Epigenetic Remodeling through Downregulation of Polycomb Repressive Complex 2 Mediates Chemotherapy Resistance in Testicular Germ Cell Tumors

 ,
,  , , ,
, , , 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
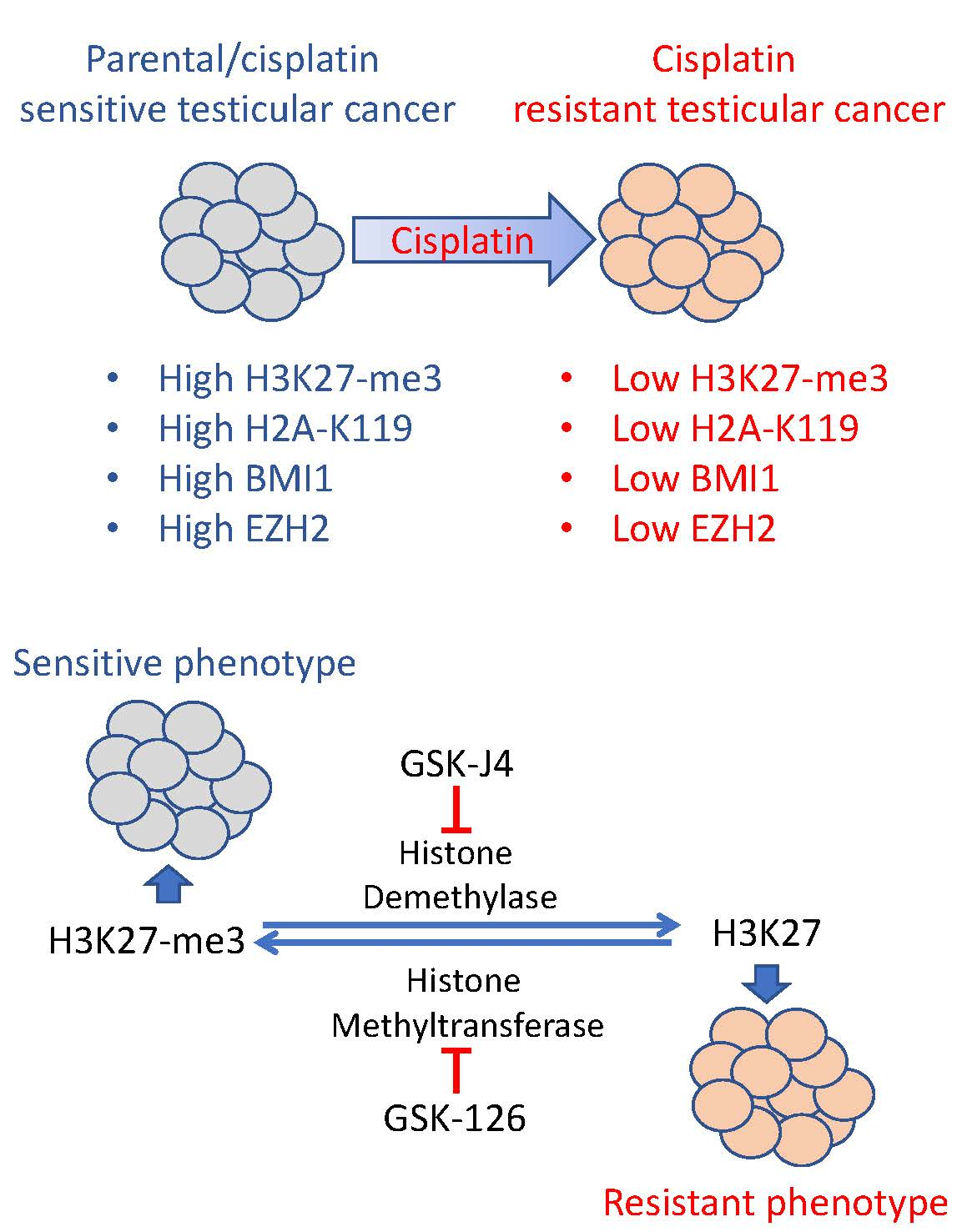
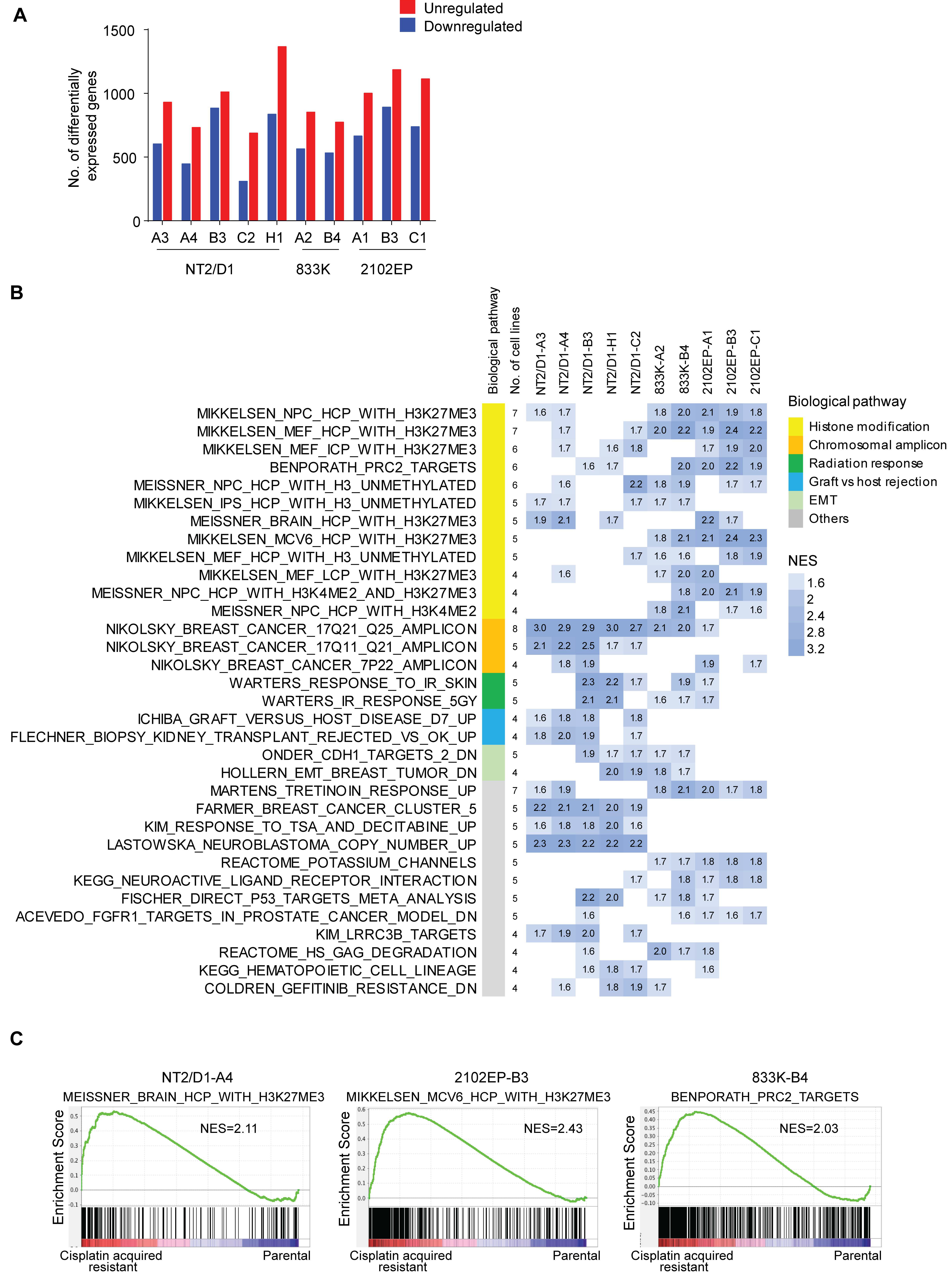
2.1. Acquired Cisplatin Resistance in Testicular Cancer Cells Is Associated with Enhanced Expression of Genes Normally Repressed by H3K27 Methylation
