Chronic Treatment with Multi-Kinase Inhibitors Causes Differential Toxicities on Skeletal and Cardiac Muscles

 and
and
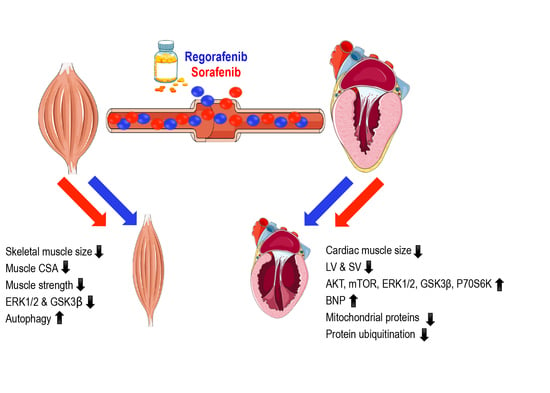
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
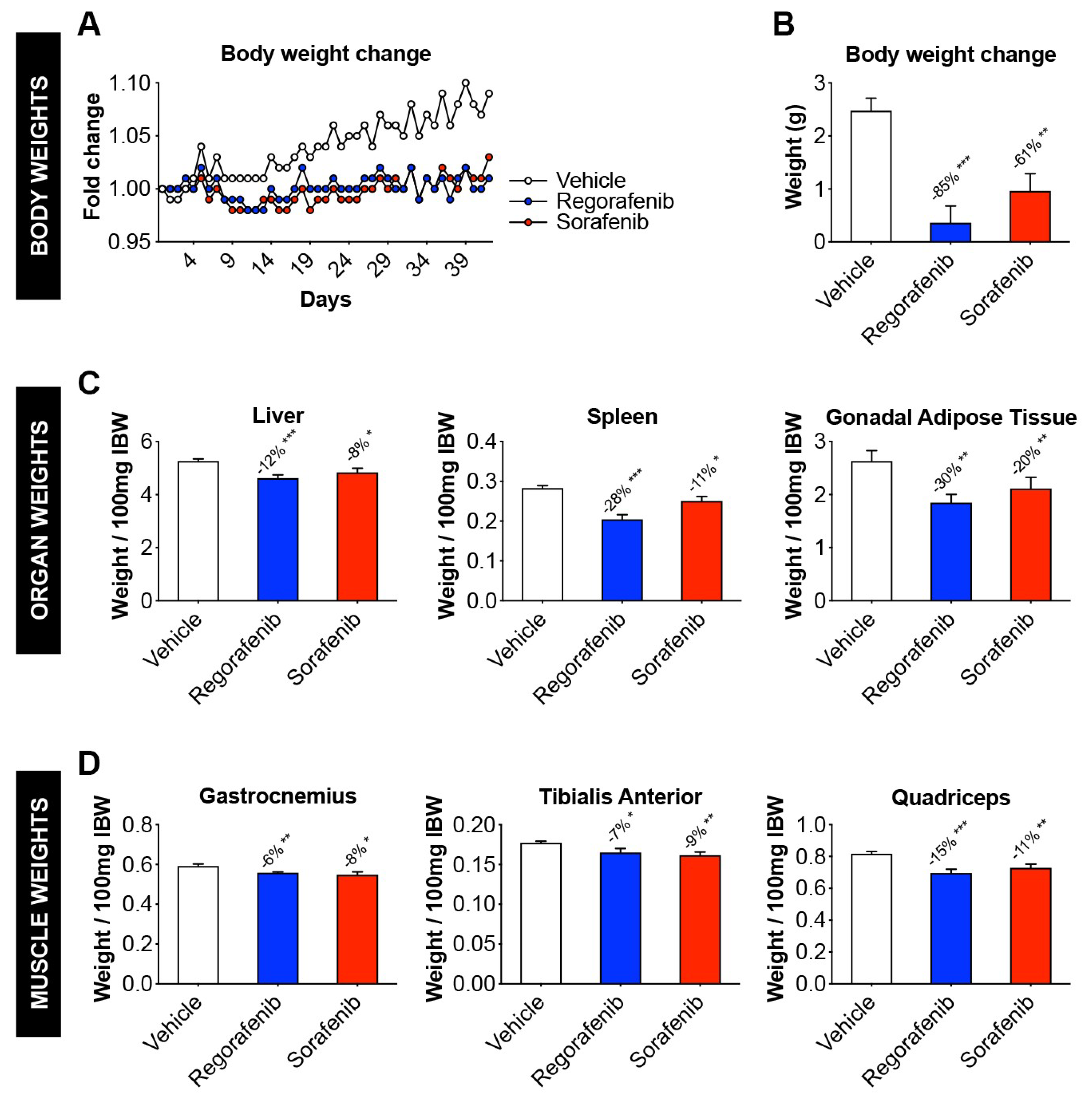
2.1. Animals Exposed to Regorafenib or Sorafenib Display Impaired Growth
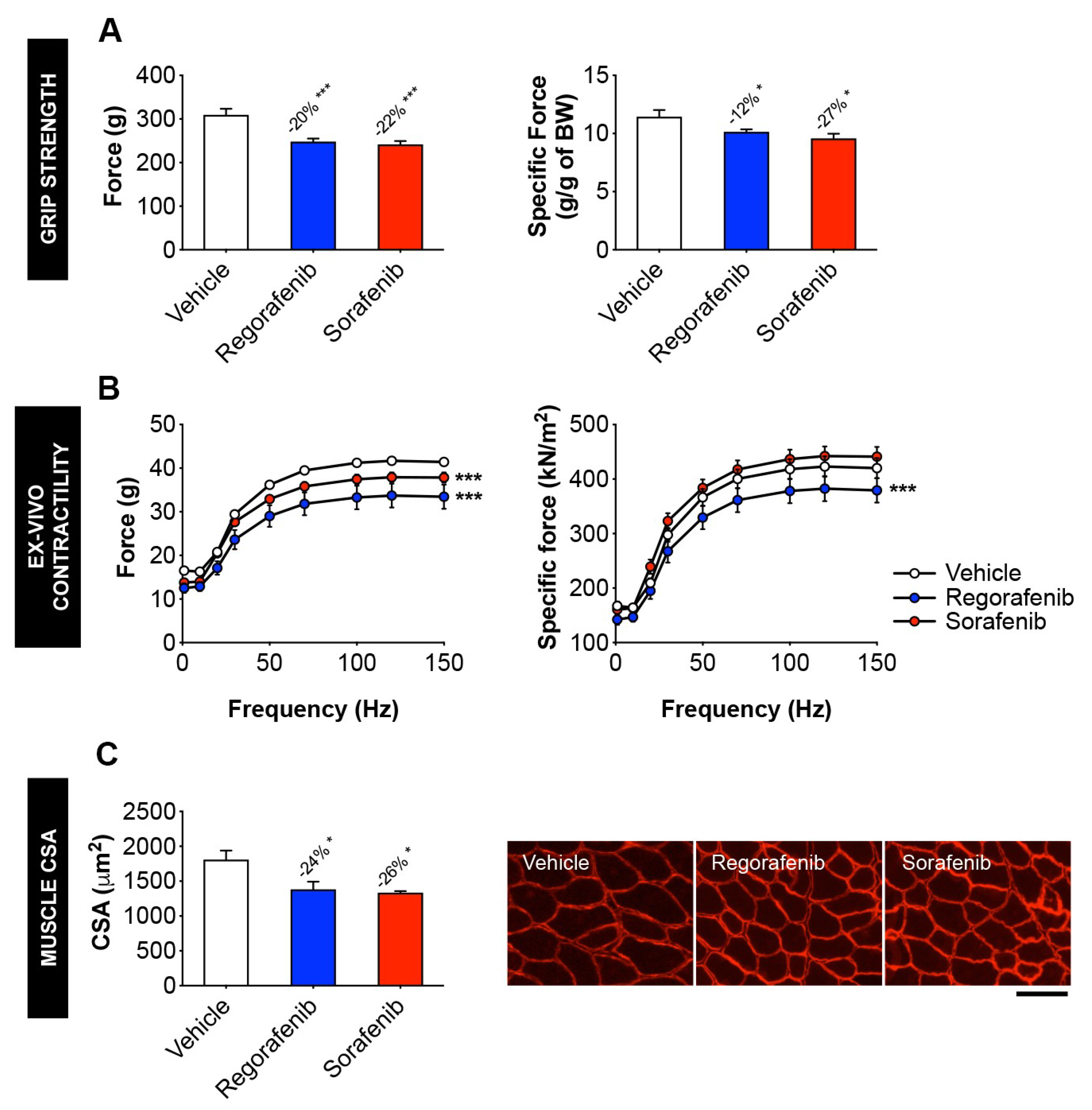
2.2. MKIs Promote Skeletal Muscle Weakness
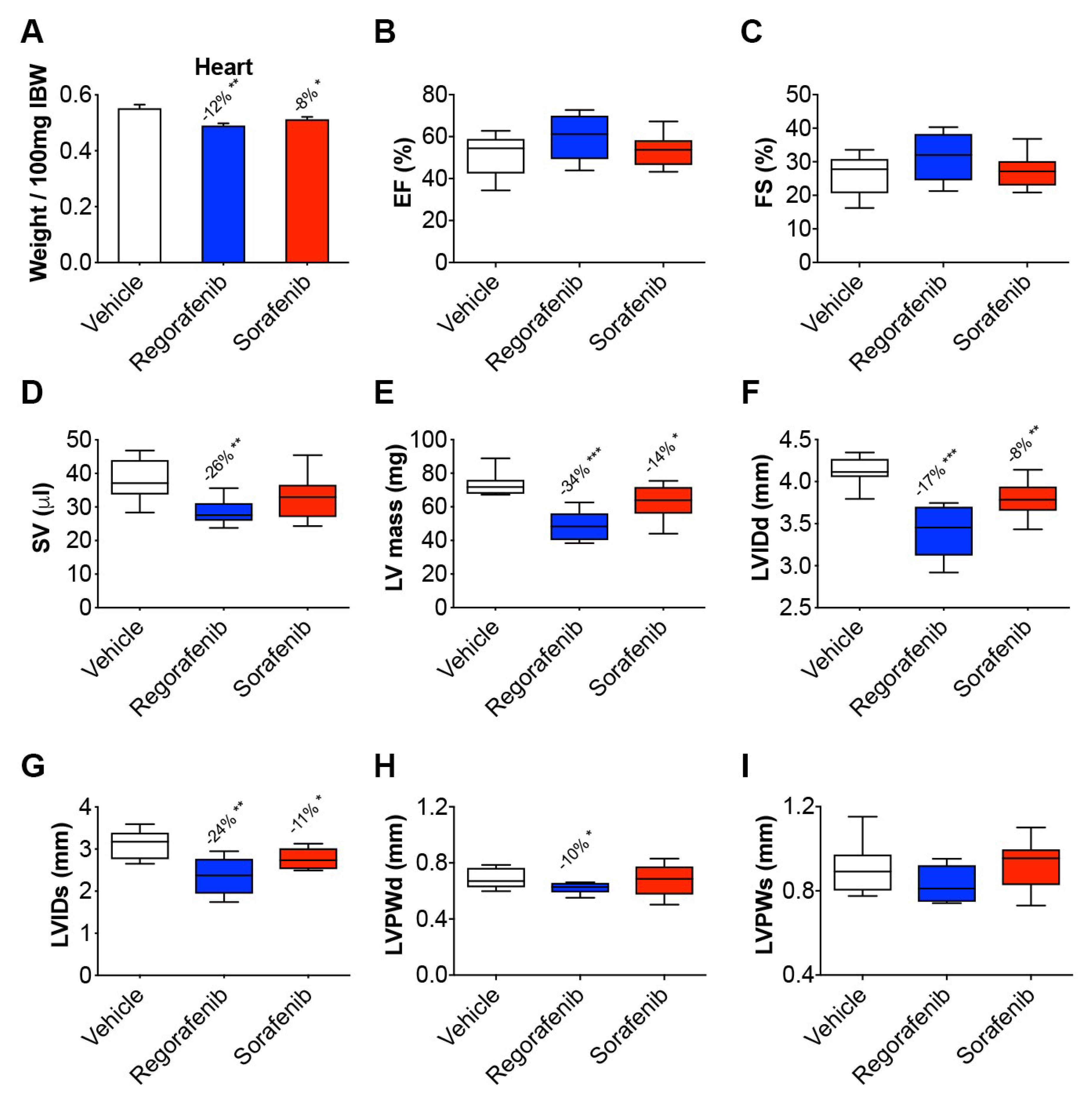
2.3. Regorafenib and Sorafenib Perturb Cardiac Muscle
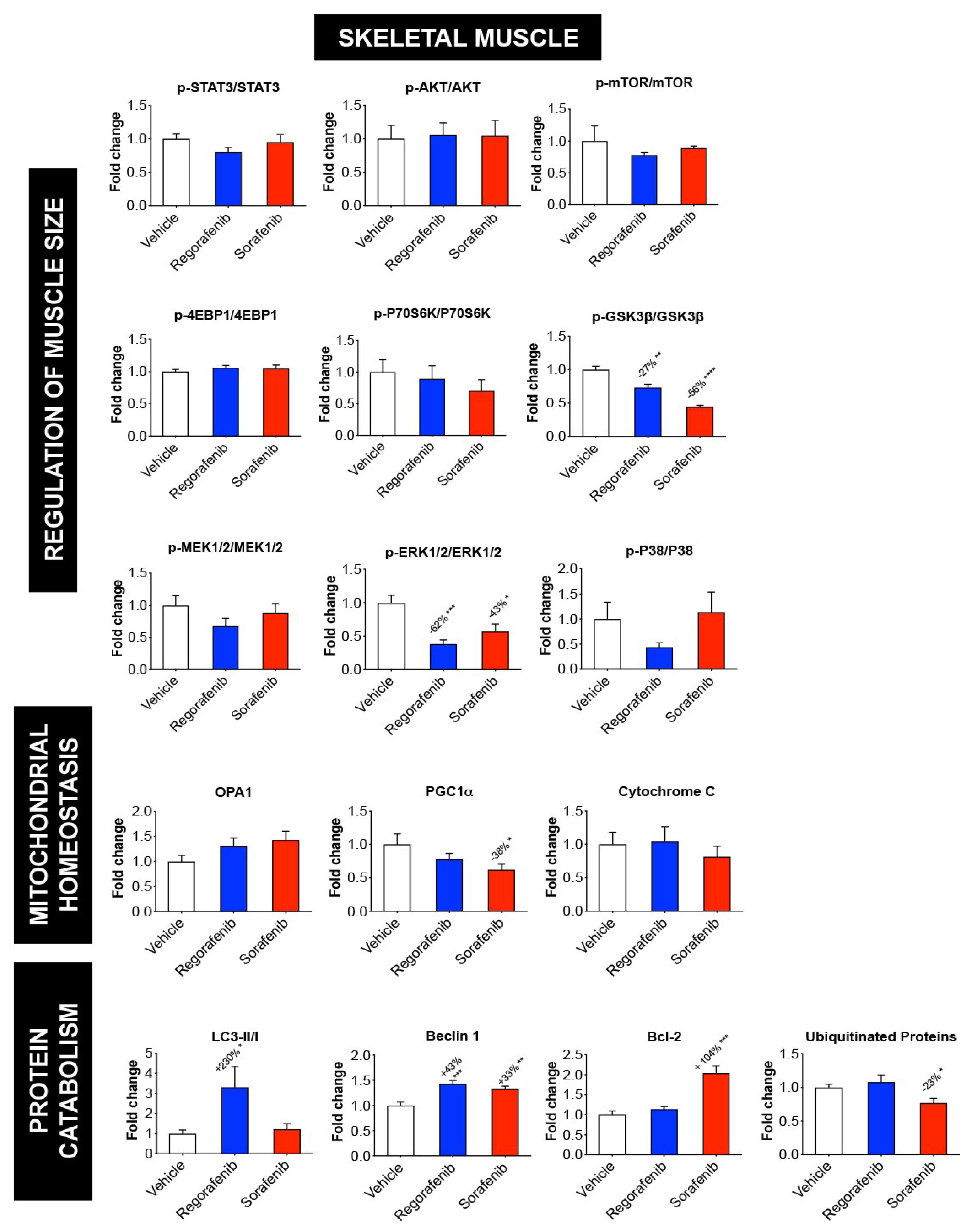
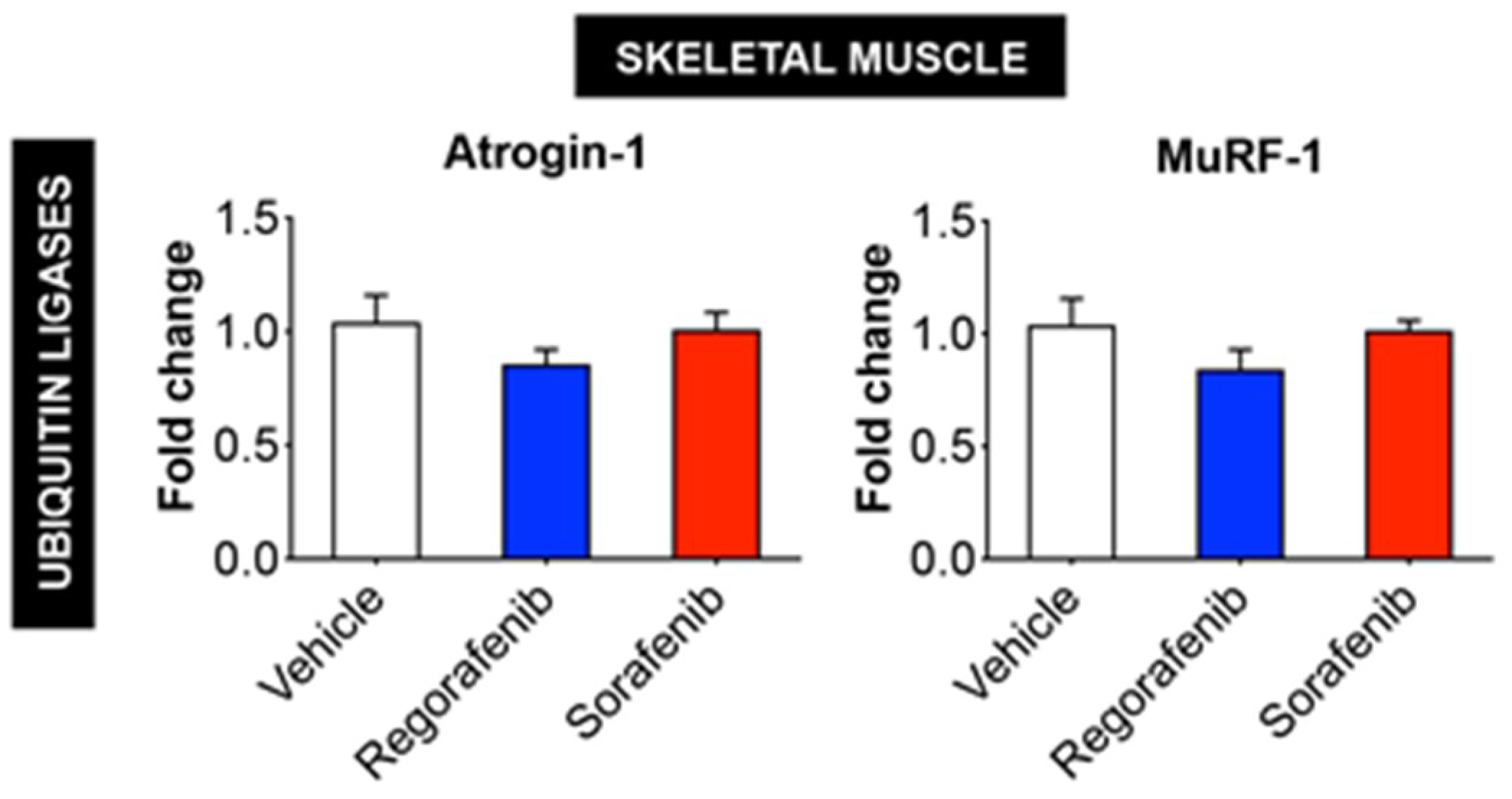
2.4. MKIs Affect Cachexia-Related Pathways in Skeletal Muscle
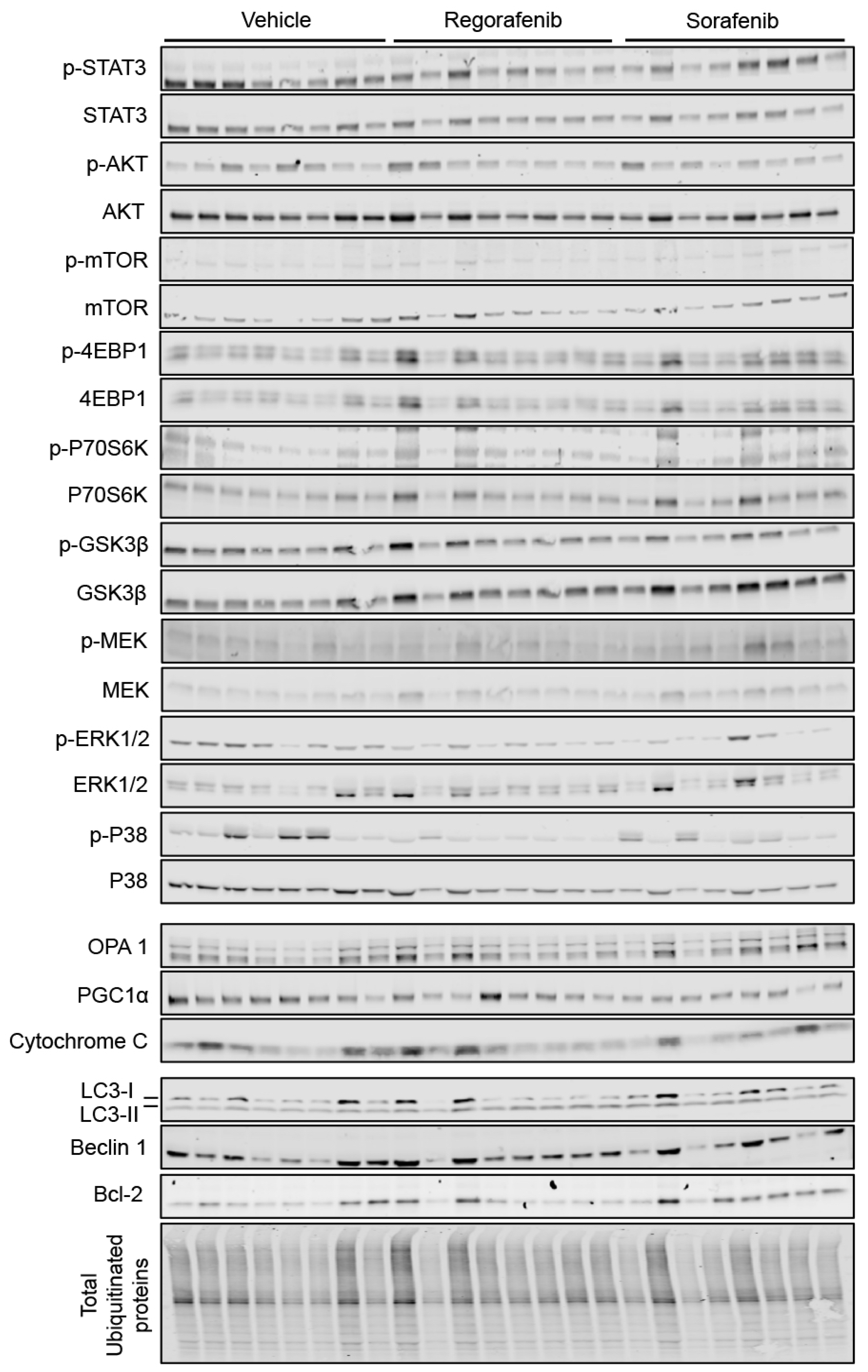
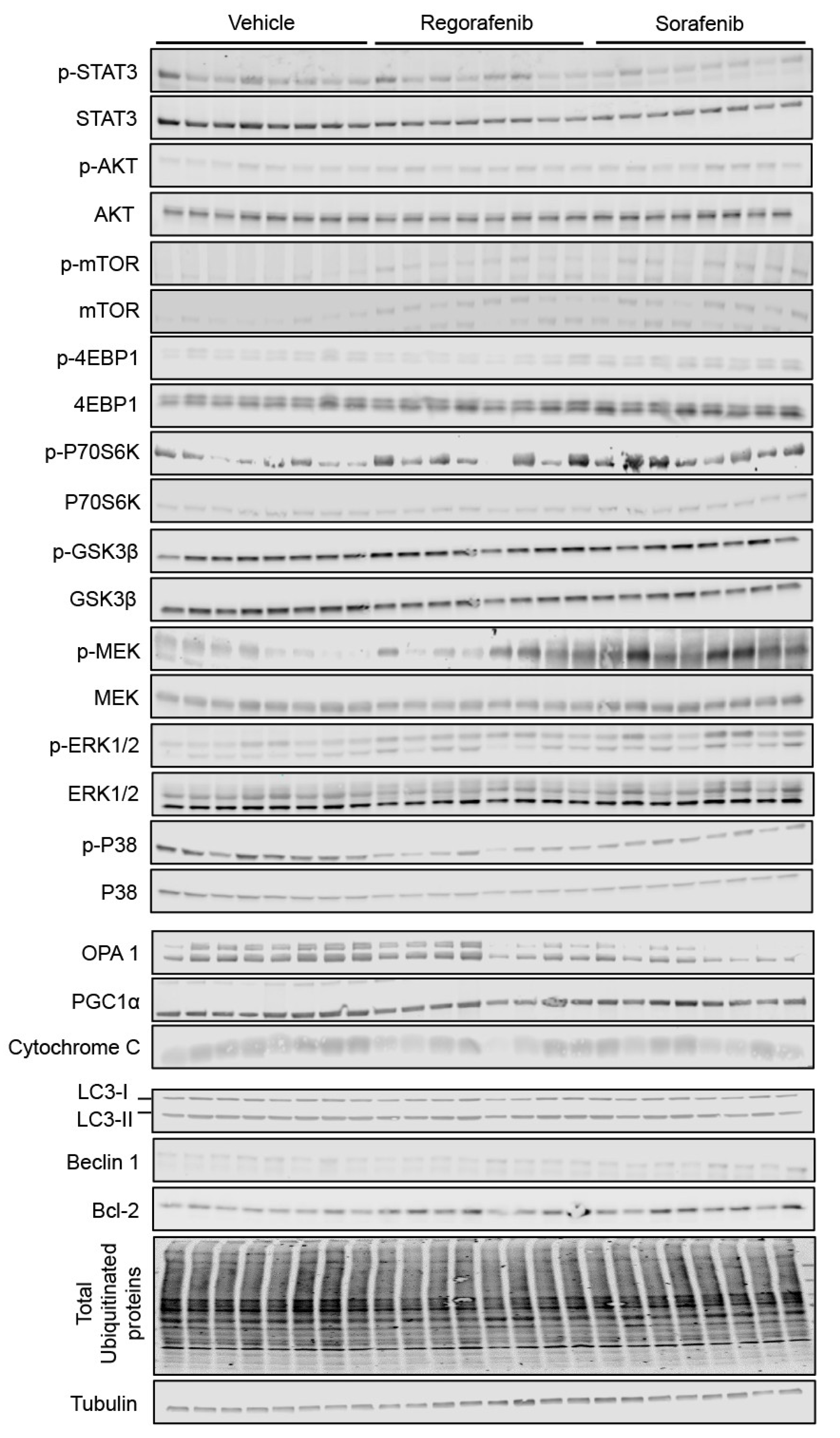
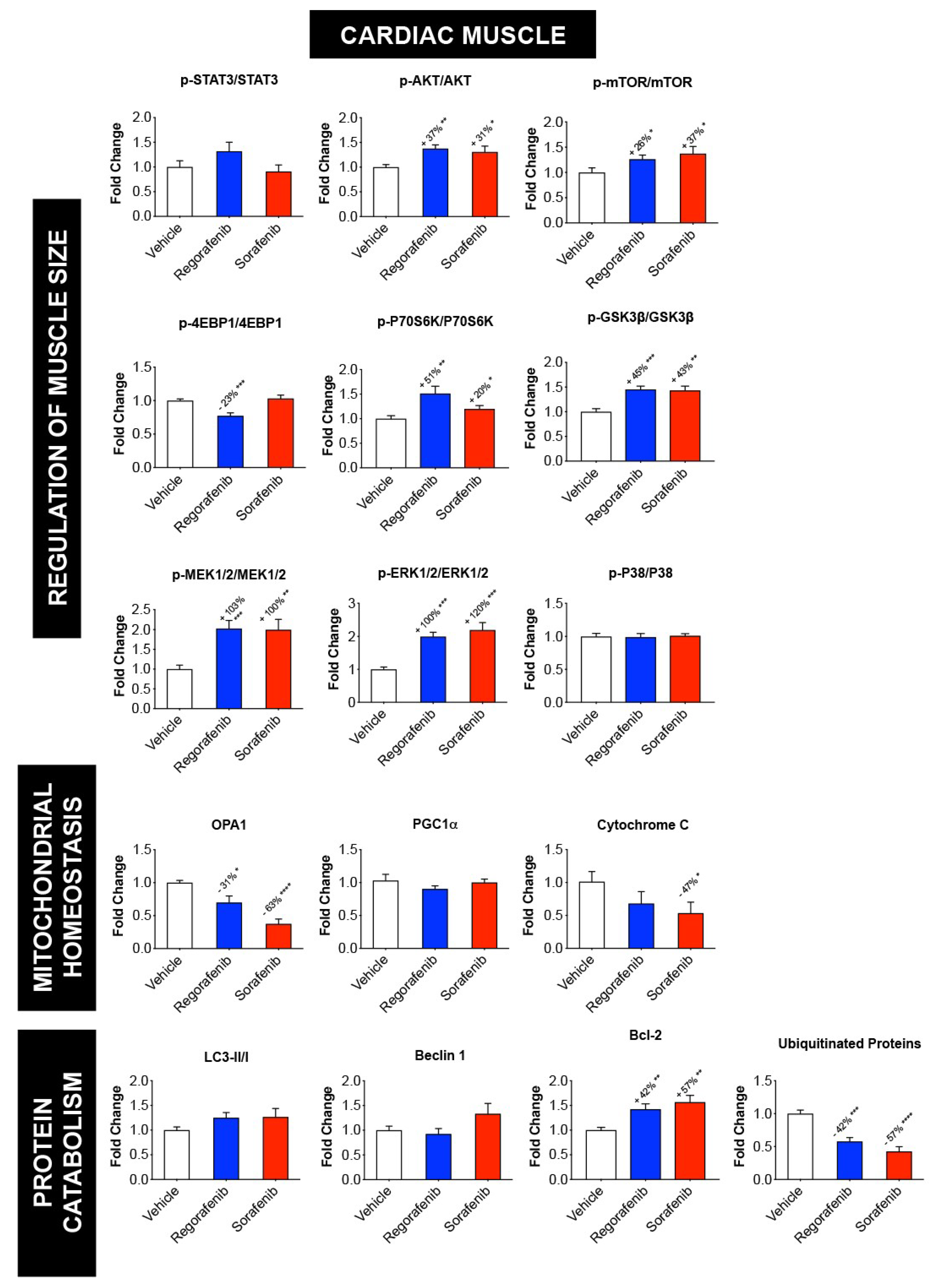
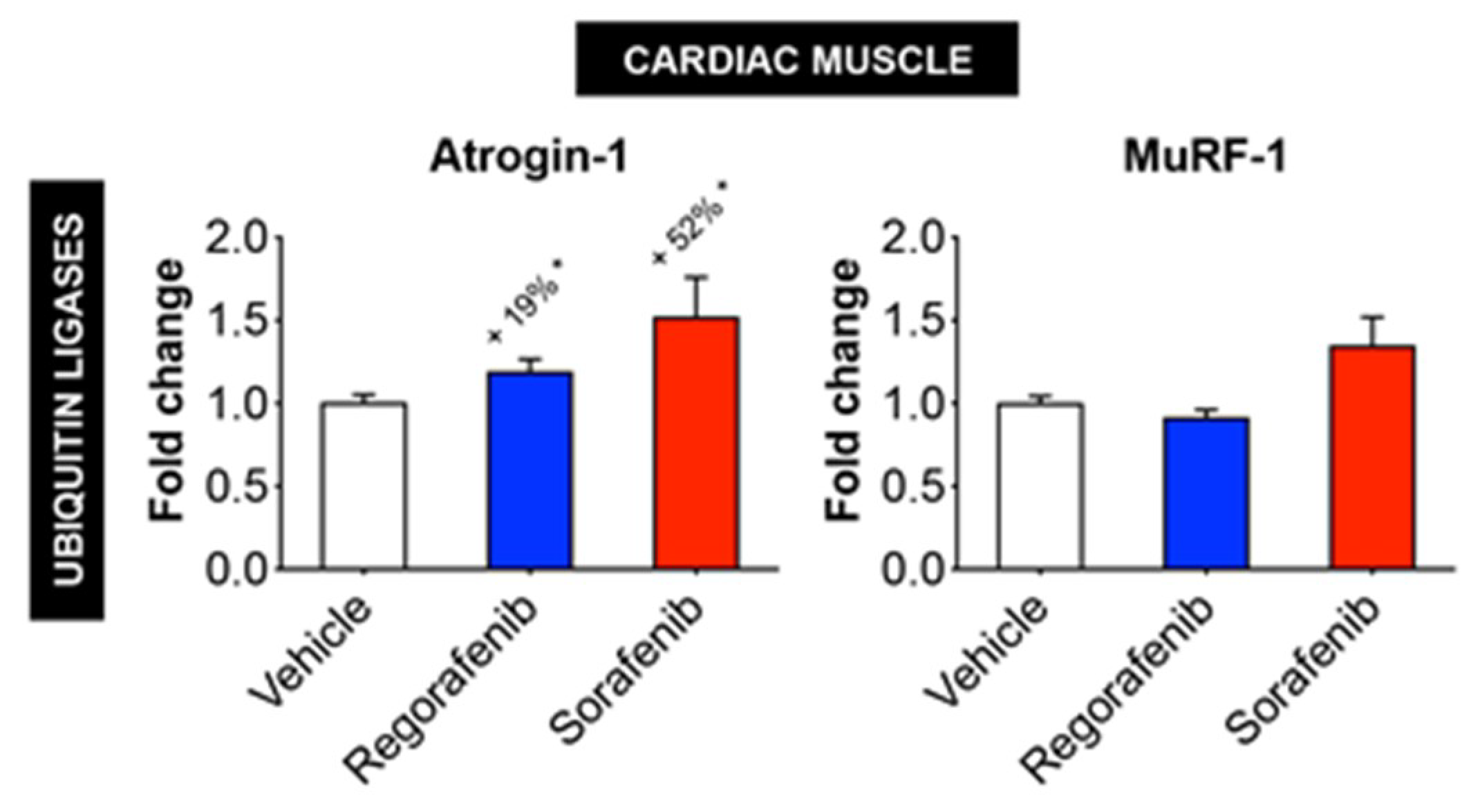
2.5. Regorafenib and Sorafenib Alter Cachexia-Associated Pathways in Cardiac Muscle
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Grip Strength Measurement
4.3. Muscle Cross-Sectional Area (CSA)
4.4. Whole Muscle Contractility
4.5. Echocardiography
4.6. Western Blotting
4.7. Real-Time Quantitative Polymerase Chain Reaction (qRT-PCR)
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MKI | multi-kinase inhibitor |
| ERK1/2 | extracellular signal-regulated kinase 1/2 |
| STAT3 | signal transducer and activator of transcription 3 |
| GSK3b | glycogen synthase kinase-3 beta |
| AKT | protein kinase b |
| mTOR | mechanistic target of rapamycin |
| p70S6K | 70 kDa ribosomal protein S6 kinase |
| 4EBP1 | eukaryotic initiation factor 4E binding protein |
| MEK1/2 | mitogen-activated protein kinase kinase |
| OPA1 | optic atrophy protein 1 |
| PGC1a | peroxisome proliferator-activated receptor gamma co-activator 1 alpha |
| LC3 | microtubule-associated protein 1A/1B-light chain 3 |
| Bcl-2 | b-cell lymphoma 2 |
| BNP | brain natriuretic peptide |
| TBP | TATA-binding protein |
| CSA | cross-sectional area |
| SV | stroke volume |
| LV | left ventricle |
| LVIDd | left ventricular internal diameter during diastole |
