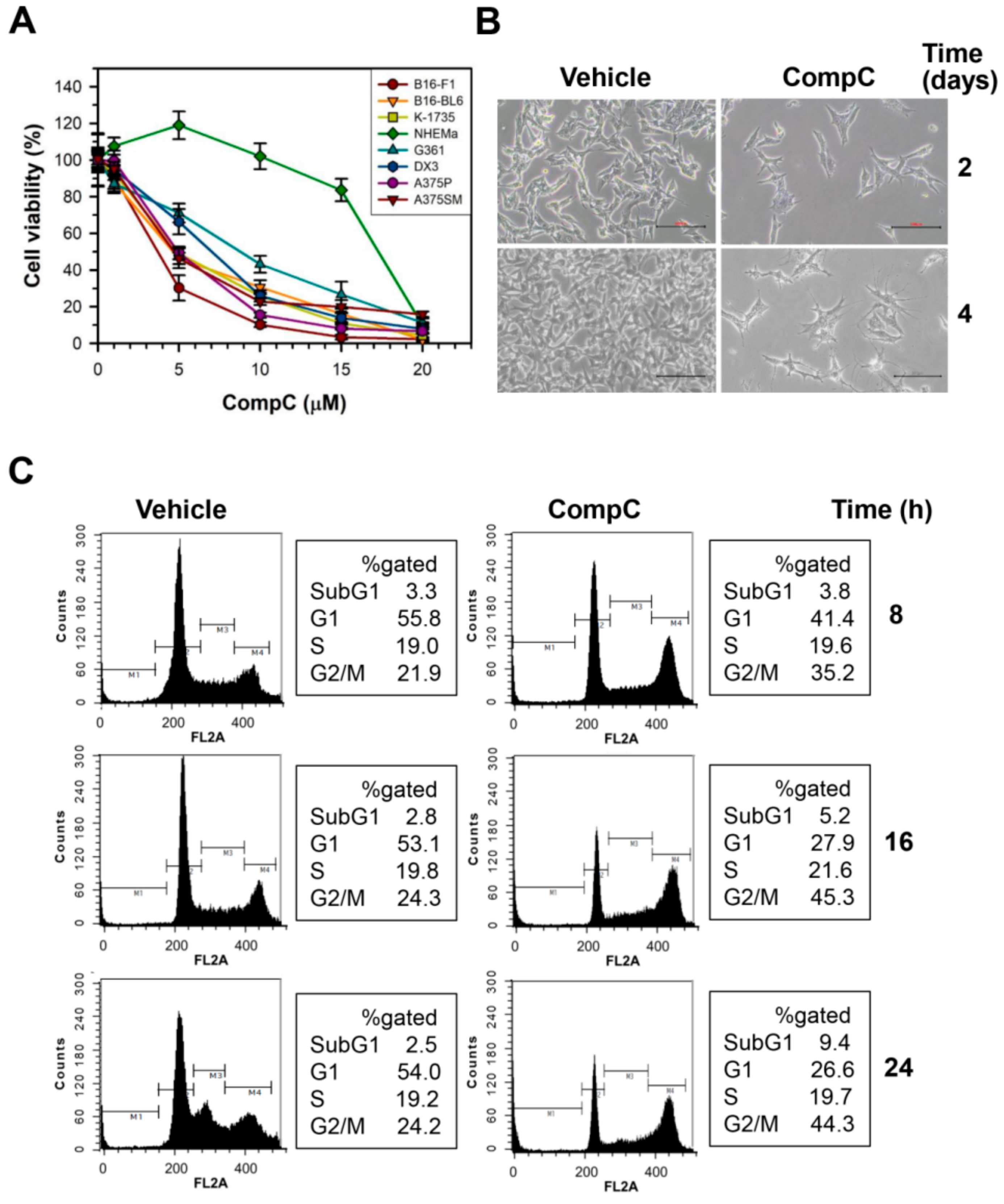
2.1. CompC Reduces the Viability of B16-F1 Melanoma Cells via G2/M Cell Cycle Arrest
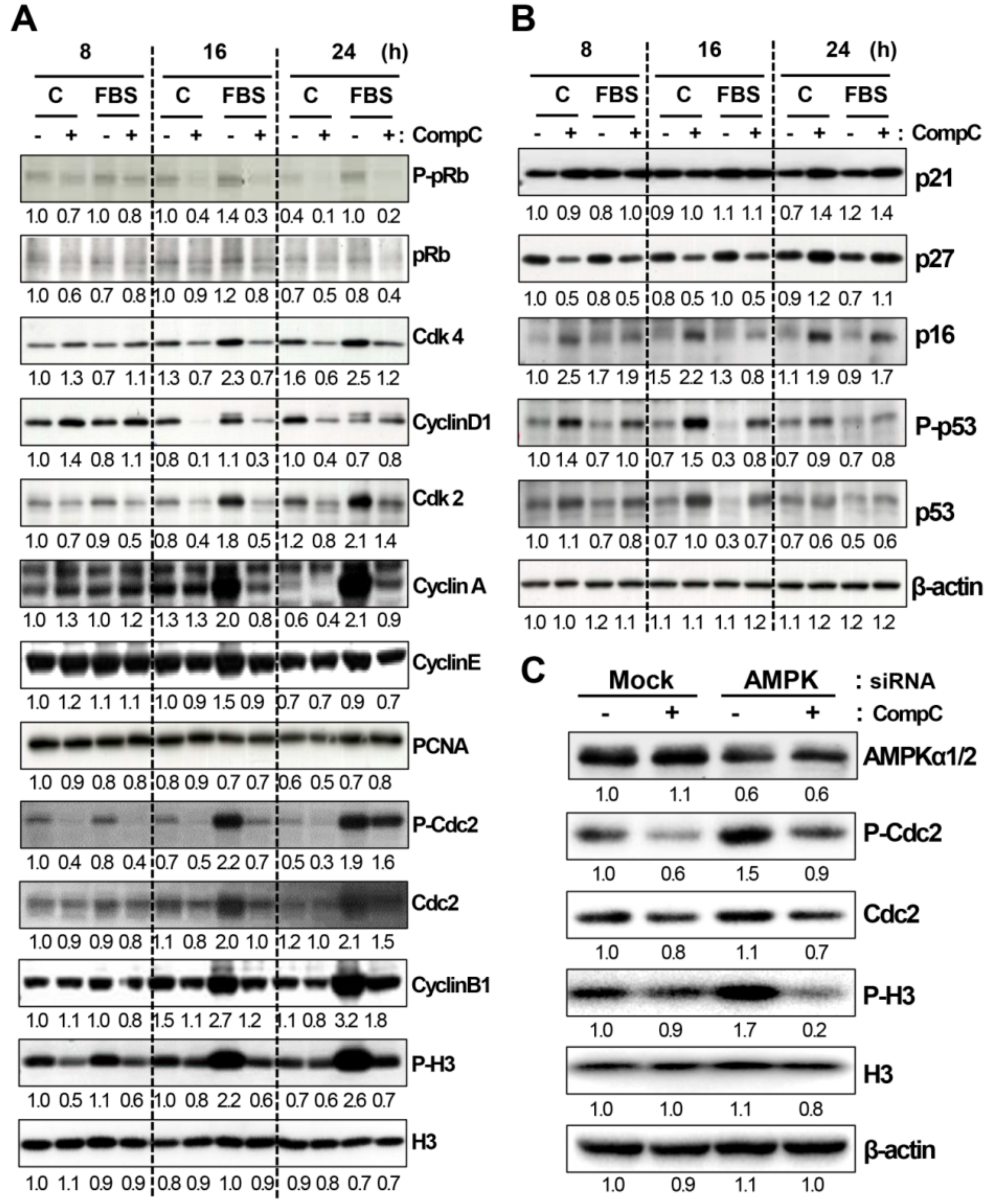
2.2. CompC Reduces the Levels of Cdks and Cyclins and Increases the Levels of Cdk Inhibitors in B16-F1 Melanoma Cells
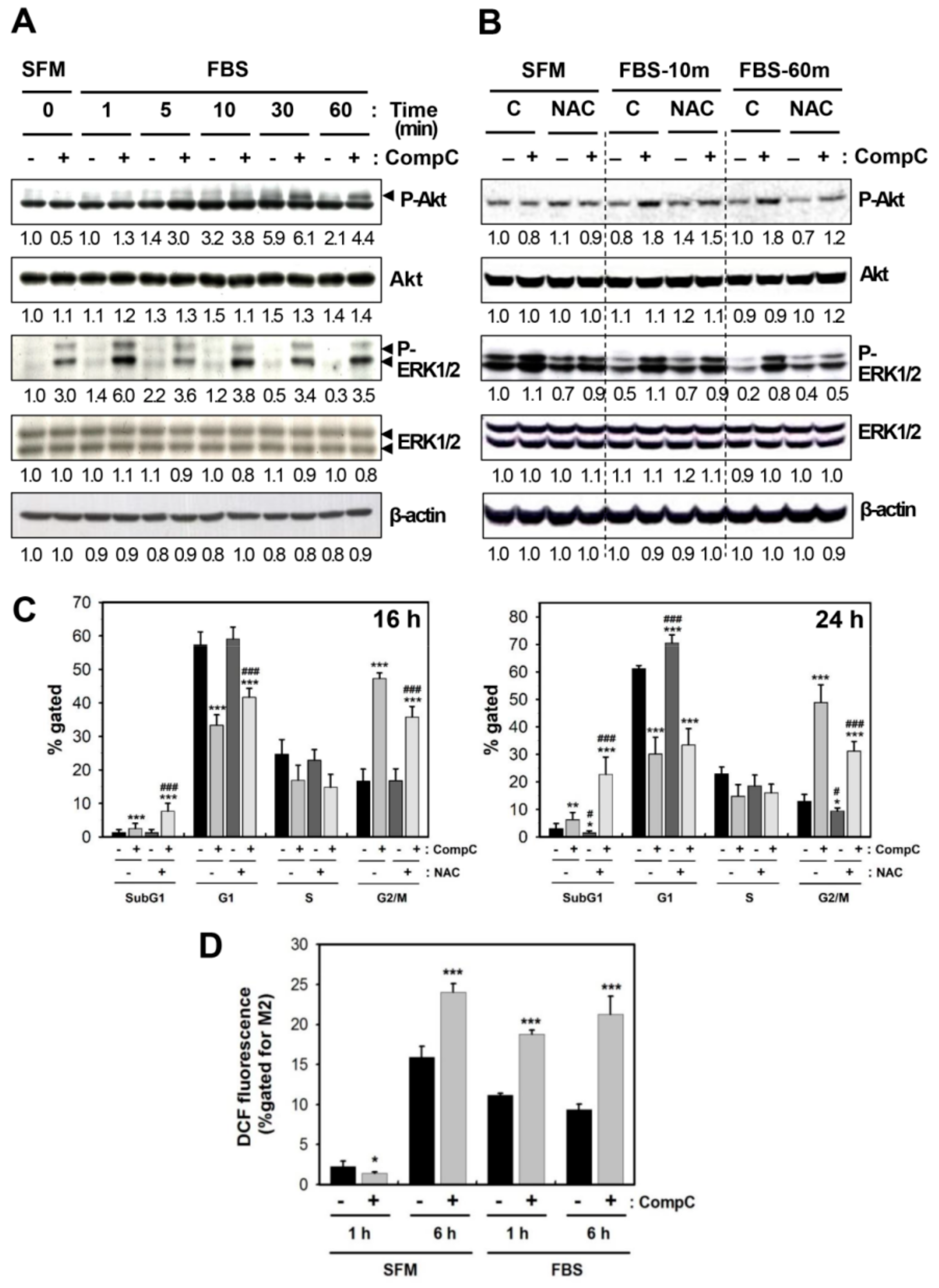
2.3. CompC Enhances the Level of Phosphorylated Akt and ERK1/2 via ROS Production in B16-F1
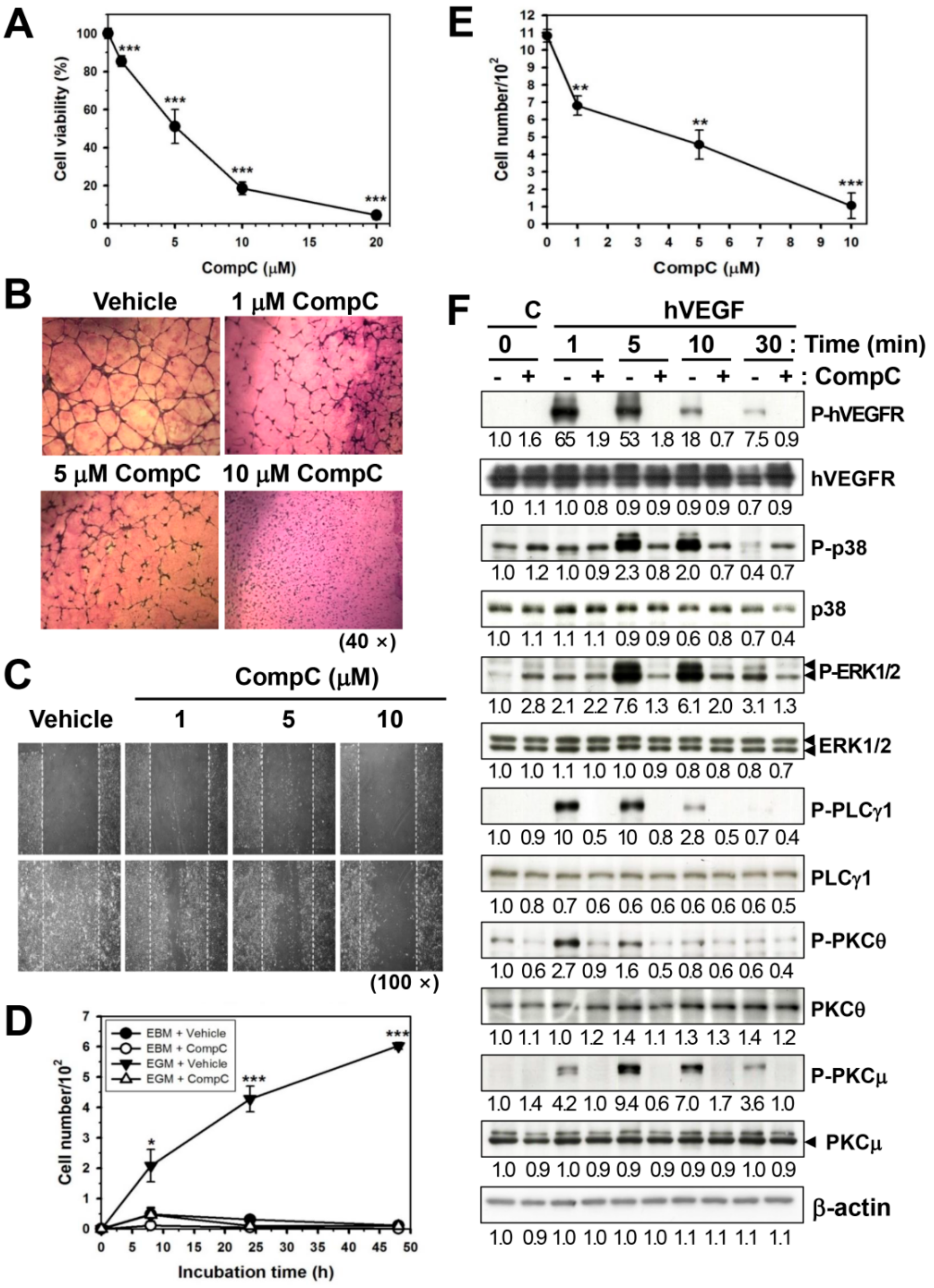
2.4. CompC Inhibits HUVEC Cell Viability, Tube Formation, and Cell Migration via the Inhibition of VEGF-Induced Signal Transduction
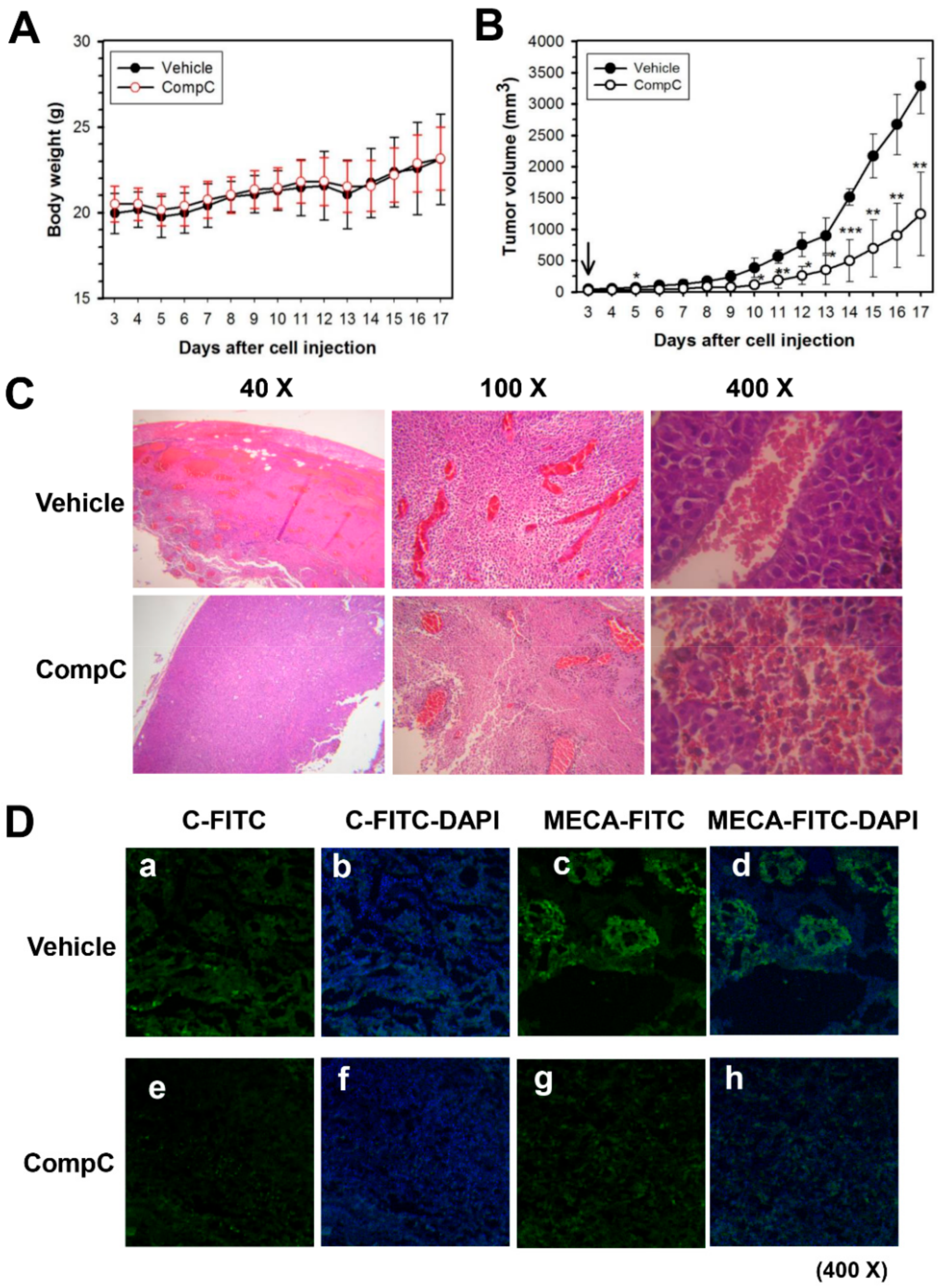
2.5. CompC Inhibits B16-F1 Tumor Growth and Angiogenesis in C57BL/6 Syngeneic Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. RNA Interference (siRNA)
4.4. MTT Assay
4.5. Flow Cytometry
4.6. Western Blot Analysis
4.7. Measurement of ROS
4.8. Tube Forming Assay
4.9. HUVEC Cell Migration
4.10. In Vivo Xenograft Experiment
4.11. H&E Staining
4.12. Immunofluorescence Staining and Confocal Microscopy
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kwon, H.J.; Kim, G.E.; Lee, Y.T.; Jeong, M.S.; Kang, I.; Yang, D.; Yeo, E.J. Inhibition of platelet-derived growth factor receptor tyrosine kinase and downstream signaling pathways by Compound C. Cell. Signal. 