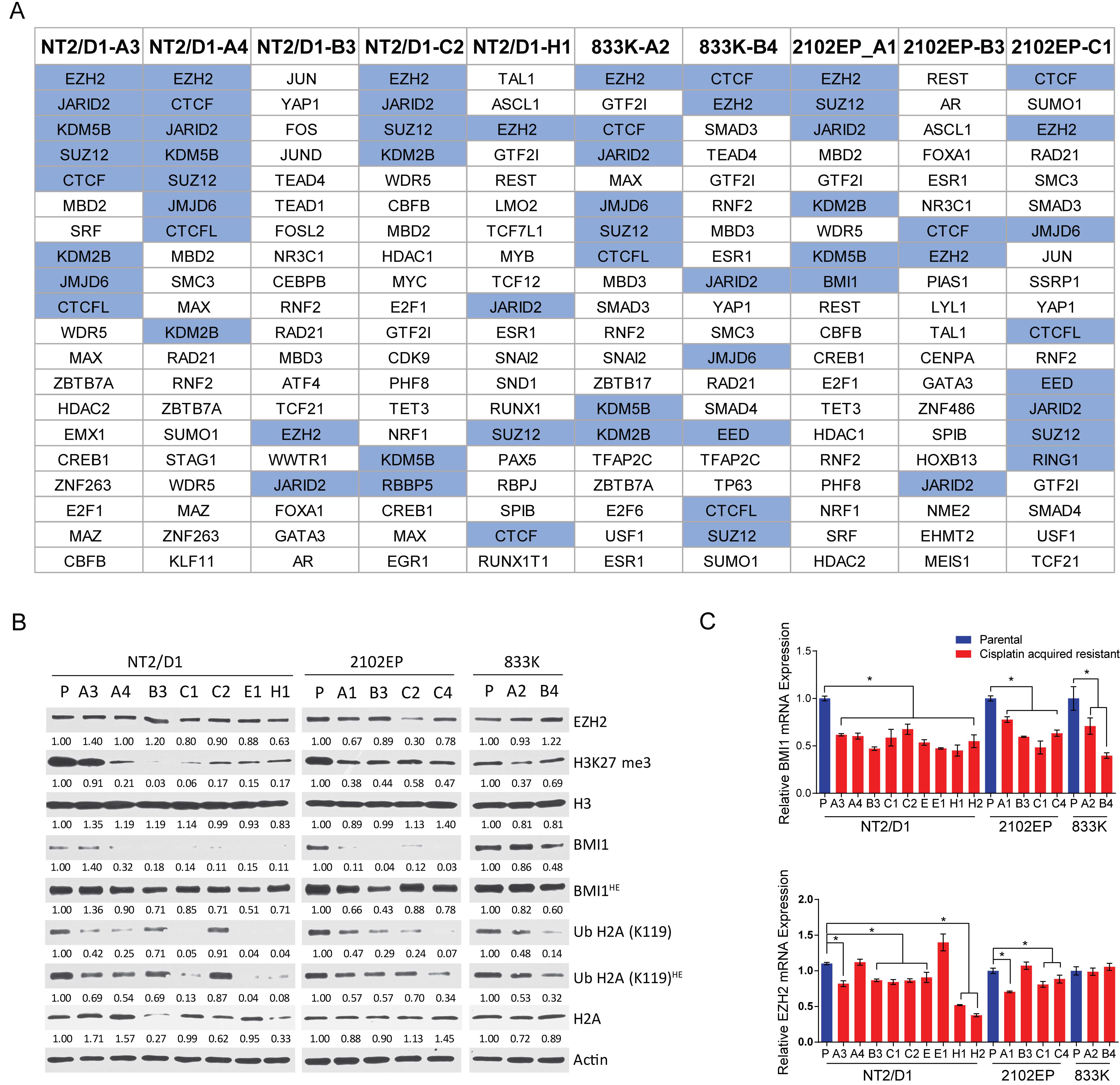
2.2. Cisplatin Resistance in Testicular Cancer Cells Is Associated with Decreased H3K27 Methylation, H2A-K119 Ubiquitination and Decreased Expression of BMI1 and EZH2.