| LVIDs | left ventricular internal diameter during systole |
| LVPWd | left ventricular posterior wall thickness during diastole |
| LVPWs | left ventricular posterior wall thickness during systole |
References
- Dewys, W.D.; Begg, C.; Lavin, P.T.; Band, P.R.; Bennett, J.M.; Bertino, J.R.; Cohen, M.H.; Douglass, H.O., Jr.; Engstrom, P.F.; Ezdinli, E.Z.; et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am. J. Med. 1980, 69, 491–497. [Google Scholar] [CrossRef]
- Barreto, R.; Mandili, G.; Witzmann, F.A.; Novelli, F.; Zimmers, T.A.; Bonetto, A. Cancer and Chemotherapy Contribute to Muscle Loss by Activating Common Signaling Pathways. Front. Physiol. 2016, 7, 472. [Google Scholar] [CrossRef]
- Barreto, R.; Waning, D.L.; Gao, H.; Liu, Y.; Zimmers, T.A.; Bonetto, A. Chemotherapy-related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget 2016, 7, 43442–43460. [Google Scholar] [CrossRef]
- Pin, F.; Barreto, R.; Couch, M.E.; Bonetto, A.; O’Connell, T.M. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J. Cachexia Sarcopenia Muscle 2019. [Google Scholar] [CrossRef]
- Ohnuma, T.; Adigun, R. Cancer, Anorexia and Cachexia; StatPearls: Treasure Island, FL, USA, 2018. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Shingina, A.; Hashim, A.M.; Haque, M.; Suen, M.; Yoshida, E.M.; Gill, S.; Donnellan, F.; Weiss, A.A. In a ‘real-world’, clinic-based community setting, sorafenib dose of 400 mg/day is as effective as standard dose of 800 mg/day in patients with advanced hepatocellular carcimona, with better tolerance and similar survival. Can. J. Gastroenterol. 2013, 27, 393–396. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Arrieta, O.; De la Torre-Vallejo, M.; Lopez-Macias, D.; Orta, D.; Turcott, J.; Macedo-Perez, E.O.; Sanchez-Lara, K.; Ramirez-Tirado, L.A.; Baracos, V.E. Nutritional Status, Body Surface, and Low Lean Body Mass/Body Mass Index Are Related to Dose Reduction and Severe Gastrointestinal Toxicity Induced by Afatinib in Patients with Non-Small Cell Lung Cancer. Oncologist 2015, 20, 967–974. [Google Scholar] [CrossRef]
