A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials


Abstract
1. Introduction
2. Immune Surveillance in the Central Nervous System
3. Dendritic Cells
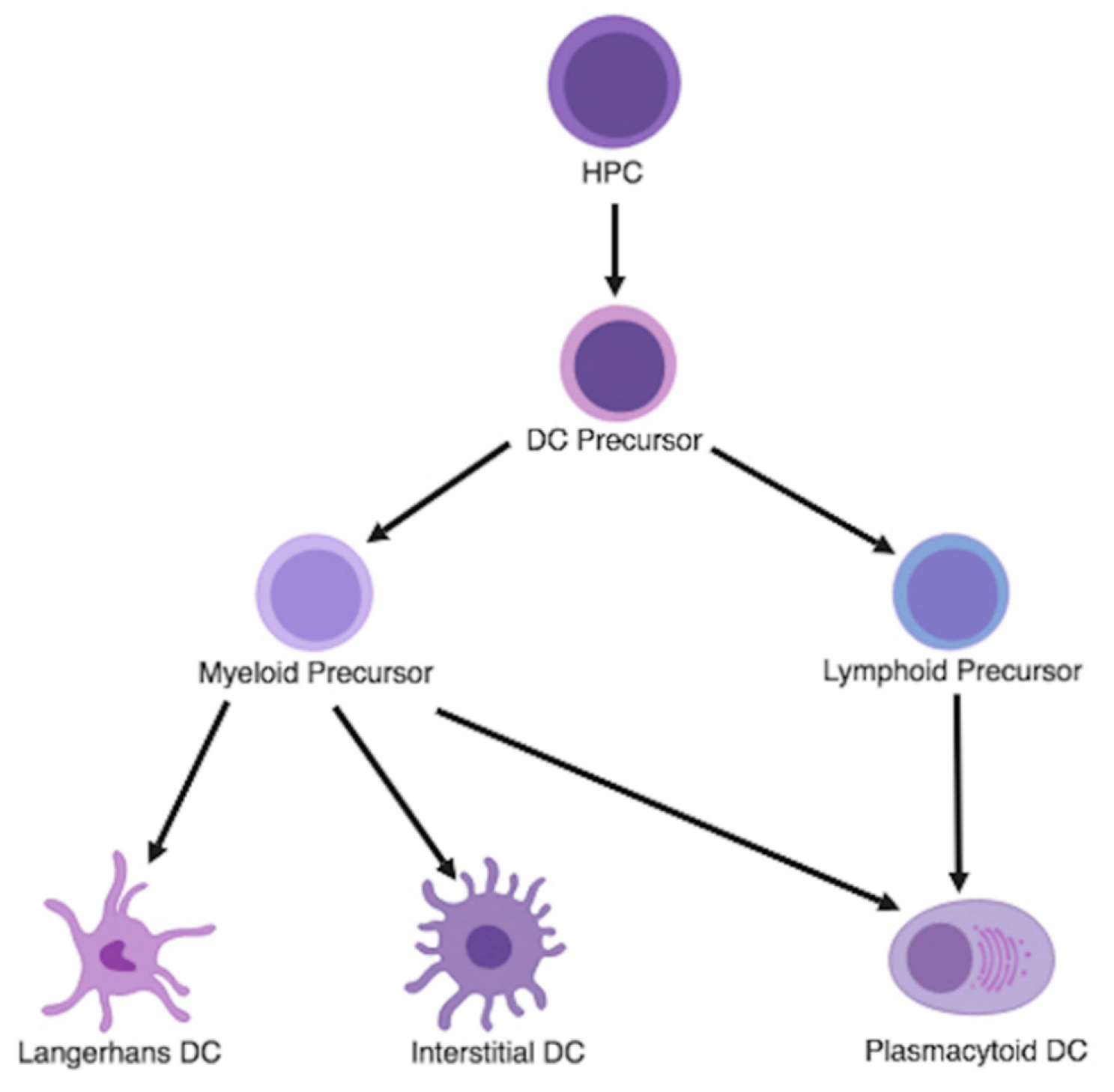
3.1. Dendritic Cell Differentiation and Classification
3.2. Dendritic Cells in Glioma
4. Preclinical Studies
4.1. Dendritic Cell Selection and Culturing
4.2. Antigen Selection
4.2.1. Tumor Cells and Lysate
4.2.2. Tumor Associated and Specific Antigens
4.2.3. Exosomes
4.3. Adjuvant Therapy
5. Dendritic Cell Vaccine Clinical Trials
5.1. GAA and GSA in Clinical Trials
5.2. Adjuvants in Clinical Trials
5.3. Safety and Toxicity
5.4. Role of DC Vaccines in Current Standard of Care
5.5. Future Approaches and Challenges to DC Vaccines
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014, 16, iv1–iv63. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Feng, E.; Sui, C.; Wang, T.; Sun, G. Temozolomide with or without radiotherapy in patients with newly diagnosed glioblastoma multiforme: A meta-analysis. Eur. Neurol. 2017, 77, 201–210. [Google Scholar] [CrossRef]
- Delgado-Lopez, P.D.; Corrales-Garcia, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Medawar, P.B. Immunity to homologous grafted skin. III. The fate of skin homographs transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 1948, 29, 58. [Google Scholar] [PubMed]
- Bentivoglio, M.; Kristensson, K. Tryps and trips: Cell trafficking across the 100-year-old blood–brain barrier. Trends Neurosci. 2014, 37, 325–333. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Blood–brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623. [Google Scholar] [CrossRef]
- Lampson, L.A. Interpreting MHC class I expression and class I/class II reciprocity in the CNS: Reconciling divergent findings. Microsc. Res. Tech. 1995, 32, 267–285. [Google Scholar] [CrossRef]
- Cullheim, S.; Thams, S. Classic major histocompatibility complex class I molecules: New actors at the neuromuscular junction. Neuroscientist 2010, 16, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Cebrián, C.; Loike, J.D.; Sulzer, D. Neuronal MHC-I expression and its implications in synaptic function, axonal regeneration and Parkinson’s and other brain diseases. Front. Neuroanat. 2014, 8, 114. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337. [Google Scholar] [CrossRef] [PubMed]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Shields, J.D.; Kourtis, I.C.; Tomei, A.A.; Roberts, J.M.; Swartz, M.A. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010, 328, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Streit, W.J. Microglia: Biology and pathology. Acta Neuropathol. 2010, 119, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Herz, J.; Filiano, A.J.; Smith, A.; Yogev, N.; Kipnis, J. Myeloid Cells in the Central Nervous System. Immunity 2017, 46, 943–956. [Google Scholar] [CrossRef]
- Gehrmann, J.; Matsumoto, Y.; Kreutzberg, G.W. Microglia: Intrinsic immuneffector cell of the brain. Brain Res. Rev. 1995, 20, 269–287. [Google Scholar] [CrossRef]
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A.; Wilcock, D.M. Assessing activation states in microglia. CNS Neurol. Disord. Targets 2010, 9, 174–191. [Google Scholar] [CrossRef]
- Brendecke, S.M.; Prinz, M. Do not judge a cell by its cover—Diversity of CNS resident, adjoining and infiltrating myeloid cells in inflammation. Semin. Immunopathol. 2015, 37, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Jorge, L.; Ramos, D.; Luppo, M.; Llombart, C.; Alexandre-Pires, G.; Nacher, V.; Melgarejo, V.; Correia, M.; Navarro, M.; Carretero, A. Scavenger function of resident autofluorescent perivascular macrophages and their contribution to the maintenance of the blood–retinal barrier. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5997–6005. [Google Scholar] [CrossRef] [PubMed]
- Carlin, L.M.; Stamatiades, E.G.; Auffray, C.; Hanna, R.N.; Glover, L.; Vizcay-Barrena, G.; Hedrick, C.C.; Cook, H.T.; Diebold, S.; Geissmann, F. Nr4a1-dependent Ly6Clow monocytes monitor endothelial cells and orchestrate their disposal. Cell 2013, 153, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Jakubzick, C.; Gautier, E.L.; Gibbings, S.L.; Sojka, D.K.; Schlitzer, A.; Johnson, T.E.; Ivanov, S.; Duan, Q.; Bala, S.; Condon, T. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 2013, 39, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Carvalho da Fonseca, A.C.; Badie, B. Microglia and macrophages in malignant gliomas: Recent discoveries and implications for promising therapies. Clin. Dev. Immunol. 2013, 2013, 264124. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Ellert-Miklaszewska, A.; Lipko, M.; Sielska, M.; Frankowska, M.; Kaminska, B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS ONE 2011, 6, e23902. [Google Scholar] [CrossRef] [PubMed]
- Ellert-Miklaszewska, A.; Dabrowski, M.; Lipko, M.; Sliwa, M.; Maleszewska, M.; Kaminska, B. Molecular definition of the pro-tumorigenic phenotype of glioma-activated microglia. Glia 2013, 61, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- de Vrij, J.; Maas, S.L.N.; Kwappenberg, K.M.C.; Schnoor, R.; Kleijn, A.; Dekker, L.; Luider, T.M.; de Witte, L.D.; Litjens, M.; van Strien, M.E. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int. J. Cancer 2015, 137, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Dendritic Cells: A Link Between Innate and Adaptive Immunity. J. Clin. Immunol. 1999, 19, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.T.; Locksley, R.M. The instructive role of innate immunity in the acquired immune response. Science 1996, 272, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. Dendritic cells: Versatile controllers of the immune system. Nat. Med. 2007, 13, 1155. [Google Scholar] [CrossRef]
- Collin, M.; McGovern, N.; Haniffa, M. Human dendritic cell subsets. Immunology 2013, 140, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Young, J.W. Human dendritic cells: Potent antigen-presenting cells at the crossroads of innate and adaptive immunity. J. Immunol. 2005, 175, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, M.F.; Masten, B.J. Dendritic cells: Immune regulators in health and disease. Physiol. Rev. 2002, 82, 97–130. [Google Scholar] [CrossRef]
- Maraskovsky, E.; Brasel, K.; Teepe, M.; Roux, E.R.; Lyman, S.D.; Shortman, K.; McKenna, H.J. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: Multiple dendritic cell subpopulations identified. J. Exp. Med. 1996, 184, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R. Glioma: Immunotherapeutic Approaches; Springer: Berlin, Germany, 2012; ISBN 9781461431466. [Google Scholar]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Bailey, S.L.; Schreiner, B.; McMahon, E.J.; Miller, S.D. CNS myeloid DCs presenting endogenous myelin peptides’ preferentially’polarize CD4+ T H-17 cells in relapsing EAE. Nat. Immunol. 2007, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Gafa, V.; Serafini, B.; Giacomini, E.; Visconti, A.; Remoli, M.E.; Severa, M.; Parmentier, M.; Ristori, G.; Salvetti, M. Plasmacytoid dendritic cells in multiple sclerosis: Intracerebral recruitment and impaired maturation in response to interferon-β. J. Neuropathol. Exp. Neurol. 2008, 67, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Doebel, T.; Voisin, B.; Nagao, K. Langerhans cells–the macrophage in dendritic cell clothing. Trends Immunol. 2017, 38, 817–828. [Google Scholar] [CrossRef]