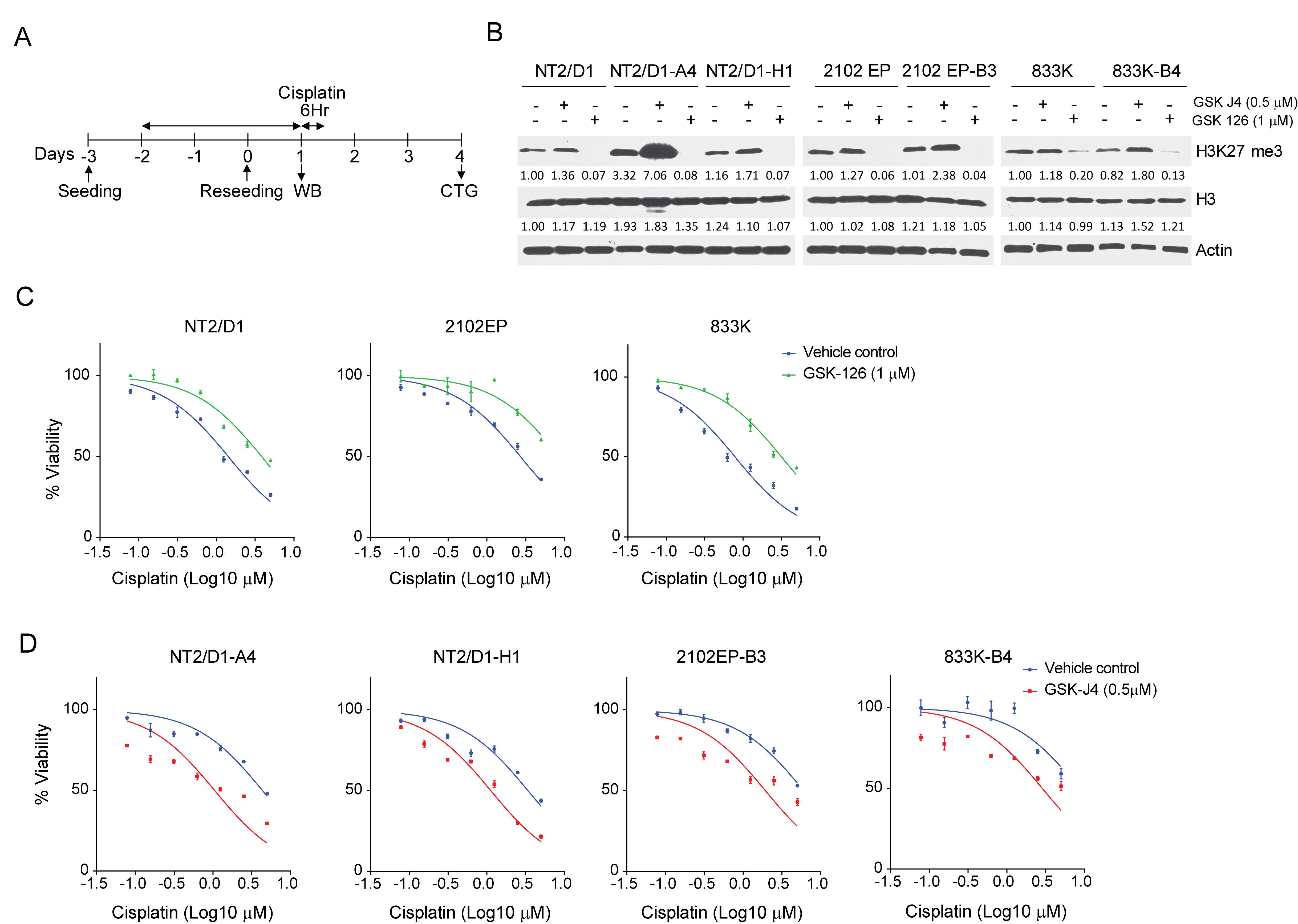
2.3. Inhibition of H3K27 Methyltransferase EZH2 Results in Cisplatin Resistance of Testicular Cancer Cells and Inhibition of H3K27 Demethylase JMJD3 Sensitizes Testicular Cancer Cells to Cisplatin
2.4. A H3K27me3-Based Gene Signature Predicts Cisplatin Sensitivity in TGCT Cells and Disease-Free Survival in TGCT Patients
3. Discussion
4. Materials and Methods
4.1. Derivation of Cisplatin Resistant Cells
4.2. Drug Treatments and Cell Viability and Proliferation Assays
4.3. Cisplatin Adduct Assay
4.4. RNA-Sequencing
4.5. Gene Set Enrichment Analysis and Gene Signature
4.6. Disease-Free Survival Analysis
4.7. Western Analysis and Real-Time PCR
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghazarian, A.A.; Kelly, S.P.; Altekruse, S.F.; Rosenberg, P.S.; McGlynn, K.A. Future of testicular germ cell tumor incidence in the United States: Forecast through 2026. Cancer 2017, 123, 2320–2328. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, D.M.; Sonne, S.B.; Ottesen, A.M.; Perrett, R.M.; Nielsen, J.E.; Almstrup, K.; Skakkebaek, N.E.; Leffers, H.; Rajpert-De Meyts, E. Origin of pluripotent germ cell tumours: The role of microenvironment during embryonic development. Mol. Cell. Endocrinol. 2008, 288, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.H.; Einhorn, L.H. Testicular cancer—Discoveries and updates. N. Engl. J. Med. 2014, 371, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, C.J.; Freemantle, S.J.; Spinella, M.J. Testicular germ cell tumors: A paradigm for the successful treatment of solid tumor stem cells. Curr. Cancer Ther. Rev. 2006, 2, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.R.; Patil, S.; Trinos, M.J.; Carousso, M.; Ginsberg, M.S.; Sheinfeld, J.; Bajorin, D.F.; Bosl, G.J.; Motzer, R.J. Progression-free and overall survival in patients with relapsed/refractory germ cell tumors treated with single-agent chemotherapy: Endpoints for clinical trial design. Cancer 2012, 118, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Porcu, P.; Bhatia, S.; Sharma, M.; Einhorn, L.H. Results of treatment after relapse from high-dose chemotherapy in germ cell tumors. J. Clin. Oncol. 2000, 18, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Koychev, D.; Oechsle, K.; Bokemeyer, C.; Honecker, F. Treatment of patients with relapsed and/or cisplatin-refractory metastatic germ cell tumours: An update. Int. J. Androl. 2011, 34, 266–273. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, M.J.; Feldman, D.R.; Carver, B.S.; Sheinfeld, J. Late relapse of testicular germ cell tumors. Urol. Clin. N. Am. 2015, 42, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, A.; Vries, E.G.; Gietema, J.A.; Spierings, D.C.; de Jong, S. Testicular germ cell tumours: The paradigm of chemo-sensitive solid tumours. Int. J. Biochem. Cell Biol. 2005, 37, 2437–2456. [Google Scholar] [CrossRef] [PubMed]
