Somatic Mutations in miRNA Genes in Lung Cancer—Potential Functional Consequences of Non-Coding Sequence Variants

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
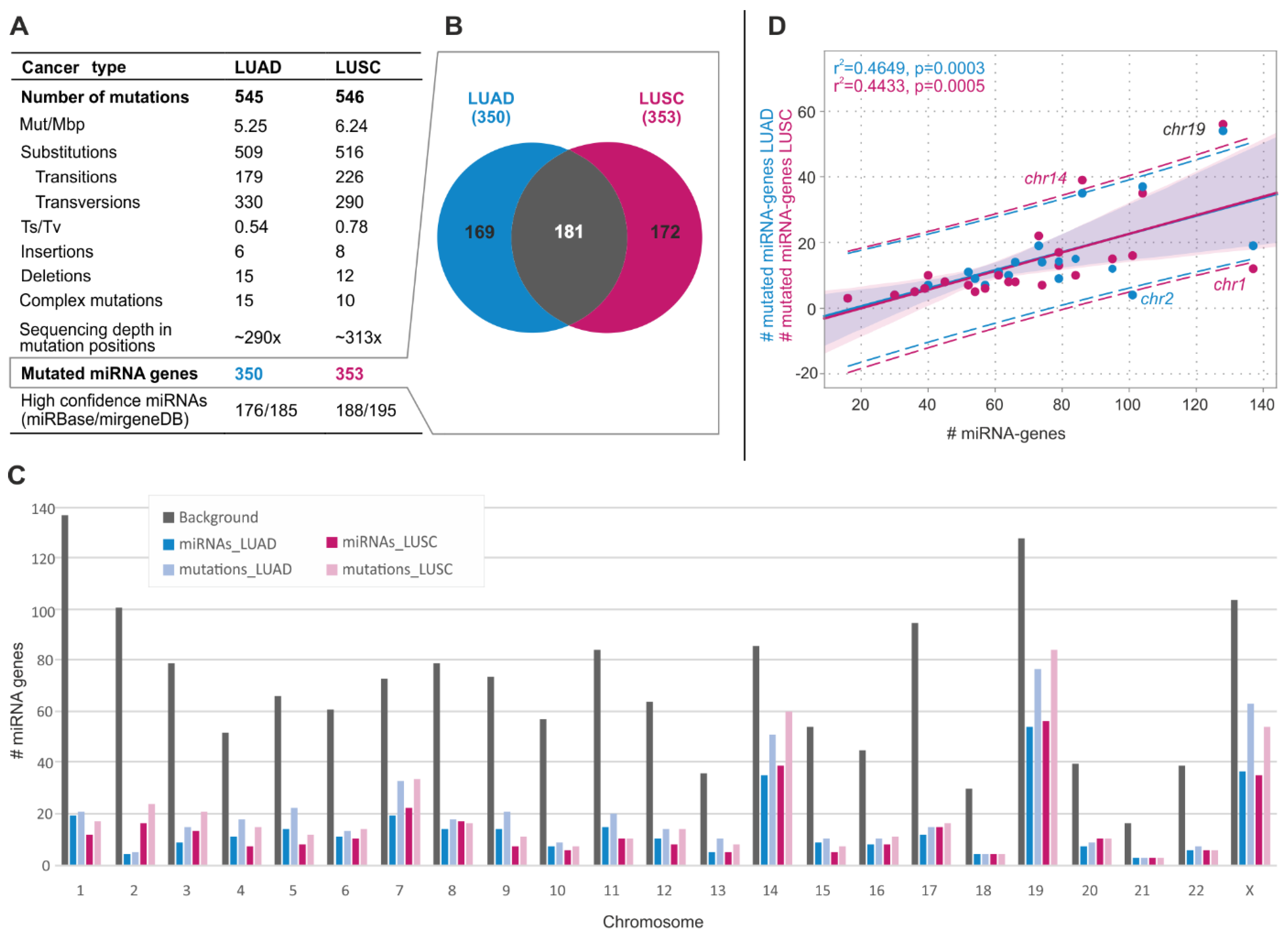
2.1. General Characteristics of the Identified Somatic Sequence Variants
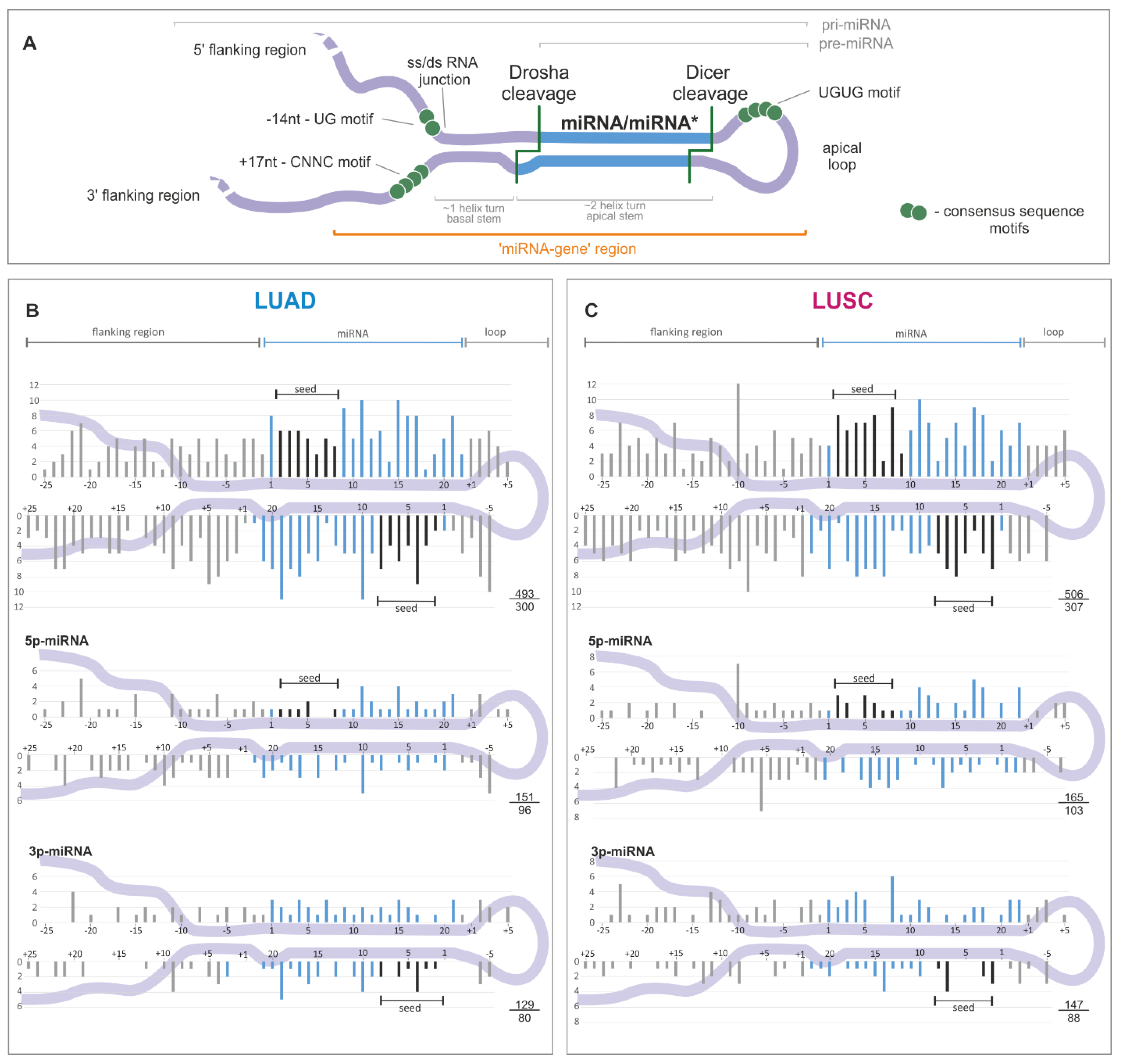
2.2. Somatic Mutations Are Not Equally Distributed Along miRNA Precursors
2.3. Mutations in miRNA Seed Regions Alter mRNA Target Recognition
2.4. Somatic Mutations May Alter Interactions of miRNA Precursors with Regulatory Proteins
2.5. Identification of Hotspot miRNA Genes
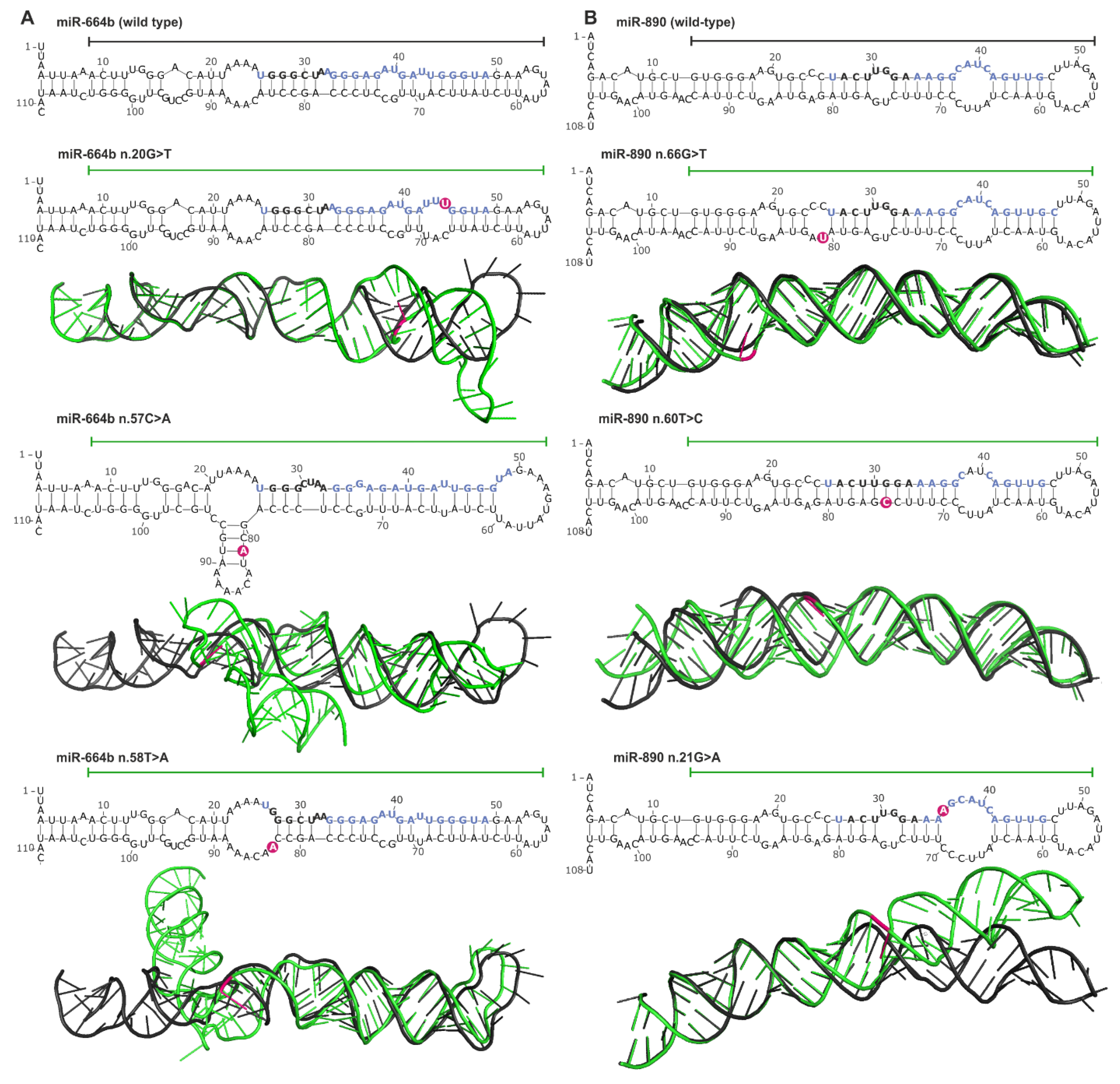
2.6. Somatic Mutations May Affect miRNA Precursor Structure
2.7. Experimental Identification of Mutations in Hotspot miRNA Genes
3. Discussion
4. Materials and Methods
4.1. Data Resources
4.2. Data Processing
4.3. Statistics
4.4. Target, Structure, and miRNA Motif predictions
4.5. Lung Sample Screening
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wong, M.C.S.; Lao, X.Q.; Ho, K.F.; Goggins, W.B.; Tse, S.L.A. Incidence and mortality of lung cancer: Global trends and association with socioeconomic status. Sci. Rep. 2017, 7, 14300. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, E.C.; McGranahan, N.; Swanton, C. Analysis of intratumor heterogeneity unravels lung cancer evolution. Mol. Cell. Oncol. 2015, 2, e985549. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, J.; Chen, Z.; Gao, Z.; Zhou, F.; Li, X.; Li, J.; Shi, S.; Feng, X.; Sun, N.; et al. Identification of somatic alterations in stage I lung adenocarcinomas by next-generation sequencing. Genes Chromosom. Cancer 2014, 53, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Morrison, C.; Wang, L.; Xiong, D.; Vedell, P.; Cui, P.; Hua, X.; Ding, F.; Lu, Y.; James, M.; et al. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis 2012, 33, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, I.S.; Devarakonda, S.; Lockwood, C.M.; Spencer, D.H.; Guebert, K.; Bredemeyer, A.J.; Al-Kateb, H.; Nguyen, T.T.; Duncavage, E.J.; Cottrell, C.E.; et al. Clinical next-generation sequencing in patients with non-small cell lung cancer. Cancer 2015, 121, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Boolell, V.; Alamgeer, M.; Watkins, D.N.; Ganju, V. The Evolution of Therapies in Non-Small Cell Lung Cancer. Cancers 2015, 7, 1815–1846. [Google Scholar] [CrossRef]
- Zhu, Q.G.; Zhang, S.M.; Ding, X.X.; He, B.; Zhang, H.Q. Driver genes in non-small cell lung cancer: Characteristics, detection methods, and targeted therapies. Oncotarget 2017, 8, 57680–57692. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef]
- Michlewski, G.; Caceres, J.F. Post-transcriptional control of miRNA biogenesis. RNA 2019, 25, 1–16. [Google Scholar] [CrossRef]
- Auyeung, V.C.; Ulitsky, I.; McGeary, S.E.; Bartel, D.P. Beyond secondary structure: Primary-sequence determinants license pri-miRNA hairpins for processing. Cell 2013, 152, 844–858. [Google Scholar] [CrossRef]
- Roden, C.; Gaillard, J.; Kanoria, S.; Rennie, W.; Barish, S.; Cheng, J.; Pan, W.; Liu, J.; Cotsapas, C.; Ding, Y.; et al. Novel determinants of mammalian primary microRNA processing revealed by systematic evaluation of hairpin-containing transcripts and human genetic variation. Genome Res. 2017, 27, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kowdley, K.V. MicroRNAs in common human diseases. Genom. Proteom. Bioinform. 2012, 10, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Slack, F.J. MicroRNA control of lifespan and metabolism. Cell Cycle 2006, 5, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Yin, Z.; Li, X.; Wu, W.; Zhou, B. Meta-analysis of human lung cancer microRNA expression profiling studies comparing cancer tissues with normal tissues. J. Exp. Clin. Cancer Res. 2012, 31, 54. [Google Scholar] [CrossRef]
- Vosa, U.; Vooder, T.; Kolde, R.; Vilo, J.; Metspalu, A.; Annilo, T. Meta-analysis of microRNA expression in lung cancer. Int. J. Cancer 2013, 132, 2884–2893. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef]
- Florczuk, M.; Szpechcinski, A.; Chorostowska-Wynimko, J. miRNAs as Biomarkers and Therapeutic Targets in Non-Small Cell Lung Cancer: Current Perspectives. Targets Oncol. 2017, 12, 179–200. [Google Scholar] [CrossRef]
- Krutovskikh, V.A.; Herceg, Z. Oncogenic microRNAs (OncomiRs) as a new class of cancer biomarkers. Bioessays 2010, 32, 894–904. [Google Scholar] [CrossRef]