- Klechevsky, E.; Morita, R.; Liu, M.; Cao, Y.; Coquery, S.; Thompson-Snipes, L.; Briere, F.; Chaussabel, D.; Zurawski, G.; Palucka, A.K. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity 2008, 29, 497–510. [Google Scholar] [CrossRef]
- Gilliet, M.; Cao, W.; Liu, Y.-J. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008, 8, 594. [Google Scholar] [CrossRef]
- Blasius, A.L.; Beutler, B. Intracellular toll-like receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Hassanzadeh-Kiabi, N.; Yáñez, A.; Dang, I.; Martins, G.A.; Underhill, D.M.; Goodridge, H.S. Autocrine Type I IFN Signaling in Dendritic Cells Stimulated with Fungal β-Glucans or Lipopolysaccharide Promotes CD8 T Cell Activation. J. Immunol. 2017, 198, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Myeloid dendritic cells: Development, functions, and role in atherosclerotic inflammation. Immunobiology 2015, 220, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Matyszak, M.K.; Perry, V.H. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience 1996, 74, 599–608. [Google Scholar] [CrossRef]
- Serot, J.-M.; Béné, M.-C.; Foliguet, B.; Faure, G.C. Monocyte-derived IL-10-secreting dendritic cells in choroid plexus epithelium. J. Neuroimmunol. 2000, 105, 115–119. [Google Scholar] [CrossRef]
- McMenamin, P.G. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J. Comp. Neurol. 1999, 405, 553–562. [Google Scholar] [CrossRef]
- D’Agostino, P.M.; Gottfried-Blackmore, A.; Anandasabapathy, N.; Bulloch, K. Brain dendritic cells: Biology and pathology. Acta Neuropathol. 2012, 124, 599–614. [Google Scholar] [CrossRef]
- Colton, C.A. Immune Heterogeneity in Neuroinflammation: Dendritic Cells in the Brain. J. Neuroimmune Pharmacol. 2013, 8, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. The instructive role of dendritic cells on T-cell responses. Arthritis Res. 2002, 4 (Suppl. S3), S127–S132. [Google Scholar] [CrossRef]
- Vieira, P.L.; de Jong, E.C.; Wierenga, E.A.; Kapsenberg, M.L.; Kaliński, P. Development of Th1-inducing capacity in myeloid dendritic cells requires environmental instruction. J. Immunol. 2000, 164, 4507–4512. [Google Scholar] [CrossRef]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Dutertre, C.-A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Sad, S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 1996, 17, 138–146. [Google Scholar] [CrossRef]
- O’Garra, A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 1998, 8, 275–283. [Google Scholar] [CrossRef]
- Du, X.; Wen, J.; Wang, Y.; Karmaus, P.W.F.; Khatamian, A.; Tan, H.; Li, Y.; Guy, C.; Nguyen, T.-L.M.; Dhungana, Y.; et al. Hippo/Mst signalling couples metabolic state and immune function of CD8α+ dendritic cells. Nature 2018, 558, 141–145. [Google Scholar] [CrossRef]
- Kaliński, P.; Schuitemaker, J.H.N.; Hilkens, C.M.U.; Kapsenberg, M.L. Prostaglandin E2 induces the final maturation of IL-12-deficient CD1a+ CD83+ dendritic cells: The levels of IL-12 are determined during the final dendritic cell maturation and are resistant to further modulation. J. Immunol. 1998, 161, 2804–2809. [Google Scholar] [PubMed]
- Liu, L.; Rich, B.E.; Inobe, J.; Chen, W.; Weiner, H.L. Induction of Th2 cell differentiation in the primary immune response: Dendritic cells isolated from adherent cell culture treated with IL-10 prime naive CD4+ T cells to secrete IL-4. Int. Immunol. 1998, 10, 1017–1026. [Google Scholar] [CrossRef]
- De Smedt, T.; Van Mechelen, M.; De Becker, G.; Urbain, J.; Leo, O.; Moser, M. Effect of interleukin-10 on dendritic cell maturation and function. Eur. J. Immunol. 1997, 27, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Vanderheyde, N.; Verhasselt, V.; Goldman, M.; Willems, F. Inhibition of human dendritic cell functions by methylprednisolone. Transplantation 1999, 67, 1342–1347. [Google Scholar] [CrossRef]
- Panina-Bordignon, P.; Mazzeo, D.; Lucia, P.D.; D’ambrosio, D.; Lang, R.; Fabbri, L.; Self, C.; Sinigaglia, F. Beta2-agonists prevent Th1 development by selective inhibition of interleukin 12. J. Clin. Investig. 1997, 100, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Snijders, A.; Kalinski, P.; Hilkens, C.M.; Kapsenberg, M.L. High-level IL-12 production by human dendritic cells requires two signals. Int. Immunol. 1998, 10, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Hilkens, C.M.U.; Kalinski, P.; de Boer, M.; Kapsenberg, M.L. Human dendritic cells require exogenous interleukin-12–inducing factors to direct the development of naive T-helper cells toward the Th1 phenotype. Blood 1997, 90, 1920–1926. [Google Scholar] [PubMed]
- Verhasselt, V.; Buelens, C.; Willems, F.; De Groote, D.; Haeffner-Cavaillon, N.; Goldman, M. Bacterial lipopolysaccharide stimulates the production of cytokines and the expression of costimulatory molecules by human peripheral blood dendritic cells: Evidence for a soluble CD14-dependent pathway. J. Immunol. 1997, 158, 2919–2925. [Google Scholar]
- Sparwasser, T.; Koch, E.; Vabulas, R.M.; Heeg, K.; Lipford, G.B.; Ellwart, J.W.; Wagner, H. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur. J. Immunol. 1998, 28, 2045–2054. [Google Scholar] [CrossRef]
- Rissoan, M.-C.; Soumelis, V.; Kadowaki, N.; Grouard, G.; Briere, F.; de Waal Malefyt, R.; Liu, Y.-J. Reciprocal control of T helper cell and dendritic cell differentiation. Science 1999, 283, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Salio, M.; Sakakibara, Y.; Langen, H.; Julkunen, I.; Lanzavecchia, A. Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J. Exp. Med. 1999, 189, 821–829. [Google Scholar] [CrossRef]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Cueto, F.J.; Martinez-Lopez, M.; Labiano, S.; Morales-Kastresana, A.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 2017, 31, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, J.P.; Reis e Sousa, C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Nabeyama, A.; Miyake, Y.; Qiu, C.-H.; Kurita, A.; Tomura, M.; Kanagawa, O.; Fujii, S.; Tanaka, M. CD169-Positive Macrophages Dominate Antitumor Immunity by Crosspresenting Dead Cell-Associated Antigens. Immunity 2011, 34, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103+/CD141+ Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef] [PubMed]
- Kastenmüller, W.; Brandes, M.; Wang, Z.; Herz, J.; Egen, J.G.; Germain, R.N. Peripheral Prepositioning and Local CXCL9 Chemokine-Mediated Guidance Orchestrate Rapid Memory CD8+ T Cell Responses in the Lymph Node. Immunity 2013, 38, 502–513. [Google Scholar] [CrossRef]
- Wendel, M.; Galani, I.E.; Suri-Payer, E.; Cerwenka, A. Natural killer cell accumulation in tumors is dependent on IFN-γ and CXCR3 ligands. Cancer Res. 2008, 68, 8437–8445. [Google Scholar] [CrossRef]