2013, 25, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem. Sci. 2004, 29, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chhipa, R.R.; Nakano, I.; Dasgupta, B. The AMPK inhibitor Compound C is a potent AMPK-independent anti-glioma agent. Mol. Cancer Ther. 2014, 13, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell. Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Sunitinib: A VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem. Biophys. Res. Commun. 2007, 356, 323–328. [Google Scholar] [CrossRef]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 2005, 109, 227–241. [Google Scholar] [CrossRef]
- Matsumoto, T.; Bohman, S.; Dixelius, J.; Berge, T.; Dimberg, A.; Magnusson, P.; Wang, L.; Wikner, C.; Qi, J.H.; Wernstedt, C.; et al. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 2005, 24, 2342–2353. [Google Scholar] [CrossRef]
- Ostman, A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine Growth Factor Rev. 2004, 15, 275–286. [Google Scholar] [CrossRef]
- Nozawa, F.; Hirota, M.; Okabe, A.; Shibata, M.; Iwamura, T.; Haga, Y.; Ogawa, M. Tumor necrosis factor alpha acts on cultured human vascular endothelial cells to increase the adhesion of pancreatic cancer cells. Pancreas 2000, 21, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Zhang, Y.; Georgescu, S.P.; Johnson, K.L.; Kong, D.; Galper, J.B. Human umbilical vein endothelial cells and human dermal microvascular endothelial