- Kasinski, A.L.; Slack, F.J. Epigenetics and genetics. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 2011, 11, 849–864. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, J.; Yang, N.; Greshock, J.; Megraw, M.S.; Giannakakis, A.; Liang, S.; Naylor, T.L.; Barchetti, A.; Ward, M.R.; et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 9136–9141. [Google Scholar] [CrossRef] [PubMed]
- Czubak, K.; Lewandowska, M.A.; Klonowska, K.; Roszkowski, K.; Kowalewski, J.; Figlerowicz, M.; Kozlowski, P. High copy number variation of cancer-related microRNA genes and frequent amplification of DICER1 and DROSHA in lung cancer. Oncotarget 2015, 6, 23399–23416. [Google Scholar] [CrossRef] [PubMed]
- Oak, N.; Ghosh, R.; Huang, K.L.; Wheeler, D.A.; Ding, L.; Plon, S.E. Framework for microRNA variant annotation and prioritization using human population and disease datasets. Hum. Mutat. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Cui, Y. SomamiR 2.0: A database of cancer somatic mutations altering microRNA-ceRNA interactions. Nucleic Acids Res. 2016, 44, D1005–D1010. [Google Scholar] [CrossRef] [PubMed]
- Hornshoj, H.; Nielsen, M.M.; Sinnott-Armstrong, N.A.; Switnicki, M.P.; Juul, M.; Madsen, T.; Sallari, R.; Kellis, M.; Orntoft, T.; Hobolth, A.; et al. Pan-cancer screen for mutations in non-coding elements with conservation and cancer specificity reveals correlations with expression and survival. NPJ Genom. Med. 2018, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Hezaveh, K.; Kloetgen, A.; Bernhart, S.H.; Mahapatra, K.D.; Lenze, D.; Richter, J.; Haake, A.; Bergmann, A.K.; Brors, B.; Burkhardt, B.; et al. Alterations of microRNA and microRNA-regulated messenger RNA expression in germinal center B-cell lymphomas determined by integrative sequencing analysis. Haematologica 2016, 101, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Kwanhian, W.; Lenze, D.; Alles, J.; Motsch, N.; Barth, S.; Doll, C.; Imig, J.; Hummel, M.; Tinguely, M.; Trivedi, P.; et al. MicroRNA-142 is mutated in about 20% of diffuse large B-cell lymphoma. Cancer Med. 2012, 1, 141–155. [Google Scholar] [CrossRef]
- Berezikov, E.; Thuemmler, F.; van Laake, L.W.; Kondova, I.; Bontrop, R.; Cuppen, E.; Plasterk, R.H. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet. 2006, 38, 1375–1377. [Google Scholar] [CrossRef]
- Berezikov, E.; Guryev, V.; van de Belt, J.; Wienholds, E.; Plasterk, R.H.; Cuppen, E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell 2005, 120, 21–24. [Google Scholar] [CrossRef]
- Quach, H.; Barreiro, L.B.; Laval, G.; Zidane, N.; Patin, E.; Kidd, K.K.; Kidd, J.R.; Bouchier, C.; Veuille, M.; Antoniewski, C.; et al. Signatures of purifying and local positive selection in human miRNAs. Am. J. Hum. Genet. 2009, 84, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.A.; Liang, H.; Li, W.H. Human polymorphism at microRNAs and microRNA target sites. Proc. Natl. Acad. Sci. USA 2007, 104, 3300–3305. [Google Scholar] [CrossRef] [PubMed]
- Marcinkowska, M.; Szymanski, M.; Krzyzosiak, W.J.; Kozlowski, P. Copy number variation of microRNA genes in the human genome. BMC Genom. 2011, 12, 183. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, F.; Li, T.; Lu, M.; Wang, L.; Yue, W.; Zhang, D. MirSNP, a database of polymorphisms altering miRNA target sites, identifies miRNA-related SNPs in GWAS SNPs and eQTLs. BMC Genom. 2012, 13, 661. [Google Scholar] [CrossRef] [PubMed]
- Kroliczewski, J.; Sobolewska, A.; Lejnowski, D.; Collawn, J.F.; Bartoszewski, R. microRNA single polynucleotide polymorphism influences on microRNA biogenesis and mRNA target specificity. Gene 2018, 640, 66–72. [Google Scholar] [CrossRef]
- Sun, G.; Yan, J.; Noltner, K.; Feng, J.; Li, H.; Sarkis, D.A.; Sommer, S.S.; Rossi, J.J. SNPs in human miRNA genes affect biogenesis and function. RNA 2009, 15, 1640–1651. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, J.; Tian, T.; Zhou, X.; Gu, H.; Xu, L.; Zeng, Y.; Miao, R.; Jin, G.; Ma, H.; et al. Genetic variants of miRNA sequences and non-small cell lung cancer survival. J. Clin. Investig. 2008, 118, 2600–2608. [Google Scholar] [CrossRef]
- Jazdzewski, K.; Murray, E.L.; Franssila, K.; Jarzab, B.; Schoenberg, D.R.; de la Chapelle, A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc. Natl. Acad. Sci. USA 2008, 105, 7269–7274. [Google Scholar] [CrossRef]
- Jazdzewski, K.; Liyanarachchi, S.; Swierniak, M.; Pachucki, J.; Ringel, M.D.; Jarzab, B.; de la Chapelle, A. Polymorphic mature microRNAs from passenger strand of pre-miR-146a contribute to thyroid cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 1502–1505. [Google Scholar] [CrossRef]