- Schmidtova, S.; Kalavska, K.; Kucerova, L. Molecular mechanisms of cisplatin chemoresistance and its circumventing in testicular germ cell tumors. Curr. Oncol. Rep. 2018, 20, 88. [Google Scholar] [CrossRef]
- Facchini, G.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Di Franco, R.; Iovane, G.; Grimaldi, G.; Piscitelli, R.; Muto, P.; Botti, G.; et al. Exploring the molecular aspects associated with testicular germ cell tumors: A review. Oncotarget 2018, 9, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- Jostes, S.; Nettersheim, D.; Schorle, H. Epigenetic drugs and their molecular targets in testicular germ cell tumours. Nat. Rev. Urol. 2019, 16, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Weiner, A.; Zack, T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016, 540, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef] [PubMed]
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic determinants of cisplatin resistance in patients with advanced germ cell tumors. J. Clin. Oncol. 2016, 34, 4000–4007. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, P.; Mathew, B.S.; Das, S.; Isaiah, R.; John, S.; Prabha, R.; Fleming, D.H.; Prabha, R.; Fleming, D.H. Cisplatin concentrations in long and short duration infusion: Implications for the optimal time of radiation delivery. J. Clin. Diagn. Res. 2016, 10, XC01–XC04. [Google Scholar] [CrossRef]
- Pasini, D.; Bracken, A.P.; Hansen, J.B.; Capillo, M.; Helin, K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell Biol. 2007, 27, 3769–3779. [Google Scholar] [CrossRef]
- Wang, Z.; Civelek, M.; Miller, C.L.; Sheffield, N.C.; Guertin, M.J.; Zang, C. BART: A transcription factor prediction tool with query gene sets or epigenomic profiles. Bioinformatics 2018, 34, 2867–2869. [Google Scholar] [CrossRef]
- Lobo, J.; Gillis, A.J.M.; Jeronimo, C.; Henrique, R.; Looijenga, L.H.J. Human germ cell tumors are developmental cancers: Impact of epigenetics on pathobiology and clinic. Int. J. Mol. Sci. 2019, 20, 258. [Google Scholar] [CrossRef]
- Honecker, F.; Wermann, H.; Mayer, F.; Gillis, A.J.; Stoop, H.; van Gurp, R.J.; Oechsle, K.; Steyerberg, E.; Hartmann, J.T.; Dinjens, W.N.; et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J. Clin. Oncol. 2009, 27, 2129–2136. [Google Scholar] [CrossRef]
- Jacobsen, C.; Honecker, F. Cisplatin resistance in germ cell tumours: Models and mechanisms. Andrology 2015, 3, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Chval, Z.; Burda, J.V. Cisplatin interaction with cysteine and methionine in aqueous solution: Computational DFT/PCM study. J. Phys. Chem. B 2009, 113, 3139–3150. [Google Scholar] [CrossRef] [PubMed]