- Kazemi-Bajestani, S.M.; Mazurak, V.C.; Baracos, V. Computed tomography-defined muscle and fat wasting are associated with cancer clinical outcomes. Semin. Cell Dev. Biol. 2016, 54, 2–10. [Google Scholar] [CrossRef]
- Prado, C.M.; Baracos, V.E.; McCargar, L.J.; Mourtzakis, M.; Mulder, K.E.; Reiman, T.; Butts, C.A.; Scarfe, A.G.; Sawyer, M.B. Body composition as an independent determinant of 5-fluorouracil-based chemotherapy toxicity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 3264–3268. [Google Scholar] [CrossRef]
- Sjoblom, B.; Gronberg, B.H.; Benth, J.S.; Baracos, V.E.; Flotten, O.; Hjermstad, M.J.; Aass, N.; Jordhoy, M. Low muscle mass is associated with chemotherapy-induced haematological toxicity in advanced non-small cell lung cancer. Lung Cancer 2015, 90, 85–91. [Google Scholar] [CrossRef]
- Tan, B.H.; Brammer, K.; Randhawa, N.; Welch, N.T.; Parsons, S.L.; James, E.J.; Catton, J.A. Sarcopenia is associated with toxicity in patients undergoing neo-adjuvant chemotherapy for oesophago-gastric cancer. Eur. J. Surg. Oncol. 2015, 41, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Kroenke, C.H.; Prado, C.M.; Meyerhardt, J.A.; Weltzien, E.K.; Xiao, J.; Cespedes Feliciano, E.M.; Caan, B.J. Muscle radiodensity and mortality in patients with colorectal cancer. Cancer 2018, 124, 3008–3015. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, G.K.; Caldeira de Oliveira, T.H.; de Oliveira Belo, N. Cardiac Hypertrophy and Brain Natriuretic Peptide Levels in an Ovariectomized Rat Model Fed a High-Fat Diet. Med. Sci. Monit. Basic. Res. 2017, 23, 380–391. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Wu, J.; Zhang, Q.; Ye, Y.; Wang, S.; Huang, J.; Liu, H.; Wang, X.; Zhang, W.; Bu, L.; et al. Differential cardiac hypertrophy and signaling pathways in pressure versus volume overload. Am. J. Physiol.Heart Circ. Physiol. 2018, 314, H552–H562. [Google Scholar] [CrossRef]