- Kotlarek, M.; Kubiak, A.; Czetwertynska, M.; Swierniak, M.; Gierlikowski, W.; Kolanowska, M.; Bakula-Zalewska, E.; Jhiang, S.M.; Jazdzewski, K.; Wojcicka, A. The rs2910164 Genetic Variant of miR-146a-3p Is Associated with Increased Overall Mortality in Patients with Follicular Variant Papillary Thyroid Carcinoma. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Ghanbari, M.; Ikram, M.A.; de Looper, H.W.; Hofman, A.; Erkeland, S.J.; Franco, O.H.; Dehghan, A. Genome-wide identification of microRNA-related variants associated with risk of Alzheimer’s disease. Sci. Rep. 2016, 6, 28387. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zeng, X.; Yang, D.; Wang, W.; Liu, Z. Effects of common polymorphism rs11614913 in Hsa-miR-196a2 on lung cancer risk. PLoS ONE 2013, 8, e61047. [Google Scholar] [CrossRef] [PubMed]
- Skalski, M.; Ustaszewski, A.; Jaskiewicz, K.; Kiwerska, K.; Wierzbicka, M.; Klimza, H.; Grenman, R.; Giefing, M. Single nucleotide polymorphism rs11614913 associated with CC genotype in miR-196a2 is overrepresented in laryngeal squamous cell carcinoma, but not salivary gland tumors in Polish population. J. Appl. Genet. 2018, 59, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.E.; Bradley, D.T.; Campbell, M.; Lechner, J.; Dash, D.P.; Simpson, D.A.; Willoughby, C.E. Mutation altering the miR-184 seed region causes familial keratoconus with cataract. Am. J. Hum. Genet. 2011, 89, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Conte, I.; Hadfield, K.D.; Barbato, S.; Carrella, S.; Pizzo, M.; Bhat, R.S.; Carissimo, A.; Karali, M.; Porter, L.F.; Urquhart, J.; et al. MiR-204 is responsible for inherited retinal dystrophy associated with ocular coloboma. Proc. Natl. Acad. Sci. USA 2015, 112, E3236–E3245. [Google Scholar] [CrossRef] [PubMed]
- Mencia, A.; Modamio-Hoybjor, S.; Redshaw, N.; Morin, M.; Mayo-Merino, F.; Olavarrieta, L.; Aguirre, L.A.; del Castillo, I.; Steel, K.P.; Dalmay, T.; et al. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat. Genet. 2009, 41, 609–613. [Google Scholar] [CrossRef]
- Iliff, B.W.; Riazuddin, S.A.; Gottsch, J.D. A single-base substitution in the seed region of miR-184 causes EDICT syndrome. Investig. Ophthalmol. Vis. Sci. 2012, 53, 348–353. [Google Scholar] [CrossRef]
- Qin, A.Y.; Zhang, X.W.; Liu, L.; Yu, J.P.; Li, H.; Wang, S.Z.; Ren, X.B.; Cao, S. MiR-205 in cancer: An angel or a devil? Eur. J. Cell Biol. 2013, 92, 54–60. [Google Scholar] [CrossRef]
- Fromm, B.; Billipp, T.; Peck, L.E.; Johansen, M.; Tarver, J.E.; King, B.L.; Newcomb, J.M.; Sempere, L.F.; Flatmark, K.; Hovig, E.; et al. A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Annu. Rev. Genet. 2015, 49, 213–242. [Google Scholar] [CrossRef]
- Urbanek-Trzeciak, M.O.; Jaworska, E.; Krzyzosiak, W.J. miRNAmotif—A Tool for the Prediction of Pre-miRNA—Protein Interactions. Int. J. Mol. Sci. 2018, 19, 4075. [Google Scholar] [CrossRef]
- Hatano, K.; Kumar, B.; Zhang, Y.; Coulter, J.B.; Hedayati, M.; Mears, B.; Ni, X.; Kudrolli, T.A.; Chowdhury, W.H.; Rodriguez, R.; et al. A functional screen identifies miRNAs that inhibit DNA repair and sensitize prostate cancer cells to ionizing radiation. Nucleic Acids Res. 2015, 43, 4075–4086. [Google Scholar] [CrossRef] [PubMed]
- Pashaei, E.; Pashaei, E.; Ahmady, M.; Ozen, M.; Aydin, N. Meta-analysis of miRNA expression profiles for prostate cancer recurrence following radical prostatectomy. PLoS ONE 2017, 12, e0179543. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Tang, L.; Xu, Y.; Xu, J.; Zhang, W.; Xie, H.; Wang, S.; Guan, X. PARP inhibitor increases chemosensitivity by upregulating miR-664b-5p in BRCA1-mutated triple-negative breast cancer. Sci. Rep. 2017, 7, 42319. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, Z.; Li, X.; Tang, X.; He, J.; Lu, S. MicroRNA-1297 contributes to tumor growth of human breast cancer by targeting PTEN/PI3K/AKT signaling. Oncol. Rep. 2017, 38, 2435–2443. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.L.; Peng, X.; Zhou, H.F.; Wang, X. miR-1297 mediates PTEN expression and contributes to cell progression in LSCC. Biochem. Biophys. Res. Commun. 