- Mittal, D.; Vijayan, D.; Putz, E.M.; Aguilera, A.R.; Markey, K.A.; Straube, J.; Kazakoff, S.; Nutt, S.L.; Takeda, K.; Hill, G.R.; et al. Interleukin-12 from CD103 + Batf3-Dependent Dendritic Cells Required for NK-Cell Suppression of Metastasis. Cancer Immunol. Res. 2017, 5, 1098–1108. [Google Scholar] [CrossRef]
- Hochrein, H.; O’Keeffe, M.; Luft, T.; Vandenabeele, S.; Grumont, R.J.; Maraskovsky, E.; Shortman, K. Interleukin (Il)-4 Is a Major Regulatory Cytokine Governing Bioactive IL-12 Production by Mouse and Human Dendritic Cells. J. Exp. Med. 2000, 192, 823–834. [Google Scholar] [CrossRef]
- Gardner, A.; Ruffell, B. Dendritic cells and cancer immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L.; Jegathesan, M.; Darnell, R.B. Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat. Immunol. 2001, 2, 1010. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Steinman, R.M.; Krasovsky, J.; Munz, C.; Bhardwaj, N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J. Exp. Med. 2001, 193, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Dieu, M.-C.; Vanbervliet, B.; Vicari, A.; Bridon, J.-M.; Oldham, E.; Aït-Yahia, S.; Brière, F.; Zlotnik, A.; Lebecque, S.; Caux, C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 1998, 188, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Mailliard, R.B.; Wankowicz-Kalinska, A.; Cai, Q.; Wesa, A.; Hilkens, C.M.; Kapsenberg, M.L.; Kirkwood, J.M.; Storkus, W.J.; Kalinski, P. α-type-1 polarized dendritic cells: A novel immunization tool with optimized CTL-inducing activity. Cancer Res. 2004, 64, 5934–5937. [Google Scholar] [CrossRef]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Tumor-derived factors modulating dendritic cell function. Cancer Immunol. Immunother. 2016, 65, 821–833. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Tsumura, H.; Miwa, M.; Inaba, K. Contrasting effects of TGF-β1 and TNF-α on the development of dendritic cells from progenitors in mouse bone marrow. Stem Cells 1997, 15, 144–153. [Google Scholar] [CrossRef]
- Platten, M.; Wick, W.; Weller, M. Malignant glioma biology: Role for TGF-β in growth, motility, angiogenesis, and immune escape. Microsc. Res. Tech. 2001, 52, 401–410. [Google Scholar] [CrossRef]
- Nitta, T.; Hishii, M.; Sato, K.; Okumura, K. Selective expression of interleukin-10 gene within glioblastoma multiforme. Brain Res. 1994, 649, 122–128. [Google Scholar] [CrossRef]
- Ludewig, P.; Gallizioli, M.; Urra, X.; Behr, S.; Brait, V.H.; Gelderblom, M.; Magnus, T.; Planas, A.M. Dendritic cells in brain diseases. Biochim. Biophys. Acta 2016, 1862, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S. Impaired IFN-α production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.-Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2, 3-dioxygenase. J. Clin. Investig. 2007, 117, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Faget, J.; Bendriss-Vermare, N.; Gobert, M.; Durand, I.; Olive, D.; Biota, C.; Bachelot, T.; Treilleux, I.; Goddard-Leon, S.; Lavergne, E. ICOS-ligand expression on plasmacytoid dendritic cells supports breast cancer progression by promoting the accumulation of immunosuppressive CD4+ T cells. Cancer Res. 2012, 72, 6130–6141. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Gregorio, J.; Wang, Y.-H.; Ito, T.; Meller, S.; Hanabuchi, S.; Anderson, S.; Atkinson, N.; Ramirez, P.T.; Liu, Y.-J. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3+ T-regulatory cells. Cancer Res. 2012, 72, 5240–5249. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, P.; Xin, S.; Wang, Z.; Li, J. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp. Cell Res. 2017, 360, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.O.; Cornforth, A.N.; Depriest, C.; McClay, E.F.; Amatruda, T.T.; de Leon, C.; Ellis, R.E.; Mayorga, C.; Carbonell, D.; Cubellis, J.M. Tumor stem cell antigens as consolidative active specific immunotherapy: A randomized phase II trial of dendritic cells versus tumor cells in patients with metastatic melanoma. J. Immunother. 2012, 35, 641–649. [Google Scholar] [CrossRef]
- Nakai, N.; Asai, J.; Ueda, E.; Takenaka, H.; Katoh, N.; Kishimoto, S. Vaccination of Japanese patients with advanced melanoma with peptide, tumor lysate or both peptide and tumor lysate-pulsed mature, monocyte-derived dendritic cells. J. Dermatol. 2006, 33, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Oshita, C.; Takikawa, M.; Kume, A.; Miyata, H.; Ashizawa, T.; Iizuka, A.; Kiyohara, Y.; Yoshikawa, S.; Tanosaki, R.; Yamazaki, N.; et al. Dendritic cell-based vaccination in metastatic melanoma patients: Phase II clinical trial. Oncol. Rep. 2012, 28, 1131–1138. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef]
- Small, E.J.; Fratesi, P.; Reese, D.M.; Strang, G.; Laus, R.; Peshwa, M.V.; Valone, F.H. Immunotherapy of Hormone-Refractory Prostate Cancer with Antigen-Loaded Dendritic Cells. J. Clin. Oncol. 2000, 18, 3894–3903. [Google Scholar] [CrossRef]
- Siesjö, P.; Visse, E.; Sjögren, H.O. Cure of established, intracerebral rat gliomas induced by therapeutic immunizations with tumor cells and purified APC or adjuvant IFN-gamma treatment. J. Immunother. Emphas. Tumor Immunol. 1996, 19, 334–345. [Google Scholar] [CrossRef]
- Liau, L.M.; Black, K.L.; Prins, R.M.; Sykes, S.N.; DiPatre, P.L.; Cloughesy, T.F.; Becker, D.P.; Bronstein, J.M. Treatment of intracranial gliomas with bone marrow-derived dendritic cells pulsed with tumor antigens. J. Neurosurg. 1999, 90, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, Y.; Hirao, M.; Robbins, P.D.; Lotze, M.T.; Tahara, H.; Kapsenberg, M.L.; Kirkwood, J.M.; Storkus, W.J.; Kalinski, P. Induction of systemic and therapeutic antitumor immunity using intratumoral injection of dendritic cells genetically modified to express interleukin 12. Cancer Res. 1999, 59, 4035–4041. [Google Scholar] [PubMed]
- Belmans, J.; Van Woensel, M.; Creyns, B.; Dejaegher, J.; Bullens, D.M.; Van Gool, S.W. Immunotherapy with subcutaneous immunogenic autologous tumor lysate increases murine glioblastoma survival. Sci. Rep. 2017, 7, 13902. [Google Scholar] [CrossRef] [PubMed]
- Jouanneau, E.; Poujol, D.; Gulia, S.; Le Mercier, I.; Blay, J.Y.; Belin, M.F.; Puisieux, I. Dendritic cells are essential for priming but inefficient for boosting antitumour immune response in an orthotopic murine glioma model. Cancer Immunol. Immunother. 2006, 55, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Aarntzen, E.H.J.G.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.G.; van Rossum, M.; et al. Natural Human Plasmacytoid Dendritic Cells Induce Antigen-Specific T-Cell Responses in Melanoma Patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef]