cells offer new insights into the relationship between lipid metabolism and angiogenesis. Stem Cell Rev. 2006, 2, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Htay, A.; Dos Santos, W.; Gillies, G.T.; Fillmore, H.L.; Sholley, M.M.; Broaddus, W.C. In vitro angiogenesis by human umbilical vein endothelial cells (HUVEC) induced by three-dimensional co-culture with glioblastoma cells. J. Neurooncol. 2009, 92, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.Y.; Choi, J.H.; Yoon, H.; Hwang, K.Y.; Noh, M.H.; Choe, W.; Yoon, K.S.; Ha, J.; Yeo, E.J.; Kang, I. Luteolin induces apoptosis through endoplasmic reticulum stress and mitochondrial dysfunction in Neuro-2a mouse neuroblastoma cells. Eur. J. Pharmacol. 2011, 668, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar] [PubMed]
- Namiki, A.; Brogi, E.; Kearney, M.; Kim, E.A.; Wu, T.; Couffinhal, T.; Varticovski, L.; Isner, J.M. Hypoxia induces vascular endothelial growth factor in cultured human endothelial cells. J. Biol. Chem. 1995, 270, 31189–31195. [Google Scholar] [CrossRef] [PubMed]
- Vucicevic, L.; Misirkic, M.; Janjetovic, K.; Harhaji-Trajkovic, L.; Prica, M.; Stevanovic, D.; Isenovic, E.; Sudar, E.; Sumarac-Dumanovic, M.; Micic, D.; et al. AMP-activated protein kinase-dependent and -independent mechanisms underlying in vitro antiglioma action of compound C. Biochem. Pharmacol. 