2012, 427, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wang, B.L.; Pan, B.S.; Guo, W. MiR-1297 regulates the growth, migration and invasion of colorectal cancer cells by targeting cyclo-oxygenase-2. Asian Pac. J. Cancer Prev. 2014, 15, 9185–9190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chi, Y.L.; Wang, P.Y.; Wang, Y.Q.; Zhang, Y.X.; Deng, J.; Lv, C.J.; Xie, S.Y. miR-511 and miR-1297 inhibit human lung adenocarcinoma cell proliferation by targeting oncogene TRIB2. PLoS ONE 2012, 7, e46090. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xue, J.; Kuang, H.; Zhou, X.; Liao, L.; Yin, F. microRNA-1297 Inhibits the Growth and Metastasis of Colorectal Cancer by Suppressing Cyclin D2 Expression. DNA Cell Biol. 2017, 36, 991–999. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, H.; Jiang, X. MiR-1297 promotes apoptosis and inhibits the proliferation and invasion of hepatocellular carcinoma cells by targeting HMGA2. Int. J. Mol. Med. 2015, 36, 1345–1352. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, Y.; Pan, Y.; Zhu, H.; Yu, C.; Sun, C. MiR-1297 suppresses pancreatic cancer cell proliferation and metastasis by targeting MTDH. Mol. Cell. Probes 2018, 40, 19–26. [Google Scholar] [CrossRef]
- Xu, M.; Qin, S.; Cao, F.; Ding, S.; Li, M. MicroRNA-379 inhibits metastasis and epithelial-mesenchymal transition via targeting FAK/AKT signaling in gastric cancer. Int. J. Oncol. 2017, 51, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Li, H.S.; Huang, J.Q.; Dong, S.H.; Huang, Z.J.; Yi, W.; Zhan, G.F.; Feng, J.T.; Sun, J.C.; Huang, X.H. MicroRNA-379-5p inhibits tumor invasion and metastasis by targeting FAK/AKT signaling in hepatocellular carcinoma. Cancer Lett. 2016, 375, 73–83. [Google Scholar] [CrossRef]
- Li, L.; Zhang, H. MicroRNA-379 inhibits cell proliferation and invasion in glioma via targeting metadherin and regulating PTEN/AKT pathway. Mol. Med. Rep. 2018, 17, 4049–4056. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Niu, X.; Tao, J.; Li, P.; Lu, Q.; Xu, A.; Chen, W.; Wang, Z. MicroRNA-379-5p plays a tumor-suppressive role in human bladder cancer growth and metastasis by directly targeting MDM2. Oncol. Rep. 2017, 37, 3502–3508. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Brougham, C.L.; Ryan, J.; Sahrudin, A.; O’Neill, G.; Wall, D.; Curran, C.; Newell, J.; Kerin, M.J.; Dwyer, R.M. miR-379 regulates cyclin B1 expression and is decreased in breast cancer. PLoS ONE 2013, 8, e68753. [Google Scholar] [CrossRef] [PubMed]
- Pollari, S.; Leivonen, S.K.; Perala, M.; Fey, V.; Kakonen, S.M.; Kallioniemi, O. Identification of microRNAs inhibiting TGF-beta-induced IL-11 production in bone metastatic breast cancer cells. PLoS ONE 2012, 7, e37361. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.J.; Hao, H.J.; Ding, Y.H.; Wen, H.; Li, X.F.; Wang, Q.R.; Zhang, B.B. Suppression of EIF4G2 by miR-379 potentiates the cisplatin chemosensitivity in nonsmall cell lung cancer cells. FEBS Lett. 2017, 591, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Shen, J.; Chan, M.T.; Wu, W.K. MicroRNA-379 suppresses osteosarcoma progression by targeting PDK1. J. Cell. Mol. Med. 2017, 21, 315–323. [Google Scholar] [CrossRef]
- Gururajan, M.; Josson, S.; Chu, G.C.; Lu, C.L.; Lu, Y.T.; Haga, C.L.; Zhau, H.E.; Liu, C.; Lichterman, J.; Duan, P.; et al. miR-154* and miR-379 in the DLK1-DIO3 microRNA mega-cluster regulate epithelial to mesenchymal transition and bone metastasis of prostate cancer. Clin. Cancer Res. 2014, 20, 6559–6569. [Google Scholar] [CrossRef]
- O’Brien, K.P.; Khan, S.; Gilligan, K.E.; Zafar, H.; Lalor, P.; Glynn, C.; O’Flatharta, C.; Ingoldsby, H.; Dockery, P.; De Bhulbh, A.; et al. Employing mesenchymal stem cells to support tumor-targeted delivery of extracellular vesicle (EV)-encapsulated microRNA-379. Oncogene 2018, 37, 2137–2149. [Google Scholar] [CrossRef] [PubMed]
- Laddha, S.V.; Nayak, S.; Paul, D.; Reddy, R.; Sharma, C.; Jha, P.; Hariharan, M.; Agrawal, A.; Chowdhury, S.; Sarkar, C.; et al. Genome-wide analysis reveals downregulation of miR-379/miR-656 cluster in human cancers. Biol. Direct 2013, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016, 7, 11215. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Lu, G.; Luo, Z.; Gui, F.; Wu, J.; Zhang, D.; Ni, Y. CircRNA circ_0067934 promotes tumor growth and metastasis in hepatocellular carcinoma through regulation of miR-1324/FZD5/Wnt/beta-catenin axis. Biochem. Biophys. Res. Commun. 2018, 497, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.L.; Cao, J.; Xu, B.; Yang, P.; Shen, F.; Sun, Z.; Li, W.L.; Wang, Q.; Liu, F. miR-892a regulated PPP2R2A expression and promoted cell proliferation of human colorectal cancer cells. Biomed. Pharmacother. 