- Pin, F.; Couch, M.E.; Bonetto, A. Preservation of muscle mass as a strategy to reduce the toxic effects of cancer chemotherapy on body composition. Curr. Opin. Support Palliat. Care 2018, 12, 420–426. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Mendez-Blanco, C.; Fondevila, F.; Garcia-Palomo, A.; Gonzalez-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 134. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schutz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer J. Int. Du Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
- Bonetto, A.; Aydogdu, T.; Kunzevitzky, N.; Guttridge, D.C.; Khuri, S.; Koniaris, L.G.; Zimmers, T.A. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 2011, 6, e22538. [Google Scholar] [CrossRef]
- Blumenschein, G.R., Jr.; Gatzemeier, U.; Fossella, F.; Stewart, D.J.; Cupit, L.; Cihon, F.; O’Leary, J.; Reck, M. Phase II, multicenter, uncontrolled trial of single-agent sorafenib in patients with relapsed or refractory, advanced non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 4274–4280. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J. Risk of regorafenib-induced cardiovascular events in patients with solid tumors: A systematic review and meta-analysis. Medicine 2018, 97, e12705. [Google Scholar] [CrossRef]
- Tovoli, F.; Negrini, G.; Benevento, F.; Faggiano, C.; Goio, E.; Granito, A. Systemic treatments for hepatocellular carcinoma: Challenges and future perspectives. Hepat. Oncol. 2018, 5, HEP01. [Google Scholar] [CrossRef]
- Yin, X.; Yin, Y.; Shen, C.; Chen, H.; Wang, J.; Cai, Z.; Chen, Z.; Zhang, B. Adverse events risk associated with regorafenib in the treatment of advanced solid tumors: Meta-analysis of randomized controlled trials. Onco Targets Ther. 2018, 11, 6405–6414. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Hiyoshi, Y.; Akiyama, T.; Kiyozumi, Y.; Eto, K.; Yohei, N.; Iwagami, S.; Baba, Y.; Yoshida, N.; Baba, H. Low Skeletal Muscle Mass before Salvage-Line Chemotherapy Is a Poor Prognostic Factor in Patients with Refractory Metastatic Colorectal Cancer. Digestion 2019, 99, 79–85. [Google Scholar] [CrossRef]