- Prue, R.L.; Vari, F.; Radford, K.J.; Tong, H.; Hardy, M.Y.; D’Rozario, R.; Waterhouse, N.J.; Rossetti, T.; Coleman, R.; Tracey, C.; et al. A phase I clinical trial of CD1c (BDCA-1)+ dendritic cells pulsed with HLA-A*0201 peptides for immunotherapy of metastatic hormone refractory prostate cancer. J. Immunother. 2015, 38, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.J.G.; Duiveman-de Boer, T.; van de Rakt, M.W.M.M.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Effective Clinical Responses in Metastatic Melanoma Patients after Vaccination with Primary Myeloid Dendritic Cells. Clin. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Schreibelt, G.; Gerritsen, W.R.; de Vries, I.J.M.; Figdor, C.G. Dendritic Cell-Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.M.; Butterfield, L.H. Dendritic Cell-Based Cancer Vaccines. J. Immunol. 2018, 200, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Nava, S.; Lisini, D.; Pogliani, S.; Dossena, M.; Bersano, A.; Pellegatta, S.; Parati, E.; Finocchiaro, G.; Frigerio, S. Safe and Reproducible Preparation of Functional Dendritic Cells for Immunotherapy in Glioblastoma Patients. Stem Cells Transl. Med. 2015, 4, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Ashley, D.M.; Faiola, B.; Nair, S.; Hale, L.P.; Bigner, D.D.; Gilboa, E. Bone marrow-generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J. Exp. Med. 1997, 186, 1177–1182. [Google Scholar] [CrossRef]
- Bregy, A.; Wong, T.M.; Shah, A.H.; Goldberg, J.M.; Komotar, R.J. Active immunotherapy using dendritic cells in the treatment of glioblastoma multiforme. Cancer Treat. Rev. 2013, 39, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Grauer, O.M.; Sutmuller, R.P.M.; van Maren, W.; Jacobs, J.F.M.; Bennink, E.; Toonen, L.W.J.; Nierkens, S.; Adema, G.J. Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate-pulsed dendritic cells in a murine glioma model. Int. J. Cancer 2007, 122, 1794–1802. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Crotty, L.E.; Archer, G.E.; McLendon, R.E.; Friedman, A.; Dranoff, G.; Bigner, D.D.; Sampson, J.H. Bone marrow-derived dendritic cells pulsed with tumor homogenate induce immunity against syngeneic intracerebral glioma. J. Neuroimmunol. 2000, 103, 16–25. [Google Scholar] [CrossRef]
- Ni, H.T.; Spellman, S.R.; Jean, W.C.; Hall, W.A.; Low, W.C. Immunization with dendritic cells pulsed with tumor extract increases survival of mice bearing intracranial gliomas. J. Neurooncol. 2001, 51, 1–9. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Crotty, L.E.; Archer, G.E.; Hess, K.R.; Wikstrand, C.J.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Epidermal Growth Factor Receptor VIII Peptide Vaccination Is Efficacious against Established Intracerebral Tumors. Clin. Cancer Res. 2003, 9, 4247–4254. [Google Scholar] [PubMed]
- Li, J.; Wang, F.; Wang, G.; Sun, Y.; Cai, J.; Liu, X.; Zhang, J.; Lu, X.; Li, Y.; Chen, M.; et al. Combination epidermal growth factor receptor variant III peptide-pulsed dendritic cell vaccine with miR-326 results in enhanced killing on EGFRvIII-positive cells. Oncotarget 2017, 8, 26256–26268. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kane, J.R.; Panek, W.K.; Young, J.S.; Rashidi, A.; Yu, D.; Kanojia, D.; Hasan, T.; Miska, J.; Gómez-Lim, M.A.; et al. A Dendritic Cell-Targeted Adenoviral Vector Facilitates Adaptive Immune Response Against Human Glioma Antigen (CMV-IE) and Prolongs Survival in a Human Glioma Tumor Model. Neurotherapeutics 2018, 15, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Prins, R.M.; Odesa, S.K.; Liau, L.M. Immunotherapeutic targeting of shared melanoma-associated antigens in a murine glioma model. Cancer Res. 2003, 63, 8487–8491. [Google Scholar] [PubMed]
- Kim, C.-H.; Woo, S.-J.; Park, J.-S.; Kim, H.-S.; Park, M.-Y.; Park, S.-D.; Hong, Y.-K.; Kim, T.-G. Enhanced antitumour immunity by combined use of temozolomide and TAT-survivin pulsed dendritic cells in a murine glioma. Immunology 2007, 122, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.G.; Eguchi, J.; Kruse, C.A.; Gomez, G.G.; Fakhrai, H.; Schroter, S.; Ma, W.; Hoa, N.; Minev, B.; Delgado, C.; et al. Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics. Clin. Cancer Res. 2007, 13, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Tian, G.; Li, P.; Chen, Q.; Zhang, R.; Ke, Y.; Xiao, Z.; Jiang, X. Anti-glioma response of autologous T cells stimulated by autologous dendritic cells electrofused with CD133+ or CD133− glioma cells. J. Neuroimmunol. 2012, 242, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Cobbs, C.S.; Harkins, L.; Samanta, M.; Gillespie, G.Y.; Bharara, S.; King, P.H.; Nabors, L.B.; Cobbs, C.G.; Britt, W.J. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002, 62, 3347–3350. [Google Scholar]
- Scheurer, M.E.; Bondy, M.L.; Aldape, K.D.; Albrecht, T.; El-Zein, R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol. 2008, 116, 79–86. [Google Scholar] [CrossRef]
- Ding, D.; Han, S.; Wang, Z.; Guo, Z.; Wu, A. Does the existence of HCMV components predict poor prognosis in glioma? J. Neurooncol. 2014, 116, 515–522. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Xie, W.; Schmittling, R.; Learn, C.; Friedman, A.; McLendon, R.E.; Sampson, J.H. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 2008, 10, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; De Leon, G.; Boczkowski, D.; Schmittling, R.; Xie, W.; Staats, J.; Liu, R.; Johnson, L.A.; Weinhold, K.; Archer, G.E.; et al. Recognition and Killing of Autologous, Primary Glioblastoma Tumor Cells by Human Cytomegalovirus pp65-Specific Cytotoxic T Cells. Clin. Cancer Res. 2014, 20, 2684–2694. [Google Scholar] [CrossRef]
- Wong, A.J.; Ruppert, J.M.; Bigner, S.H.; Grzeschik, C.H.; Humphrey, P.A.; Bigner, D.S.; Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 1992, 89, 2965–2969. [Google Scholar] [CrossRef]
- Bu, N.; Wu, H.; Sun, B.; Zhang, G.; Zhan, S.; Zhang, R.; Zhou, L. Exosome-loaded dendritic cells elicit tumor-specific CD8+ cytotoxic T cells in patients with glioma. J. Neurooncol. 2011, 104, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, L.; Liu, J.; Meng, H.; Zhang, R.; Ma, L.; Wu, L.; Yu, S.; Shi, F.; Li, Y.; et al. Co-delivery of tumor-derived exosomes with alpha-galactosylceramide on dendritic cell-based immunotherapy for glioblastoma. Cancer Lett. 2017, 411, 182–190. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.-N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Candolfi, M.; King, G.D.; Yagiz, K.; Curtin, J.F.; Mineharu, Y.; Muhammad, A.G.; Foulad, D.; Kroeger, K.M.; Barnett, N.; Josien, R.; et al. Plasmacytoid Dendritic Cells in the Tumor Microenvironment: Immune Targets for Glioma Therapeutics. Neoplasia 2012, 14, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, R.; Luksik, A.S.; Garzon-Muvdi, T.; Lim, M. The Potential of Cellular- and Viral-Based Immunotherapies for Malignant Glioma–Dendritic Cell Vaccines, Adoptive Cell Transfer, and Oncolytic Viruses. Curr. Neurol. Neurosci. Rep. 2017, 17, 50. [Google Scholar] [CrossRef] [PubMed]
- Bassiri, H.; Das, R.; Guan, P.; Barrett, D.M.; Brennan, P.J.; Banerjee, P.P.; Wiener, S.J.; Orange, J.S.; Brenner, M.B.; Grupp, S.A.; et al. iNKT cell cytotoxic responses control T-lymphoma growth in vitro and in vivo. Cancer Immunol. Res. 2014, 2, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Mohrs, M.; Reinhardt, R.L.; Baron, J.L.; Wang, Z.-E.; Gapin, L.; Kronenberg, M.; Locksley, R.M. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J. Exp. Med. 2003, 198, 1069–1076. [Google Scholar] [CrossRef]