- Gerwing, M.; Jacobsen, C.; Dyshlovoy, S.; Hauschild, J.; Rohlfing, T.; Oing, C.; Venz, S.; Oldenburg, J.; Oechsle, K.; Bokemeyer, C.; et al. Cabazitaxel overcomes cisplatin resistance in germ cell tumour cells. J. Cancer Res. Clin. Oncol. 2016, 142, 1979–1994. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Seidel, C.; Bokemeyer, C. Therapeutic approaches for refractory germ cell cancer. Expert Rev. Anticancer Ther. 2018, 18, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Albany, C.; Hever-Jardine, M.P.; von Herrmann, K.M.; Yim, C.Y.; Tam, J.; Warzecha, J.M.; Shin, L.; Bock, S.E.; Curran, B.S.; Chaudhry, A.S.; et al. Refractory testicular germ cell tumors are highly sensitive to the second generation DNA methylation inhibitor guadecitabine. Oncotarget 2017, 8, 2949–2959. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Veneti, Z.; Gkouskou, K.K.; Eliopoulos, A.G. Polycomb repressor complex 2 in genomic instability and cancer. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Maganti, H.B.; Jrade, H.; Cafariello, C.; Manias Rothberg, J.L.; Porter, C.J.; Yockell-Lelievre, J.; Battaion, H.L.; Khan, S.T.; Howard, J.P.; Li, Y.; et al. Targeting the mtf2-mdm2 axis sensitizes refractory acute myeloid leukemia to chemotherapy. Cancer Discov. 2018, 8, 1376–1389. [Google Scholar] [CrossRef]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Hojfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef]
- Gollner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Pekmezci, M.; Cuevas-Ocampo, A.K.; Perry, A.; Horvai, A.E. Significance of H3K27me3 loss in the diagnosis of malignant peripheral nerve sheath tumors. Mod. Pathol. 2017, 30, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Beyrouthy, M.J.; Garner, K.M.; Hever, M.P.; Freemantle, S.J.; Eastman, A.; Dmitrovsky, E.; Spinella, M.J. High DNA methyltransferase 3B expression mediates 5-aza-deoxycytidine hypersensitivity in testicular germ cell tumors. Cancer Res. 2009, 69, 9360–9366. [Google Scholar] [CrossRef] [PubMed]
- Wermann, H.; Stoop, H.; Gillis, A.J.; Honecker, F.; van Gurp, R.J.; Ammerpohl, O.; Richter, J.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Global DNA methylation in fetal human germ cells and germ cell tumours: Association with differentiation and cisplatin resistance. J. Pathol. 2010, 221, 433–442. [Google Scholar] [PubMed]
- Oing, C.; Verem, I.; Mansour, W.Y.; Bokemeyer, C.; Dyshlovoy, S.; Honeckerm, F. 5-Azacitidine Exerts Prolonged Pro-Apoptotic Effects and Overcomes Cisplatin-Resistance in Non-Seminomatous Germ Cell Tumor Cells. Int. J. Mol. Sci. 2018, 20. [Google Scholar] [CrossRef] [PubMed]
- Abbosh, P.H.; Montgomery, J.S.; Starkey, J.A.; Novotny, M.; Zuhowski, E.G.; Egorin, M.J.; Moseman, A.P.; Golas, A.; Brannon, K.M.; Balch, C.; et al. Dominant-negative histone H3 lysine 27 mutant derepresses silenced tumor suppressor genes and reverses the drug-resistant phenotype in cancer cells. Cancer Res. 2006, 66, 5582–5591. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Yu, L.; Li, Z.; Shen, Y.; Wang, J.; Cai, J.; Xiao, L.; Wang, Z. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol. Ther. 2010, 10, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Tang, J.; Wang, J.; Duan, G.; Zhou, L.; Zhou, X. MiR-138 acts as a tumor suppressor by targeting ezh2 and enhances cisplatin-induced apoptosis in osteosarcoma cells. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Rijlaarsdam, M.A.; Tax, D.M.; Gillis, A.J.; Dorssers, L.C.; Koestler, D.C.; de Ridder, J.; Looijenga, L.H. Genome wide DNA methylation profiles provide clues to the origin and pathogenesis of germ cell tumors. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, A.B.; Gu, H.; Bartels, S.J.; Zhang, Y.; Matarese, F.; Simmer, F.; Marks, H.; Bock, C.; Gnirke, A.; Meissner, A.; et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012, 22, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Saksouk, N.; Barth, T.K.; Ziegler-Birling, C.; Olova, N.; Nowak, A.; Rey, E.; Mateos-Langerak, J.; Urbach, S.; Reik, W.; Torres-Padilla, M.E.; et al. Redundant mechanisms to form silent chromatin at pericentromeric regions rely on BEND3 and DNA methylation. Mol. Cell 2014, 56, 580–594. [Google Scholar] [CrossRef]
- Van Mierlo, G.; Dirks, R.A.M.; De Clerck, L.; Brinkman, A.B.; Huth, M.; Kloet, S.L.; Saksouk, N.; Kroeze, L.I.; Willems, S.; Farlik, M.; et al. Integrative Proteomic Profiling Reveals PRC2-Dependent Epigenetic Crosstalk Maintains Ground-State Pluripotency. Cell Stem Cell 2019, 24, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Srivastava, A.K.; Cui, T.; Wang, Q.E.; Wani, A.A. Differential DNA lesion formation and repair in heterochromatin and euchromatin. Carcinogenesis 2016, 37, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, R.; Fazal, Z.; Corbet, A.K.; Bikorimana, E.; Rodriguez, J.C.; Khan, E.M.; Shahid, K.; Freemantle, S.J.; Spinella, M.J. Epigenetic Remodeling through Downregulation of Polycomb Repressive Complex 2 Mediates Chemotherapy Resistance in Testicular Germ Cell Tumors. Cancers 2019, 11, 796. https://doi.org/10.3390/cancers11060796
Singh R, Fazal Z, Corbet AK, Bikorimana E, Rodriguez JC, Khan EM, Shahid K, Freemantle SJ, Spinella MJ. Epigenetic Remodeling through Downregulation of Polycomb Repressive Complex 2 Mediates Chemotherapy Resistance in Testicular Germ Cell Tumors. Cancers. 2019; 11(6):796. https://doi.org/10.3390/cancers11060796
Chicago/Turabian StyleSingh, Ratnakar, Zeeshan Fazal, Andrea K. Corbet, Emmanuel Bikorimana, Jennifer C. Rodriguez, Ema M. Khan, Khadeeja Shahid, Sarah J. Freemantle, and Michael J. Spinella. 2019. "Epigenetic Remodeling through Downregulation of Polycomb Repressive Complex 2 Mediates Chemotherapy Resistance in Testicular Germ Cell Tumors" Cancers 11, no. 6: 796. https://doi.org/10.3390/cancers11060796
APA StyleSingh, R., Fazal, Z., Corbet, A. K., Bikorimana, E., Rodriguez, J. C., Khan, E. M., Shahid, K., Freemantle, S. J., & Spinella, M. J. (2019). Epigenetic Remodeling through Downregulation of Polycomb Repressive Complex 2 Mediates Chemotherapy Resistance in Testicular Germ Cell Tumors. Cancers, 11(6), 796. https://doi.org/10.3390/cancers11060796