- Stuhlmiller, T.J.; Zawistowski, J.S.; Chen, X.; Sciaky, N.; Angus, S.P.; Hicks, S.T.; Parry, T.L.; Huang, W.; Beak, J.Y.; Willis, M.S.; et al. Kinome and Transcriptome Profiling Reveal Broad and Distinct Activities of Erlotinib, Sunitinib, and Sorafenib in the Mouse Heart and Suggest Cardiotoxicity from Combined Signal Transducer and Activator of Transcription and Epidermal Growth Factor Receptor Inhibition. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Antoun, S.; Birdsell, L.; Sawyer, M.B.; Venner, P.; Escudier, B.; Baracos, V.E. Association of skeletal muscle wasting with treatment with sorafenib in patients with advanced renal cell carcinoma: Results from a placebo-controlled study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 1054–1060. [Google Scholar] [CrossRef]
- Damaraju, V.L.; Kuzma, M.; Cass, C.E.; Putman, C.T.; Sawyer, M.B. Multitargeted kinase inhibitors imatinib, sorafenib and sunitinib perturb energy metabolism and cause cytotoxicity to cultured C2C12 skeletal muscle derived myotubes. Biochem. Pharmcol. 2018, 155, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.C.; Wang, M.H.; Tsai, J.J.; Kuo, Y.C.; Liu, Y.C.; Hsu, F.T.; Wang, H.E. Regorafenib inhibits tumor progression through suppression of ERK/NF-kappaB activation in hepatocellular carcinoma bearing mice. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M.; Penna, F.; Busquets, S.; Lopez-Soriano, F.J.; Argiles, J.M. Distinct behaviour of sorafenib in experimental cachexia-inducing tumours: The role of STAT3. PLoS ONE 2014, 9, e113931. [Google Scholar] [CrossRef]
- Narayanan, K.R.; Bansal, D.; Walia, R.; Sachdeva, N.; Bhansali, A.; Varma, N.; Marwaha, R.K. Growth failure in children with chronic myeloid leukemia receiving imatinib is due to disruption of GH/IGF-1 axis. Pediatr. Blood Cancer 2013, 60, 1148–1153. [Google Scholar] [CrossRef]
- Duran, J.M.; Makarewich, C.A.; Trappanese, D.; Gross, P.; Husain, S.; Dunn, J.; Lal, H.; Sharp, T.E.; Starosta, T.; Vagnozzi, R.J.; et al. Sorafenib cardiotoxicity increases mortality after myocardial infarction. Circ. Res. 2014, 114, 1700–1712. [Google Scholar] [CrossRef]