- Carbone, E.; Ruggiero, G.; Terrazzano, G.; Palomba, C.; Manzo, C.; Fontana, S.; Spits, H.; Kärre, K.; Zappacosta, S. A new mechanism of NK cell cytotoxicity activation: The CD40-CD40 ligand interaction. J. Exp. Med. 1997, 185, 2053–2060. [Google Scholar] [CrossRef]
- Haraguchi, K.; Takahashi, T.; Nakahara, F.; Matsumoto, A.; Kurokawa, M.; Ogawa, S.; Oda, H.; Hirai, H.; Chiba, S. CD1d expression level in tumor cells is an important determinant for anti-tumor immunity by natural killer T cells. Leuk. Lymphoma 2006, 47, 2218–2223. [Google Scholar] [CrossRef] [PubMed]
- Hermans, I.F.; Silk, J.D.; Gileadi, U.; Salio, M.; Mathew, B.; Ritter, G.; Schmidt, R.; Harris, A.L.; Old, L.; Cerundolo, V. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J. Immunol. 2003, 171, 5140–5147. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Asgharzadeh, S.; Salo, J.; Engell, K.; Wu, H.W.; Sposto, R.; Ara, T.; Silverman, A.M.; DeClerck, Y.A.; Seeger, R.C.; et al. Vα24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J. Clin. Investig. 2009, 119, 1524–1536. [Google Scholar] [CrossRef]
- De Santo, C.; Salio, M.; Masri, S.H.; Lee, L.Y.-H.; Dong, T.; Speak, A.O.; Porubsky, S.; Booth, S.; Veerapen, N.; Besra, G.S.; et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J. Clin. Investig. 2008, 118, 4036–4048. [Google Scholar] [CrossRef]
- Shimizu, K.; Kurosawa, Y.; Taniguchi, M.; Steinman, R.M.; Fujii, S. Cross-presentation of glycolipid from tumor cells loaded with α-galactosylceramide leads to potent and long-lived T cell–mediated immunity via dendritic cells. J. Exp. Med. 2007, 204, 2641–2653. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.-I.; Shimizu, K.; Smith, C.; Bonifaz, L.; Steinman, R.M. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J. Exp. Med. 2003, 198, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.R.; Sika-Paotonu, D.; Knight, D.A.; Dickgreber, N.; Farrand, K.J.; Ronchese, F.; Hermans, I.F. Potent anti-tumor responses to immunization with dendritic cells loaded with tumor tissue and an NKT cell ligand. Immunol. Cell Biol. 2010, 88, 596–604. [Google Scholar] [CrossRef]
- Dhodapkar, K.M.; Cirignano, B.; Chamian, F.; Zagzag, D.; Miller, D.C.; Finlay, J.L.; Steinman, R.M. Invariant natural killer T cells are preserved in patients with glioma and exhibit antitumor lytic activity following dendritic cell-mediated expansion. Int. J. Cancer 2004, 109, 893–899. [Google Scholar] [CrossRef]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Mathios, D.; Kim, J.E.; Mangraviti, A.; Phallen, J.; Park, C.-K.; Jackson, C.M.; Garzon-Muvdi, T.; Kim, E.; Theodros, D.; Polanczyk, M.; et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 2016, 8, 370ra180. [Google Scholar] [CrossRef] [PubMed]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Orpilla, J.; Moughon, D.; Shin, N.; Sedighim, S.; Yong, W.H.; Li, G.; Cloughesy, T.F.; et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2016, 1, e87059. [Google Scholar] [CrossRef] [PubMed]
- Garzon-Muvdi, T.; Theodros, D.; Luksik, A.S.; Maxwell, R.; Kim, E.; Jackson, C.M.; Belcaid, Z.; Ganguly, S.; Tyler, B.; Brem, H.; et al. Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget 2018, 9, 20681–20697. [Google Scholar] [CrossRef] [PubMed]
- Schnare, M.; Barton, G.M.; Holt, A.C.; Takeda, K.; Akira, S.; Medzhitov, R. Toll-like receptors control activation of adaptive immune responses. Nat. Immunol. 2001, 2, 947–950. [Google Scholar] [CrossRef] [PubMed]
- Mattei, F.; Schiavoni, G.; Belardelli, F.; Tough, D.F. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J. Immunol. 2001, 167, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.L.; Kadima, A.N.; Cole, D.J.; Gillanders, W.E. Defining the antigen-specific T-cell response to vaccination and poly(I:C)/TLR3 signaling: Evidence of enhanced primary and memory CD8 T-cell responses and antitumor immunity. J. Immunother. 2005, 28, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-F.; Li, K.; Zhang, Q.; Wang, C.; Yue, Y.; Chen, Z.; Yuan, S.-J.; Liu, X.; Wen, Y.; Han, M.; et al. Dendritic cell-mediated delivery of doxorubicin-polyglycerol-nanodiamond composites elicits enhanced anti-cancer immune response in glioblastoma. Biomaterials 2018, 181, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-F.; Xu, Y.-H.; Li, K.; Wang, C.; Liu, X.; Yue, Y.; Chen, Z.; Yuan, S.-J.; Wen, Y.; Zhang, Q.; et al. Doxorubicin-polyglycerol-nanodiamond composites stimulate glioblastoma cell immunogenicity through activation of autophagy. Acta Biomater. 2019, 86, 381–394. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Krombach, J.; Hennel, R.; Brix, N.; Orth, M.; Schoetz, U.; Ernst, A.; Schuster, J.; Zuchtriegel, G.; Reichel, C.A.; Bierschenk, S.; et al. Priming anti-tumor immunity by radiotherapy: Dying tumor cell-derived DAMPs trigger endothelial cell activation and recruitment of myeloid cells. Oncoimmunology 2019, 8, e1523097. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Muili, K.; Bolyard, C.; Russell, L.; Lee, T.J.; Banasavadi-Siddegowda, Y.; Yoo, J.Y.; Yan, Y.; Ballester, L.Y.; Bockhorst, K.H.; et al. Suppression of HMGB1 Released in the Glioblastoma Tumor Microenvironment Reduces Tumoral Edema. Mol. Ther. Oncolytics 2019, 12, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.-C.; Chou, M.-H.; Chien, C.-Y.; Chuang, J.-H.; Liu, Y.-L. Triggering TLR3 pathway promotes tumor growth and cisplatin resistance in head and neck cancer cells. Oral Oncol. 2018, 86, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Lv, X.; Zhang, X.; Li, T.; Zang, G.; Yang, N.; Wang, X.; Wu, J.; Chen, W.; Liu, Y.-J.; et al. An effective dendritic cell-based vaccine containing glioma stem-like cell lysate and CpG adjuvant for an orthotopic mouse model of glioma. Int. J. Cancer 2019, 144, 2867–2879. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, G.; Yuan, X.; Xu, M.; Wang, H.; Ji, J.; Konda, B.; Black, K.L.; Yu, J.S. Antigen-Specific T-Cell Response from Dendritic Cell Vaccination Using Cancer Stem-Like Cell-Associated Antigens. Stem Cells 2009, 27, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Pellegatta, S.; Poliani, P.L.; Corno, D.; Menghi, F.; Ghielmetti, F.; Suarez-Merino, B.; Caldera, V.; Nava, S.; Ravanini, M.; Facchetti, F.; et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 2006, 66, 10247–10252. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yao, Y.; Hua, W.; Wu, Z.; Zhong, P.; Mao, Y.; Zhou, L.; Luo, F.; Chu, Y. Mouse glioma immunotherapy mediated by A2B5+ GL261 cell lysate-pulsed dendritic cells. J. Neurooncol. 2014, 116, 497–504. [Google Scholar] [CrossRef]
- Hartmann, G.; Weiner, G.J.; Krieg, A.M. CpG DNA: A potent signal for growth, activation, and maturation of human dendritic cells. Proc. Natl. Acad. Sci. USA 1999, 96, 9305–9310. [Google Scholar] [CrossRef]
- Kadowaki, N.; Antonenko, S.; Liu, Y.J. Distinct CpG DNA and polyinosinic-polycytidylic acid double-stranded RNA, respectively, stimulate CD11c- type 2 dendritic cell precursors and CD11c+ dendritic cells to produce type I IFN. J. Immunol. 2001, 166, 2291–2295. [Google Scholar] [CrossRef]