2015, 72, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Tan, L.; Jin, Z.; Jiao, Y.; Fu, Y.; Liu, Y. MiR-892a Promotes Hepatocellular Carcinoma Cells Proliferation and Invasion Through Targeting CD226. J. Cell. Biochem. 2017, 118, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, N.; Yin, D.; Li, Y.K.; Guo, L.; Shi, L.P.; Huang, X. Changes in the Expression of Serum MiR-887-5p in Patients with Endometrial Cancer. Int. J. Gynecol. Cancer 2016, 26, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Mullany, L.E.; Herrick, J.S.; Wolff, R.K.; Stevens, J.R.; Slattery, M.L. Association of cigarette smoking and microRNA expression in rectal cancer: Insight into tumor phenotype. Cancer Epidemiol. 2016, 45, 98–107. [Google Scholar] [CrossRef]
- Stegeman, S.; Moya, L.; Selth, L.A.; Spurdle, A.B.; Clements, J.A.; Batra, J. A genetic variant of MDM4 influences regulation by multiple microRNAs in prostate cancer. Endocr. Relat. Cancer 2015, 22, 265–276. [Google Scholar] [CrossRef]
- Gao, F.; Xiong, X.; Pan, W.; Yang, X.; Zhou, C.; Yuan, Q.; Zhou, L.; Yang, M. A Regulatory MDM4 Genetic Variant Locating in the Binding Sequence of Multiple MicroRNAs Contributes to Susceptibility of Small Cell Lung Cancer. PLoS ONE 2015, 10, e0135647. [Google Scholar] [CrossRef]
- Fite, K.; Elkhadragy, L.; Gomez-Cambronero, J. A Repertoire of MicroRNAs Regulates Cancer Cell Starvation by Targeting Phospholipase D in a Feedback Loop That Operates Maximally in Cancer Cells. Mol. Cell. Biol. 2016, 36, 1078–1089. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, W.; Qiao, C.; Luo, H.; Zhang, X.; Liu, D.; Zang, S.; Zhang, L.; Bai, J. The Tumor Suppressive Role of MiRNA-509-5p by Targeting FOXM1 in Non-Small Cell Lung Cancer. Cell. Physiol. Biochem. 2016, 38, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Robertson, G.; Pedersen, L.; Lim, E.; Hernandez-Herrera, A.; Rowat, A.C.; Patil, S.L.; Chan, C.K.; Wen, Y.; Zhang, X.; et al. miR-509-3p is clinically significant and strongly attenuates cellular migration and multi-cellular spheroids in ovarian cancer. Oncotarget 2016, 7, 25930–25948. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, J.; Zhang, W.; Zhang, J.; Sun, S.; Li, G.; Song, H.; Wan, D. MicroRNA-509-3p Inhibits Cancer Cell Proliferation and Migration via Upregulation of XIAP in Gastric Cancer Cells. Oncol. Res. 2017, 25, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Chen, D.; Zhang, E.; Li, Y.; Yu, Z.; Shi, M.; Jiang, Z.; Ni, L.; Yang, S.; Gui, Y.; et al. MicroRNA-509-3p inhibits cancer cell proliferation and migration by targeting the mitogen-activated protein kinase kinase kinase 8 oncogene in renal cell carcinoma. Mol. Med. Rep. 2015, 12, 1535–1543. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, X.H.; Lu, Y.; Liang, J.J.; Cao, J.X.; Jin, Y.Q.; An, G.S.; Ni, J.H.; Jia, H.T.; Li, S.Y. MiR-509-3-5p causes aberrant mitosis and anti-proliferative effect by suppression of PLK1 in human lung cancer A549 cells. Biochem. Biophys. Res. Commun. 2016, 478, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, Z.; Sheng, J.; Yu, Z.; Yao, B.; Huang, K.; Zhou, L.; Qiu, Z.; Huang, C. miR-509-3-5P inhibits the invasion and lymphatic metastasis by targeting PODXL and serves as a novel prognostic indicator for gastric cancer. Oncotarget 2017, 8, 34867–34883. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Calleja, L.; Jacques, C.; Lamoureux, F.; Baud’huin, M.; Tellez Gabriel, M.; Quillard, T.; Sahay, D.; Perrot, P.; Amiaud, J.; Charrier, C.; et al. DeltaNp63alpha Silences a miRNA Program to Aberrantly Initiate a Wound-Healing Program That Promotes TGFbeta-Induced Metastasis. Cancer Res. 2016, 76, 3236–3251. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Rao, P.; Li, W. MiR-592 represses FOXO3 expression and promotes the proliferation of prostate cancer cells. Int. J. Clin. Exp. Med. 2015, 8, 15246–15253. [Google Scholar]
- Fu, Q.; Du, Y.; Yang, C.; Zhang, D.; Zhang, N.; Liu, X.; Cho, W.C.; Yang, Y. An oncogenic role of miR-592 in tumorigenesis of human colorectal cancer by targeting Forkhead Box O3A (FoxO3A). Expert Opin. Ther. Targets 2016, 20, 771–782. [Google Scholar] [CrossRef]
- Liu, M.; Zhi, Q.; Wang, W.; Zhang, Q.; Fang, T.; Ma, Q. Up-regulation of miR-592 correlates with tumor progression and poor prognosis in patients with colorectal cancer. Biomed. Pharmacother. 2015, 69, 214–220. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W.; Zhou, L.; Yue, D.; Su, X. MicroRNA-592 targets DEK oncogene and suppresses cell growth in the hepatocellular carcinoma cell line HepG2. Int. J. Clin. Exp. Pathol. 2015, 8, 12455–12463. [Google Scholar] [PubMed]