2009, 77, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Hashimoto, T.; Shimizu, K.; Yoshida, T.; Sakai, T.; Sowa, Y.; Komoto, A.; Kanazawa, K. Fucoxanthin induces cell cycle arrest at G0/G1 phase in human colon carcinoma cells through up-regulation of p21WAF1/Cip1. Biochim. Biophys. Acta 2005, 1726, 328–335. [Google Scholar] [CrossRef]
- De Boer, L.; Oakes, V.; Beamish, H.; Giles, N.; Stevens, F.; Somodevilla-Torres, M.; Desouza, C.; Gabrielli, B. Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene 2008, 27, 4261–4268. [Google Scholar] [CrossRef]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef]
- Enserink, J.M.; Kolodner, R.D. An overview of Cdk1-controlled targets and processes. Cell Div. 2010, 5, 11–41. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Moberg, K.; Boyd, S.D.; Taya, Y.; Jacks, T.; Lees, J.A. Inhibition of cyclin-dependent kinase 2 by p21 is necessary for retinoblastoma protein-mediated G1 arrest after gamma-irradiation. Proc. Natl. Acad. Sci. USA 1999, 96, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Luciani, M.G.; Hutchins, J.R.; Zheleva, D.; Hupp, T.R. The C-terminal regulatory domain of p53 contains a functional docking site for cyclin A. J. Mol. Biol. 2000, 300, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Ababneh, M.; Gotz, C.; Montenarh, M. Downregulation of the cdc2/cyclin B protein kinase activity by binding of p53 to p34(cdc2). Biochem. Biophys. Res. Commun. 