- Yi, A.K.; Klinman, D.M.; Martin, T.L.; Matson, S.; Krieg, A.M. Rapid immune activation by CpG motifs in bacterial DNA. Systemic induction of IL-6 transcription through an antioxidant-sensitive pathway. J. Immunol. 1996, 157, 5394–5402. [Google Scholar]
- Krieg, A.M.; Yi, A.K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549. [Google Scholar] [CrossRef]
- Yu, J.S.; Wheeler, C.J.; Zeltzer, P.M.; Ying, H.; Finger, D.N.; Lee, P.K.; Yong, W.H.; Incardona, F.; Thompson, R.C.; Riedinger, M.S.; et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001, 61, 842–847. [Google Scholar] [PubMed]
- Kikuchi, T.; Akasaki, Y.; Irie, M.; Homma, S.; Abe, T.; Ohno, T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol. Immunother. 2001, 50, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Akasaki, Y.; Abe, T.; Fukuda, T.; Saotome, H.; Ryan, J.L.; Kufe, D.W.; Ohno, T. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J. Immunother. 2004, 27, 452–459. [Google Scholar] [CrossRef]
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.-W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin. Cancer Res. 2005, 11, 5515–5525. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, S.; De Vleeschouwer, S.; Kaempgen, E.; Wolff, J.E.A.; Kühl, J.; Demaerel, P.; Warmuth-Metz, M.; Flamen, P.; Van Calenbergh, F.; Plets, C. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br. J. Cancer 2004, 91, 1656. [Google Scholar] [CrossRef]
- Yamanaka, R.; Homma, J.; Yajima, N.; Tsuchiya, N.; Sano, M.; Kobayashi, T.; Yoshida, S.; Abe, T.; Narita, M.; Takahashi, M. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: Results of a clinical phase I/II trial. Clin. Cancer Res. 2005, 11, 4160–4167. [Google Scholar] [CrossRef]
- Yu, J.S.; Liu, G.; Ying, H.; Yong, W.H.; Black, K.L.; Wheeler, C.J. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004, 64, 4973–4979. [Google Scholar] [CrossRef]
- De Vleeschouwer, S.; Fieuws, S.; Rutkowski, S.; Van Calenbergh, F.; Van Loon, J.; Goffin, J.; Sciot, R.; Wilms, G.; Demaerel, P.; Warmuth-Metz, M. Postoperative adjuvant dendritic cell–based immunotherapy in patients with relapsed glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 3098–3104. [Google Scholar] [CrossRef] [PubMed]
- Caruso, D.A.; Orme, L.M.; Neale, A.M.; Radcliff, F.J.; Amor, G.M.; Maixner, W.; Downie, P.; Hassall, T.E.; Tang, M.L.K.; Ashley, D.M. Results of a phase 1 study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children and young adults with brain cancer. Neuro Oncol. 2004, 6, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Laherty, R.; Tomlinson, F.H.; Chuah, T.; Schmidt, C. Results of a phase I dendritic cell vaccine trial for malignant astrocytoma: Potential interaction with adjuvant chemotherapy. J. Clin. Neurosci. 2008, 15, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Lieberman, F.S.; Walter, K.A.; Lunsford, L.D.; Kondziolka, D.S.; Bejjani, G.K.; Hamilton, R.L.; Torres-Trejo, A.; Kalinski, P.; Cai, Q. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. J. Transl. Med. 2007, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Prins, R.M.; Soto, H.; Konkankit, V.; Odesa, S.K.; Eskin, A.; Yong, W.H.; Nelson, S.F.; Liau, L.M. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin. Cancer Res. 2011, 17, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Ardon, H.; De Vleeschouwer, S.; Van Calenbergh, F.; Claes, L.; Kramm, C.M.; Rutkowski, S.; Wolff, J.E.A.; Van Gool, S.W. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr. Blood Cancer 2010, 54, 519–525. [Google Scholar] [CrossRef]
- Sampson, J.H.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; Herndon, J.E.; Lally-Goss, D.; McGehee-Norman, S.; Paolino, A.; Reardon, D.A.; Friedman, A.H. An epidermal growth factor receptor variant III–targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol. Cancer Ther. 2009, 8, 2773–2779. [Google Scholar] [CrossRef]
- Okada, H.; Kalinski, P.; Ueda, R.; Hoji, A.; Kohanbash, G.; Donegan, T.E.; Mintz, A.H.; Engh, J.A.; Bartlett, D.L.; Brown, C.K.; et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 2011, 29, 330–336. [Google Scholar] [PubMed]
- Phuphanich, S.; Wheeler, C.J.; Rudnick, J.D.; Mazer, M.; Wang, H.; Nuno, M.A.; Richardson, J.E.; Fan, X.; Ji, J.; Chu, R.M. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 2013, 62, 125–135. [Google Scholar] [CrossRef]
- Akiyama, Y.; Oshita, C.; Kume, A.; Iizuka, A.; Miyata, H.; Komiyama, M.; Ashizawa, T.; Yagoto, M.; Abe, Y.; Mitsuya, K.; et al. α-type-1 polarized dendritic cell-based vaccination in recurrent high-grade glioma: A phase I clinical trial. BMC Cancer 2012, 12, 623. [Google Scholar] [CrossRef] [PubMed]
- Prins, R.M.; Wang, X.; Soto, H.; Young, E.; Lisiero, D.N.; Fong, B.; Everson, R.; Yong, W.H.; Lai, A.; Li, G. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J. Immunother. 2013, 36, 152. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Abe, T.; Yajima, N.; Tsuchiya, N.; Homma, J.; Kobayashi, T.; Narita, M.; Takahashi, M.; Tanaka, R. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: Results of a clinical phase I/II trial. Br. J. Cancer 2003, 89, 1172. [Google Scholar] [CrossRef]
- Wheeler, C.J.; Black, K.L.; Liu, G.; Mazer, M.; Zhang, X.; Pepkowitz, S.; Goldfinger, D.; Ng, H.; Irvin, D.; John, S.Y. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008, 68, 5955–5964. [Google Scholar] [CrossRef] [PubMed]
- Fadul, C.E.; Fisher, J.L.; Hampton, T.H.; Lallana, E.C.; Li, Z.; Gui, J.; Szczepiorkowski, Z.M.; Tosteson, T.D.; Rhodes, C.H.; Wishart, H.A.; et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J. Immunother. 2011, 34, 382–389. [Google Scholar] [CrossRef]
- Chang, C.-N.; Huang, Y.-C.; Yang, D.-M.; Kikuta, K.; Wei, K.-J.; Kubota, T.; Yang, W.-K. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J. Clin. Neurosci. 2011, 18, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.-Y.; Yang, W.-K.; Lee, H.-C.; Hsu, D.-M.; Lin, H.-L.; Lin, S.-Z.; Chen, C.-C.; Harn, H.-J.; Liu, C.-L.; Lee, W.-Y.; et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: A phase II clinical trial. World Neurosurg. 2012, 77, 736–744. [Google Scholar] [CrossRef]
- Lasky, J.L.; Panosyan, E.H.; Plant, A.; Davidson, T.; Yong, W.H.; Prins, R.M.; Liau, L.M.; Moore, T.B. Autologous tumor lysate-pulsed dendritic cell immunotherapy for pediatric patients with newly diagnosed or recurrent high-grade gliomas. Anticancer Res. 2013, 33, 2047–2056. [Google Scholar]
- Jie, X.; Hua, L.; Jiang, W.; Feng, F.; Feng, G.; Hua, Z. Clinical application of a dendritic cell vaccine raised against heat-shocked glioblastoma. Cell Biochem. Biophys. 2012, 62, 91–99. [Google Scholar] [CrossRef]
- Ardon, H.; Van Gool, S.; Lopes, I.S.; Maes, W.; Sciot, R.; Wilms, G.; Demaerel, P.; Bijttebier, P.; Claes, L.; Goffin, J.; et al. Integration of autologous dendritic cell-based immunotherapy in the primary treatment for patients with newly diagnosed glioblastoma multiforme: A pilot study. J. Neurooncol. 2010, 99, 261–272. [Google Scholar] [CrossRef]