- Mutlak, M.; Kehat, I. Extracellular signal-regulated kinases 1/2 as regulators of cardiac hypertrophy. Front. Pharmcol. 2015, 6, 149. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chen, J.Y.; Kuo, C.H.; Pai, P.Y.; Ho, T.J.; Chen, T.S.; Tsai, F.J.; Padma, V.V.; Kuo, W.W.; Huang, C.Y. Mitochondrial ROS-induced ERK1/2 activation and HSF2-mediated AT1 R upregulation are required for doxorubicin-induced cardiotoxicity. J. Cell. Physiol. 2018, 233, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, D.; Melo, T.; Deng, Z.; Almeida, C.; Zhao, W. ERK/GSK3beta/Snail signaling mediates radiation-induced alveolar epithelial-to-mesenchymal transition. Free Radic. Biol. Med. 2012, 52, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Xia, W.; Liu, J.C.; Yang, J.Y.; Lee, D.F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef]
- Pin, F.; Barreto, R.; Kitase, Y.; Mitra, S.; Erne, C.E.; Novinger, L.J.; Zimmers, T.A.; Couch, M.E.; Bonewald, L.F.; Bonetto, A. Growth of ovarian cancer xenografts causes loss of muscle and bone mass: A new model for the study of cancer cachexia. J. Cachexia Sarcopenia Muscle 2018, 9, 685–700. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Parys, J.B.; Bultynck, G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells 2012, 1, 284–312. [Google Scholar] [CrossRef]
- Ogasawara, S.; Cheng, X.W.; Inoue, A.; Hu, L.; Piao, L.; Yu, C.; Goto, H.; Xu, W.; Zhao, G.; Lei, Y.; et al. Cathepsin K activity controls cardiotoxin-induced skeletal muscle repair in mice. J. Cachexia Sarcopenia Muscle 2018, 9, 160–175. [Google Scholar] [CrossRef]
- Jensen, B.C.; Parry, T.L.; Huang, W.; Beak, J.Y.; Ilaiwy, A.; Bain, J.R.; Newgard, C.B.; Muehlbauer, M.J.; Patterson, C.; Johnson, G.L.; et al. Effects of the kinase inhibitor sorafenib on heart, muscle, liver and plasma metabolism in vivo using non-targeted metabolomics analysis. Br. J. Pharmcol. 2017, 174, 4797–4811. [Google Scholar] [CrossRef]
- Pachon, R.E.; Scharf, B.A.; Vatner, D.E.; Vatner, S.F. Best anesthetics for assessing left ventricular systolic function by echocardiography in mice. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1525–H1529. [Google Scholar] [CrossRef]