2001, 283, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Slingerland, J.M. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003, 2, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, G.G.; Mahimainathan, L.; Das, F.; Venkatesan, B.; Ghosh-Choudhury, N. c-Src couples PI 3 kinase/Akt and MAPK signaling to PDGF-induced DNA synthesis in mesangial cells. Cell. Signal. 2006, 18, 1854–1864. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Matsui, T.; Rosenzweig, A. Convergent signal transduction pathways controlling cardiomyocyte survival and function: The role of PI 3-kinase and Akt. J. Mol. Cell Cardiol. 2005, 38, 63–71. [Google Scholar] [CrossRef]
- Chen, E.Y.; Mazure, N.M.; Cooper, J.A.; Giaccia, A.J. Hypoxia activates a platelet-derived growth factor receptor/phosphatidylinositol 3-kinase/Akt pathway that results in glycogen synthase kinase-3 inactivation. Cancer Res. 2001, 61, 2429–2433. [Google Scholar]
- Hers, I.; Vincent, E.E.; Tavare, J.M. Akt signalling in health and disease. Cell. Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Nakamura, N.; Vazquez, F.; Batt, D.B.; Perera, S.; Roberts, T.M.; Sellers, W.R. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 2110–2115. [Google Scholar] [CrossRef] [PubMed]