- Sakai, K.; Shimodaira, S.; Maejima, S.; Udagawa, N.; Sano, K.; Higuchi, Y.; Koya, T.; Ochiai, T.; Koide, M.; Uehara, S.; et al. Dendritic cell-based immunotherapy targeting Wilms’ tumor 1 in patients with recurrent malignant glioma. J. Neurosurg. 2015, 123, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Hunn, M.K.; Bauer, E.; Wood, C.E.; Gasser, O.; Dzhelali, M.; Ancelet, L.R.; Mester, B.; Sharples, K.J.; Findlay, M.P.; Hamilton, D.A.; et al. Dendritic cell vaccination combined with temozolomide retreatment: Results of a phase I trial in patients with recurrent glioblastoma multiforme. J. Neurooncol. 2015, 121, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Vik-Mo, E.O.; Nyakas, M.; Mikkelsen, B.V.; Moe, M.C.; Due-Tønnesen, P.; Suso, E.M.I.; Sæbøe-Larssen, S.; Sandberg, C.; Brinchmann, J.E.; Helseth, E.; et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 2013, 62, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E.; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef]
- Inogés, S.; Tejada, S.; de Cerio, A.L.-D.; Gállego Pérez-Larraya, J.; Espinós, J.; Idoate, M.A.; Domínguez, P.D.; de Eulate, R.G.; Aristu, J.; Bendandi, M.; et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J. Transl. Med. 2017, 15, 104. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed]
- Iwami, K.; Shimato, S.; Ohno, M.; Okada, H.; Nakahara, N.; Sato, Y.; Yoshida, J.; Suzuki, S.; Nishikawa, H.; Shiku, H.; et al. Peptide-pulsed dendritic cell vaccination targeting interleukin-13 receptor α2 chain in recurrent malignant glioma patients with HLA-A*24/A*02 allele. Cytotherapy 2012, 14, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Black, K.L.; Martin, N.A.; Sykes, S.N.; Bronstein, J.M.; Jouben-Steele, L.; Mischel, P.S.; Belldegrun, A.; Cloughesy, T.F. Treatment of a glioblastoma patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class I–matched tumor peptides: Case report. Neurosurg. Focus 2000, 9, 1–5. [Google Scholar] [CrossRef]
- Prins, R.M.; Craft, N.; Bruhn, K.W.; Khan-Farooqi, H.; Koya, R.C.; Stripecke, R.; Miller, J.F.; Liau, L.M. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: Relation to central nervous system antitumor immunity. J. Immunol. 2006, 176, 157–164. [Google Scholar] [CrossRef] [PubMed]
- van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; de Vries, I.J.M.; Bol, K.F. Dendritic Cell Cancer Therapy: Vaccinating the Right Patient at the Right Time. Front. Immunol. 2018, 9, 2265. [Google Scholar] [CrossRef]
- Wheeler, C.J.; Das, A.; Liu, G.; Yu, J.S.; Black, K.L. Clinical Responsiveness of Glioblastoma Multiforme to Chemotherapy after Vaccination. Clin. Cancer Res. 2004, 10, 5316–5326. [Google Scholar] [CrossRef]
- Choi, C.W.; Jeong, M.H.; Park, Y.-S.; Son, C.-H.; Lee, H.-R.; Koh, E.-K. Combination Treatment of Stereotactic Body Radiation Therapy and Immature Dendritic Cell Vaccination for Augmentation of Local and Systemic Effects. Cancer Res. Treat. 2018. [Google Scholar] [CrossRef]
- Rodríguez-Ruiz, M.E.; Perez-Gracia, J.L.; Rodríguez, I.; Alfaro, C.; Oñate, C.; Pérez, G.; Gil-Bazo, I.; Benito, A.; Inogés, S.; López-Diaz de Cerio, A.; et al. Combined immunotherapy encompassing intratumoral poly-ICLC, dendritic-cell vaccination and radiotherapy in advanced cancer patients. Ann. Oncol. 2018, 29, 1312–1319. [Google Scholar] [CrossRef]
- de Vries, I.J.M.; Krooshoop, D.J.E.B.; Scharenborg, N.M.; Lesterhuis, W.J.; Diepstra, J.H.S.; van Muijen, G.N.P.; Strijk, S.P.; Ruers, T.J.; Boerman, O.C.; Oyen, W.J.G. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003, 63, 12–17. [Google Scholar]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl. Med. 2014, 6, 232ra51. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Study | Phase | Year | Patients | Antigen | Adjuvant Therapy | Clinical Efficacy | Immunologic Response |
|---|---|---|---|---|---|---|---|
| Yu et al. [183] | I | 2001 | 7 GBM 2 AA | Autologous glioma peptides | Vaccine group: OS 455 days Control group: OS 257 days | Four out of seven patients demonstrated increased cytotoxic T cell activity; Two out of four patients who underwent re-operation showed increased infiltration of CD8+ and CD45RO+ T cells. | |
| Kikuchi et al. [184] | I | 2001 | 5 GBM 2 AA 1 AO | Glioma cells | Two patients had partial response | Post immunization PBMC showed reactivity against autologous glioma or U87MG cells. | |
| Kikuchi et al. [185] | I | 2004 | 6 GBM 2 AOA 7 AA | Glioma cells | Four patients had partial response. One patient had mixed response. Two patients with stable disease. The rest of the patients progressed. | Two out of seven patients had cytolytic activities against glioma cells post immunization. | |
| Liau et al. [186] | I | 2005 | 12 GBM | Tumor associated antigen | Vaccine group: PFS 19.9 months, OS 35.9 months Historical control group: PFS 8.2 months, OS 18.3 months. | Six patients developed peripheral cytotoxic tumor-specific activity. Systemic cytotoxic activity and tumor lymphocytic infiltration were associated with response. | |
| Rutkowski et al. [187] | 1 | 2004 | 10 GBM 1 PXA 1 ALL | Tumor lysate | Four out of 12 patients had partial response. Two out of six patients with complete resection had survival >35 months. | Six out of eight patients who underwent DTH skin test had a positive test after the third vaccination. | |
| Yamanaka et al. [188] | I/II | 2005 | 18 GBM 2 AA 2 AOA 2 AG | Tumor lysate | One partial responder and three minor responders. Vaccine group: OS 480 days Control group: OS 400 days | Presence of tumor lysate specific T cell response after vaccination was associated with longer OS. | |
| Yu et al. [189] | I | 2004 | 10 GBM 4 AA | Tumor lysate | Vaccine group: OS 133 weeks Matched control group: OS 30 weeks. | Eleven out of 14 patients showed evidence of cytotoxic T cell activities. Four out of nine patients studied showed cytotoxic T cells specific against tumor antigens post vaccination. | |
| De Vleeschouwer et al. [190] | I/II | 2008 | 56 GBM | Tumor lysate | Improved PFS in a cohort of patients who received weekly vaccination. | Nine out of 21 patients demonstrated positive DTH response post immunization. | |
| Caruso et al. [191] | I | 2004 | 2 GBM 3 EPM 1 AA 1 PXA | Tumor RNA | One partial responder in AA group. All GBM patients progressed on therapy. | No statistically significant cell-mediated anti-tumor responses in either an IFN-γ-producing assay or T cell proliferation assay. Modest increase in anti-tumor antibodies in two patients. | |
| Walker et al. [192] | I | 2008 | 9 GBM 4 AA | Irradiated glioma cells | Two partial responders in GBM group. One partial and one complete responder in AA group. | Increase in tumor T cell infiltration in three out of four patients who underwent re-operation post vaccination. | |
| Okada et al. [193] | I | 2007 | 6 GBM 1 AA | Tumor cell | TFG-hIL4-Neo-TK | Initial radiographic improvement, but ultimate progression of disease. | Local infiltration of CD4+ and CD8+ T cells with associated IFN-γ response to EphA2883-891. |