- Carr, B.I.; Cavallini, A.; Lippolis, C.; D’Alessandro, R.; Messa, C.; Refolo, M.G.; Tafaro, A. Fluoro-Sorafenib (Regorafenib) effects on hepatoma cells: Growth inhibition, quiescence, and recovery. J. Cell. Physiol. 2013, 228, 292–297. [Google Scholar] [CrossRef]
- Prieto-Dominguez, N.; Ordonez, R.; Fernandez, A.; Garcia-Palomo, A.; Muntane, J.; Gonzalez-Gallego, J.; Mauriz, J.L. Modulation of Autophagy by Sorafenib: Effects on Treatment Response. Front. Pharmcol. 2016, 7, 151. [Google Scholar] [CrossRef]
- Tavallai, M.; Hamed, H.A.; Roberts, J.L.; Cruickshanks, N.; Chuckalovcak, J.; Poklepovic, A.; Booth, L.; Dent, P. Nexavar/Stivarga and viagra interact to kill tumor cells. J. Cell. Physiol. 2015, 230, 2281–2298. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, M.A.; Gonzalez, R.; de la Rosa, A.J.; Gallego, P.; Ordonez, R.; Navarro-Villaran, E.; Contreras, L.; Rodriguez-Arribas, M.; Gonzalez-Gallego, J.; Alamo-Martinez, J.M.; et al. Molecular characterization of autophagic and apoptotic signaling induced by sorafenib in liver cancer cells. J. Cell. Physiol. 2018, 234, 692–708. [Google Scholar] [CrossRef]
- Vaidya, T.; Kamta, J.; Chaar, M.; Ande, A.; Ait-Oudhia, S. Systems pharmacological analysis of mitochondrial cardiotoxicity induced by selected tyrosine kinase inhibitors. J. Pharmacokinet. Pharmacodyn. 2018, 45, 401–418. [Google Scholar] [CrossRef]
- Mirantes, C.; Dosil, M.A.; Eritja, N.; Felip, I.; Gatius, S.; Santacana, M.; Matias-Guiu, X.; Dolcet, X. Effects of the multikinase inhibitors Sorafenib and Regorafenib in PTEN deficient neoplasias. Eur. J. Cancer 2016, 63, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M.; Penna, F.; Oliva, F.; Luque, M.; Betancourt, A.; Marmonti, E.; Lopez-Soriano, F.J.; Argiles, J.M.; Busquets, S. A multifactorial anti-cachectic approach for cancer cachexia in a rat model undergoing chemotherapy. J. Cachexia Sarcopenia Muscle 2016, 7, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, Y.; Yu, Y.; Pang, J.C.; Woodfield, S.E.; Tao, L.; Guan, S.; Zhang, H.; Bieerkehazhi, S.; Shi, Y.; et al. Small molecule inhibitor regorafenib inhibits RET signaling in neuroblastoma cells and effectively suppresses tumor growth in vivo. Oncotarget 2017, 8, 104090–104103. [Google Scholar] [CrossRef]