- Li, Z.; Li, B.; Niu, L.; Ge, L. miR-592 functions as a tumor suppressor in human non-small cell lung cancer by targeting SOX9. Oncol. Rep. 2017, 37, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Zhang, H.; Bai, X.; Liu, X.; Yu, Y.; Song, L.; Du, Y. Suppressive role of miR-592 in breast cancer by repressing TGF-beta2. Oncol. Rep. 2017, 38, 3447–3454. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef]
- Tuna, M.; Amos, C.I. Activating Mutations and Targeted Therapy in Cancer. In Mutations in Human Genetic Disease; IntechOpen: London, UK, 2012; pp. 279–292. [Google Scholar]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.V.; Kreil, S.; Zoi, K.; Waghorn, K.; Curtis, C.; Zhang, L.; Score, J.; Seear, R.; Chase, A.J.; Grand, F.H.; et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 2005, 106, 2162–2168. [Google Scholar] [CrossRef]
- Gan, K.A.; Carrasco Pro, S.; Sewell, J.A.; Bass, J.I.F. Identification of Single Nucleotide Non-coding Driver Mutations in Cancer. Front. Genet. 2018, 9, 16. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef]
- Jager, N.; Schlesner, M.; Jones, D.T.; Raffel, S.; Mallm, J.P.; Junge, K.M.; Weichenhan, D.; Bauer, T.; Ishaque, N.; Kool, M.; et al. Hypermutation of the inactive X chromosome is a frequent event in cancer. Cell 2013, 155, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Gallego, B.; Bao, J.; Tsafnat, G. Pan-cancer scale landscape of simple somatic mutations. BioRxiv 2017. [Google Scholar] [CrossRef]
- Hodgkinson, A.; Chen, Y.; Eyre-Walker, A. The large-scale distribution of somatic mutations in cancer genomes. Hum. Mutat. 2012, 33, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.D.; Esquela-Kerscher, A.; Stefani, G.; Byrom, M.; Kelnar, K.; Ovcharenko, D.; Wilson, M.; Wang, X.; Shelton, J.; Shingara, J.; et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007, 67, 7713–7722. [Google Scholar] [CrossRef] [PubMed]
- Hezova, R.; Kovarikova, A.; Srovnal, J.; Zemanova, M.; Harustiak, T.; Ehrmann, J.; Hajduch, M.; Sachlova, M.; Svoboda, M.; Slaby, O. MiR-205 functions as a tumor suppressor in adenocarcinoma and an oncogene in squamous cell carcinoma of esophagus. Tumor Biol. 2016, 37, 8007–8018. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.P.; Rajapakshe, K.; Hartig, S.M.; Reva, B.; McLellan, M.D.; Kandoth, C.; Ding, L.; Zack, T.I.; Gunaratne, P.H.; Wheeler, D.A.; et al. Identification of a pan-cancer oncogenic microRNA superfamily anchored by a central core seed motif. Nat. Commun. 2013, 4, 2730. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar]
- Gagliardi, P.A.; Puliafito, A.; Primo, L. PDK1: At the crossroad of cancer signaling pathways. Semin. Cancer Biol. 2018, 48, 27–35. [Google Scholar] [CrossRef]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A Role in Cancer Stem Cell Survival and Repopulation of Cancer Cells during Therapy. Stem Cells Int. 2016, 2016, 2048731. [Google Scholar] [CrossRef] [PubMed]
- McFarland, C.D.; Yaglom, J.A.; Wojtkowiak, J.W.; Scott, J.G.; Morse, D.L.; Sherman, M.Y.; Mirny, L.A. The Damaging Effect of Passenger Mutations on Cancer Progression. Cancer Res. 2017, 77, 4763–4772. [Google Scholar] [CrossRef] [PubMed]
- Castro-Giner, F.; Ratcliffe, P.; Tomlinson, I. The mini-driver model of polygenic cancer evolution. Nat. Rev. Cancer 2015, 15, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J. ‘Latent drivers’ expand the cancer mutational landscape. Curr. Opin. Struct. Biol. 2015, 32, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Starega-Roslan, J.; Witkos, T.M.; Galka-Marciniak, P.; Krzyzosiak, W.J. Sequence features of Drosha and Dicer cleavage sites affect the complexity of isomiRs. Int. J. Mol. Sci 2015, 16, 8110–8127. [Google Scholar] [CrossRef] [PubMed]
- Starega-Roslan, J.; Galka-Marciniak, P.; Krzyzosiak, W.J. Nucleotide sequence of miRNA precursor contributes to cleavage site selection by Dicer. Nucleic Acids Res. 2015, 43, 10939–10951. [Google Scholar] [CrossRef]
- Galka-Marciniak, P.; Olejniczak, M.; Starega-Roslan, J.; Szczesniak, M.W.; Makalowska, I.; Krzyzosiak, W.J. siRNA release from pri-miRNA scaffolds is controlled by the sequence and structure of RNA. Biochim. Biophys. Acta 2016, 1859, 639–649. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Arase, M.; Matsuyama, H.; Choi, Y.L.; Ueno, T.; Mano, H.; Sugimoto, K.; Miyazono, K. MCPIP1 ribonuclease antagonizes dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol. Cell 2011, 44, 424–436. [Google Scholar] [CrossRef]