- Kandel, E.S.; Skeen, J.; Majewski, N.; Di Cristofano, A.; Pandolfi, P.P.; Feliciano, C.S.; Gartel, A.; Hay, N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol. Cell. Biol. 2002, 22, 7831–7841. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Hwang, J.T.; Yun, H.; Chi, S.G.; Lee, S.J.; Kang, I.; Yoon, K.S.; Choe, W.J.; Kim, S.S.; Ha, J. Inhibition of AMP-activated protein kinase sensitizes cancer cells to cisplatin-induced apoptosis via hyper-induction of p53. J. Biol. Chem. 2008, 283, 3731–3742. [Google Scholar] [CrossRef] [PubMed]
- Persons, D.L.; Yazlovitskaya, E.M.; Pelling, J.C. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J. Biol. Chem. 2000, 275, 35778–35785. [Google Scholar] [CrossRef] [PubMed]
- Homma, K.; Katagiri, K.; Nishitoh, H.; Ichijo, H. Targeting ASK1 in ER stress-related neurodegenerative diseases. Expert Opin. Ther. Targets 2009, 13, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Forman, H.J. Redox signaling and the MAP kinase pathways. Biofactors 2003, 17, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Minireview: The AMP-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology 2003, 144, 5179–5183. [Google Scholar] [CrossRef]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef]
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Fratto, M.E.; Imperatori, M.; Vincenzi, B.; Tomao, F.; Santini, D.; Tonini, G. New perspectives: Role of sunitinib in breast cancer. Clin. Ter. 2010, 161, 475–482. [Google Scholar] [PubMed]