- Gu, Q.; Zhang, B.; Sun, H.; Xu, Q.; Tan, Y.; Wang, G.; Luo, Q.; Xu, W.; Yang, S.; Li, J.; et al. Genomic characterization of a large panel of patient-derived hepatocellular carcinoma xenograft tumor models for preclinical development. Oncotarget 2015, 6, 20160–20176. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Rupert, J.E.; Barreto, R.; Zimmers, T.A. The Colon-26 Carcinoma Tumor-bearing Mouse as a Model for the Study of Cancer Cachexia. J. Vis. Exp. JOVE 2016. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Andersson, D.C.; Waning, D.L. Assessment of muscle mass and strength in mice. Bonekey Rep. 2015, 4, 732. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huot, J.R.; Essex, A.L.; Gutierrez, M.; Barreto, R.; Wang, M.; Waning, D.L.; Plotkin, L.I.; Bonetto, A. Chronic Treatment with Multi-Kinase Inhibitors Causes Differential Toxicities on Skeletal and Cardiac Muscles. Cancers 2019, 11, 571. https://doi.org/10.3390/cancers11040571
Huot JR, Essex AL, Gutierrez M, Barreto R, Wang M, Waning DL, Plotkin LI, Bonetto A. Chronic Treatment with Multi-Kinase Inhibitors Causes Differential Toxicities on Skeletal and Cardiac Muscles. Cancers. 2019; 11(4):571. https://doi.org/10.3390/cancers11040571
Chicago/Turabian StyleHuot, Joshua R., Alyson L. Essex, Maya Gutierrez, Rafael Barreto, Meijing Wang, David L. Waning, Lilian I. Plotkin, and Andrea Bonetto. 2019. "Chronic Treatment with Multi-Kinase Inhibitors Causes Differential Toxicities on Skeletal and Cardiac Muscles" Cancers 11, no. 4: 571. https://doi.org/10.3390/cancers11040571
APA StyleHuot, J. R., Essex, A. L., Gutierrez, M., Barreto, R., Wang, M., Waning, D. L., Plotkin, L. I., & Bonetto, A. (2019). Chronic Treatment with Multi-Kinase Inhibitors Causes Differential Toxicities on Skeletal and Cardiac Muscles. Cancers, 11(4), 571. https://doi.org/10.3390/cancers11040571