- Hata, A.; Davis, B.N. Control of microRNA biogenesis by TGFbeta signaling pathway-A novel role of Smads in the nucleus. Cytokine Growth Factor Rev. 2009, 20, 517–521. [Google Scholar] [CrossRef]
- Davis, B.N.; Hilyard, A.C.; Nguyen, P.H.; Lagna, G.; Hata, A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol. Cell 2010, 39, 373–384. [Google Scholar] [CrossRef]
- Kim, K.; Nguyen, T.D.; Li, S.; Nguyen, T.A. SRSF3 recruits DROSHA to the basal junction of primary microRNAs. RNA 2018, 24, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Dobson, J.R.; Oesper, L.; Vandin, F. Identifying driver mutations in sequenced cancer genomes: Computational approaches to enable precision medicine. Genome Med. 2014, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Kamburov, A.; Tamborero, D.; Pons, T.; Grases, D.; Valencia, A.; Lopez-Bigas, N.; Getz, G.; Godzik, A. Comparison of algorithms for the detection of cancer drivers at subgene resolution. Nat. Methods 2017, 14, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LUAD | LUSC | |||||||
|---|---|---|---|---|---|---|---|---|
| N Mutations | Mut/Mbp | Fold Change | p-Value | N Mutations | Mut/Mbp | Fold Change | p-Value | |
| total | 509 | 4.90 | 1 | - | 516 | 5.90 | 1 | - |
| 5′-flanking region | 84 | 3.60 | 0.73 | 0.0010341 | 103 | 5.24 | 0.89 | 0.1845670 |
| 3′-flanking region | 104 | 4.45 | 0.91 | 0.2909070 | 114 | 5.80 | 0.98 | 0.8810662 |
| loop | 88 | 5.23 | 1.07 | 0.5342181 | 70 | 4.94 | 0.84 | 0.1142622 |
| passenger strand | 68 | 4.30 | 0.88 | 0.2699724 | 74 | 5.56 | 0.94 | 0.6339850 |
| guide strand | 170 | 6.92 | 1.41 | 0.0000008 | 155 | 7.50 | 1.27 | 0.0010096 |
| seed region | 47 | 5.89 | 1.20 | 0.2197888 | 53 | 7.90 | 1.34 | 0.0392875 |
| Binding Protein | Sequence Motif | Lost | Gained |
|---|---|---|---|
| hnRNPA1 | UAGGGAW | 0 | 0 |
| HuR | AUUUUUAUUUU | 0 | 0 |
| KSRP | GGGU | 5 | 0 |
| Lin28 | GGAG | 1 | 3 |
| MBNL1 | YGCY | 4 | 5 |
| MCPIP1 | UGC | 5 | 8 |
| DGCR8 | UGU | 14 | 9 |
| MATR3 | AUCUU | 1 | 1 |
| ZC3H7 | SMUANY | 1 | 2 |
| YBX1 | CAUC | 3 | 5 |
| TRIM71 | UAUAA | 0 | 1 |
| PTBP1/3 | UUUUUCCNUCUUU | 0 | 0 |
| DDX17 | VCAUCH | 5 | 10 |
| RBFOX | GCAUG | 2 | 3 |
| SMAD | CAGAC | 8 | 3 |
| CELF1/2 | UGUNNNNNNNUGU | 5 | 3 |
| ZC3H10 | GCAGCGC | 1 | 1 |
| Total | 55 | 54 |
| miRNA | Cancer-Related Functions | miRTarBase /Literature Validated Targets | miRBase/MiRGeneDB Confidence | Number of Mutations LUAD/LUSC * | p-Value Nominal (BH Corrected **) | Weighted Mut. Score LUAD/LUSC * | Weighted p-Value Nominal (BH Corrected **) | |
|---|---|---|---|---|---|---|---|---|
| LUAD | miR-890 chrX (−) | oncomiR; inhibits DNA repair and damage response genes; potent IR sensitizing agent due to targeting the MAD2L2, WEE1 and XPC genes, downregulated in PRC [61,62] | MAD2L2, WEE1, XPC | −/+ | 7/4 | 9.2 × 10-8 (9.5 × 10-5) | 9/4.5 | 1.3 × 10−9 (1.5 × 10−6) |
| miR-664b chrX (+) | suppressormiR; inhibits proliferation, migration, and invasion and increases the chemosensitivity of BRCA1-mutated TNBC cells by targeting CCNE2 [63] | CCNE2 | +/− | 7/0 | 1.2 × 10-7 (9.5 × 10-5) | 9.5/0 | 1.8 × 10−9 (1.5 × 10−6) | |
| miR-1297 chr13 (−) | oncomiR; promotes cell progression in BC and LUSC by targeting PTEN, resulting in activation of PTEN/PI3K/AKT signaling pathway [64,65]; suppressormiR; suppresses cell proliferation by targeting TRIB2 and further increasing C/EBPα expression in LUAD [65,66,67]; inhibits the growth and metastasis of CRC by suppressing CCND2 [68]; suppresses PTEN expression and inhibits cell progression in LUSC [65]; promotes apoptosis and inhibits the proliferation and invasion of HCC cells by targeting HMGA2 [69]; inhibits the growth, migration and invasion of CRC cells by targeting Cox-2 [66]; suppresses PAC cell proliferation and metastasis by targeting MTDH [70] | TRIB2, MALAT1, HMGA1, HMGA2, PTEN, CCND2, COX2, MTDH | −/− | 6/0 | 1.8 × 10-6 (7.0 × 10-4) | 8/0 | 2.4 × 10−8 (1.3 × 10−5) | |