- Rhim, J.H.; Jang, I.S.; Kwon, S.T.; Song, K.Y.; Yeo, E.J.; Park, S.C. Activation of wound healing in aged rats by altering the cellular mitogenic potential. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65, 704–711. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.T.; Lim, S.H.; Lee, B.; Kang, I.; Yeo, E.-J. Compound C Inhibits B16-F1 Tumor Growth in a Syngeneic Mouse Model via the Blockage of Cell Cycle Progression and Angiogenesis. Cancers 2019, 11, 823. https://doi.org/10.3390/cancers11060823
Lee YT, Lim SH, Lee B, Kang I, Yeo E-J. Compound C Inhibits B16-F1 Tumor Growth in a Syngeneic Mouse Model via the Blockage of Cell Cycle Progression and Angiogenesis. Cancers. 2019; 11(6):823. https://doi.org/10.3390/cancers11060823
Chicago/Turabian StyleLee, Yun Taek, So Hyun Lim, Boram Lee, Insug Kang, and Eui-Ju Yeo. 2019. "Compound C Inhibits B16-F1 Tumor Growth in a Syngeneic Mouse Model via the Blockage of Cell Cycle Progression and Angiogenesis" Cancers 11, no. 6: 823. https://doi.org/10.3390/cancers11060823
APA StyleLee, Y. T., Lim, S. H., Lee, B., Kang, I., & Yeo, E.-J. (2019). Compound C Inhibits B16-F1 Tumor Growth in a Syngeneic Mouse Model via the Blockage of Cell Cycle Progression and Angiogenesis. Cancers, 11(6), 823. https://doi.org/10.3390/cancers11060823