| Okada et al. [193] | I | 2007 | 5 GBM | Tumor cell | TFG-hIL4-Neo-TK + Type I DC | All patients progressed within 10 months of vaccination. | No IFN-γ activity detected. |
| Prins et al. [194] | I | 2010 | 23 GBM | Tumor lysate | Imiquimod or Poly-ICLC | Significantly increased median OS in newly diagnosed GBM compared to recurrent patients. | Patients with mesenchymal gene signatures had improved survival compared to historical data. |
| Ardon et al. [195] | I | 2010 | 22 GBM 5 AA 2 PXA 1 AOA 1 AGG 1 DIPG 5 MB 4 EPM 3 ATRT | Tumor lysate | Imiquimod DC maturation ex vivo with IL-B1 and TNF-α | Six long term survivors (>24 months) in the high grade glioma group, four of which are GBM. | |
| Mitchell et al. [148] | I/II | 2015 | 12 GBM | CMV pp65 RNA | Td toxoid | Median OS 18.5 months in DC only cohort. Three out of six patients in Td group still alive at >36 months. | Increased migration of DC to tumor site with Td toxoid administration. pp65-specific immune response was present for 6 months in long term survivors. pp65-specific IFN-γ response was correlated with PFS and OS. |
| Sampson et al. [196] | I | 2009 | 12 GBM | EGFRvIII peptide | Vaccinated group: Median OS 22.8 months. | Increased antigen-specific T cell responses post vaccination. Positive response to pulsed peptide. | |
| Okada et al. [197] | I/II | 2011 | 13 GBM 5 AA 3 AO 1 AOA | IL-13Rα2, EphA2883-891, GP100209-217, and YKL-40201-210 | Poly-ICLC | One complete responder and one partial responder in GBM group. | Eleven out of 19 patients showed tumor-associated peptide response by ELISPOT and tetramer assay. |
| Phuphanich et al. [198] | I | 2013 | 21 GBM 1 DIPG | HER2, TRP-2, gp100, MAGE-11, IL13 Rα2, and AIM-2 | Median PFS newly diagnosed GBM 16.9 months Median OS newly diagnosed GBM 38.4 months | Five of 15 GBM patients had positive immune response of >0.5-fold compared to pre vaccination. | |
| Akiyama et al. [199] | I | 2012 | 7 GBM 1 AA 1 AO | WT-1, HER2, MAGE-A3, MAGE-A1, gp100 | One patient with stable disease; eight patients with progressive disease. | Cytotoxic T cell precursors against tumor-associated peptides were detected in six evaluable cases; four patients had positive DTH tests against all peptides. | |
| Prins et al. [200] | I | 2013 | Tumor lysate: 23 GBM, 5 AA TAA: 4 GBM, 2 AA | Comparison between tumor lysate and tumor associated antigens | Tumor lysate: OS 34.4 months, PFS 18.1 months TAA: OS 14.5 months, PFS 9.6 months | Increased activated NK cell population in TAA group. Post vaccination and pre vaccination Treg ratio showed trend toward association with survival. | |
| Yamanaka et al. [201] | I/II | 2003 | 7 GBM 3 AG | Tumor lysate | Two patients with minor responses | Positive T cell-mediated immune response in two out of five tested patients. Three patients showed positive DTH | |
| Wheeler et al. [202] | II | 2008 | 34 GBM | Tumor lysate | Vaccine responder: OS 642 days Vaccine non-responder: OS 430 days Vaccine responders associated with improved OS and PFS. | Seventeen patients had >1.5 fold increase in lysate directed IFN-γ response post vaccination (vaccine responder) | |
| Fadul et al. [203] | I | 2011 | 10 GBM | Tumor lysate | Patients with high immune function measures showed improved OS trends. Four out of five patients with high immune function measures had survival >2 years. | Proportion of CD4+ and CD8+ IFN-γ producing cells showed trend of increase post vaccination. | |
| Chang et al. [204] | I/II | 2011 | 16 GBM 1 AA 2 MOG | Tumor cells | Vaccine group: OS 520 days Historical control: OS 380 days 37.5% 3-year survival rate, 18.8% 5-year survival rate | Increased diffuse tumor infiltration lymphocyte post vaccination. Increased CD8+ to CD4+ tumor-infiltrating lymphocyte ratio. | |
| Cho et al. [205] | II | 2012 | 34 GBM | Tumor lysate | Vaccine group: OS 31.9 months, PFS 8.5 months Control group: 15 months, PFS 8 months | ||
| Laskey et al. [206] | I | 2013 | 2 GBM 1 AOA | Tumor lysate | Two out of three patients alive >40 months. | No increase in infiltrating lymphocyte post vaccination in one studied patient. Increase in IL10 after vaccination in one studied patient. | |
| Jie et al. [207] | II | 2012 | 25 GBM | Tumor cells | Vaccine group: OS 17 months, PFS 11.92 months Control group: OS 10.5 months, PFS 7.75 months | Higher CD3+, CD4+, CD4+/CD8+ and NK cells levels post vaccination. | |
| Ardon et al. [208] | I | 2010 | 8 GBM | Tumor lysate | One patient free from progression >34 months. Three patients alive at follow up >34 months | Five out of eight patients showed increased antigen reactive T cell IFN-γ production post vaccination. | |
| Sakai et al. [209] | I | 2015 | 6 GBM 2 AA 1 AOA 1 OG | WT-1 antigen, tumor lysate | Median OS 26 months. One GBM patient alive > 46 months post vaccination. | Eight patients had positive DTH reactions post vaccination. Six patients demonstrated increased WT1-specific cytotoxic T lymphocytes. | |
| Hunn et al. [210] | I | 2015 | 14 GBM | Tumor lysate | Pretreatment with TMZ | Two patients had partial response. Two patients had prolonged progression-free survival. Median OS: 23 months. | Two patients demonstrated increased tumor-associated antigen response post vaccination. |
| Vik-Mo et al. [211] | I/II | 2013 | 7 GBM | Glioma mRNA | Booster vaccines | Vaccine group: OS 759 days, PFS 694 days. Historical control group: OS 585 days, PFS 236 days. | All seven patients had tumorsphere lysate-specific lymphocyte proliferation. |
| Batich et al. [212] | I | 2017 | 11 GBM | CMV pp65 mRNA with GM-CSF | Treated with TMZ | Vaccine group: OS 41.1 months; Historical control group: OS 19.2 months. | Ten out of 11 patients demonstrated increase in pp65 specific IFN-γ response. Pp65 specific CD8+ T cells increased post vaccination. |
| Inoges et al. [213] | II | 2017 | 31 GBM | Tumor lysate | OS was 23.4 months, PFS was 12.7 months. | Eight patients showed increased IFN-γ production post vaccination | |
| Liau et al. [214] | III | 2018 | 331 GBM Dcvax-L: 232 Placebo: 99 | Tumor lysate | Treated with TMZ | Intent to treat group: OS 23.1 months; 223 patients alive >30 months from surgery; 100 extended survivors of OS > 40.5 months. | |
| Iwami et al. [215] | I | 2012 | 5 GBM 1 AA 2 AO | IL-13Rα2 | Three patients with stable disease. One patient had mixed radiographic response. | Two out of three patients where immunologic studies can be conducted showed peptide-specific T cell activity post vaccination. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. https://doi.org/10.3390/cancers11040537
Srivastava S, Jackson C, Kim T, Choi J, Lim M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers. 2019; 11(4):537. https://doi.org/10.3390/cancers11040537
Chicago/Turabian StyleSrivastava, Siddhartha, Christina Jackson, Timothy Kim, John Choi, and Michael Lim. 2019. "A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials" Cancers 11, no. 4: 537. https://doi.org/10.3390/cancers11040537
APA StyleSrivastava, S., Jackson, C., Kim, T., Choi, J., & Lim, M. (2019). A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers, 11(4), 537. https://doi.org/10.3390/cancers11040537