| miR-379 chr14 (+) | suppressormiR; inhibits tumor invasion and metastasis by targeting FAK and suppressing FAK/AKT signaling in HCC and GC [71,72]; inhibits cell proliferation and invasion in glioma by targeting MTDH and inhibiting the PTEN/AKT pathway [73]; inhibits BLC growth and metastasis by targeting MDM2 [74]; inhibits cell proliferation by suppressing CCND1; downregulated in BC [75]; inhibits TGF-β-induced IL-11 production in bone metastatic BC cells [76]; suppresses EIF4G2 and potentiates cisplatin chemosensitivity in NSCLC [77]; suppresses osteosarcoma progression by targeting PDK1 [78]; inhibits EMT and bone metastasis of PRC [79]; suppresses BC progression by targeting COX2; potential therapeutic agent in BC treatment [80]; miR-379/miR-656 cluster is downregulated in multiple human cancers, especially GBM, KIRC and BC [81]; sponged by circHIPK3 [82] | IL11, PTK2, CCND1, MTDH, EIF4G2, PDK1, COX2, MDM2 | +/+ | 6/1 | 2.4 × 10-6 (7.3 × 10-4) | 8/1.5 | 3.7 × 10−8 (1.5 × 10−5) | |
| miR-1324 chr3 (+) | suppressormiR; targets FZD5 and downregulates the Wnt/β-catenin signaling pathway in HCC; inhibited by circ_0067934; the circ_0067934/miR-1324/FZD5/Wnt/β-catenin signaling pathway axis is involved in the regulation of HCC progression [83] | FZD5 | −/− | 6/2 | 6.1 × 10-6 (1.4 × 10-3) | 8/2 | 1.2 × 10−7 (4.0 × 10−5) | |
| miR-892a chrX (−) | oncomiR; upregulated in human CRC tissues and cell lines; promotes cell proliferation and colony formation by suppressing PPP2R2A expression [84]; promotes HCC cell proliferation and invasion through targeting CD226 [85] | PPP2R2A, CD226 | +/+ | 6/1 | 1.4 × 10-6 (7.0 × 10-4) | 7.5/1 | 3.7 × 10−7 (9.8 × 10−5) | |
| miR-887 chr5 (+) | oncomiR; potential biomarker for ENC; serum miR-887-5p levels are increased in patients with ENC [86]; increased levels in CRC [87]; suppressormiR; downregulates MDM4 in PRC and SCLC by having a high affinity for the MDM4 rs4245739 SNP C-allele [88,89]; inhibits the invasion of BC cells by targeting PLD2 [90] | PLD2, MDM4 | +/+ | 5/3 | 4.6 × 10-5 (8.0 × 10-3) | 5.5/3 | 1.6 × 10−4 (2.9 × 10−2) | |
| miR-509-3 chrX (−) | suppressormiR; decreases cell proliferation, migration and invasion of NSCLC cells by targeting FOXM1 [91]; attenuates cellular migration and multicellular spheroid formation in OVC by targeting YAP1 [92]; inhibits the migration and proliferation of GC cells by targeting XIAP and of KIRC by targeting MAP3K8 [93,94]; represses PLK1, causing mitotic aberration and growth arrest of LUAD A549 cell line [95]; inhibits the invasion and metastasis of GC by targeting PODXL [96] | NTRK3, CFTR, XIAP, MAP3K8, FOXM1, YAP1, PLK1, PODXL | +/+ | 5/2 | 4.0 × 10-5 8.0 × 10-3) | 5.5/3 | 5.8 × 10−7 (1.3 × 10-4) | |
| LUSC | miR-527 chr19 (+) | suppressormiR; reduces metastatic dissemination by repressing SMAD4 in osteosarcoma [97] | SMAD4 | −/+ | 1/6 | 2.7 × 10-6 (7.3 × 10-4) | 1.5/7.5 | 8.0 × 10−7 (1.6 × 10−4) |
| miR-592 chr7 (−) | oncomiR; represses FOXO3 expression and promotes the proliferation of PRC cells [98,99]; upregulation correlates with tumor progression and poor prognosis for patients with CRC [100] suppressormiR; targets the DEK oncogene and suppresses cell growth in the HCC cell line HepG2 [101]; tumor suppressor in NSCLC by targeting SOX9 [102]; inhibits cell proliferation, colony formation, migration and invasion in BC by targeting TGFβ-2 [103] | FOXO3, DEK, SOX9 | −/+ | 3/5 | 5.0 × 10-5 (8.0 × 10-3) | 4.5/5.5 | 1.8 × 10−4 (2.9 × 10−2) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galka-Marciniak, P.; Urbanek-Trzeciak, M.O.; Nawrocka, P.M.; Dutkiewicz, A.; Giefing, M.; Lewandowska, M.A.; Kozlowski, P. Somatic Mutations in miRNA Genes in Lung Cancer—Potential Functional Consequences of Non-Coding Sequence Variants. Cancers 2019, 11, 793. https://doi.org/10.3390/cancers11060793
Galka-Marciniak P, Urbanek-Trzeciak MO, Nawrocka PM, Dutkiewicz A, Giefing M, Lewandowska MA, Kozlowski P. Somatic Mutations in miRNA Genes in Lung Cancer—Potential Functional Consequences of Non-Coding Sequence Variants. Cancers. 2019; 11(6):793. https://doi.org/10.3390/cancers11060793
Chicago/Turabian StyleGalka-Marciniak, Paulina, Martyna Olga Urbanek-Trzeciak, Paulina Maria Nawrocka, Agata Dutkiewicz, Maciej Giefing, Marzena Anna Lewandowska, and Piotr Kozlowski. 2019. "Somatic Mutations in miRNA Genes in Lung Cancer—Potential Functional Consequences of Non-Coding Sequence Variants" Cancers 11, no. 6: 793. https://doi.org/10.3390/cancers11060793
APA StyleGalka-Marciniak, P., Urbanek-Trzeciak, M. O., Nawrocka, P. M., Dutkiewicz, A., Giefing, M., Lewandowska, M. A., & Kozlowski, P. (2019). Somatic Mutations in miRNA Genes in Lung Cancer—Potential Functional Consequences of Non-Coding Sequence Variants. Cancers, 11(6), 793. https://doi.org/10.3390/cancers11060793