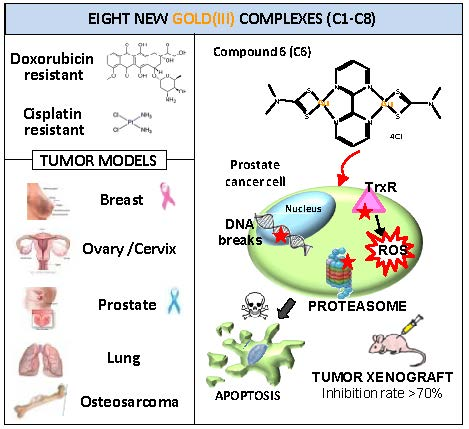
Potent In Vitro and In Vivo Anticancer Activity of New Bipyridine and Bipyrimidine Gold (III) Dithiocarbamate Derivatives

 ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results
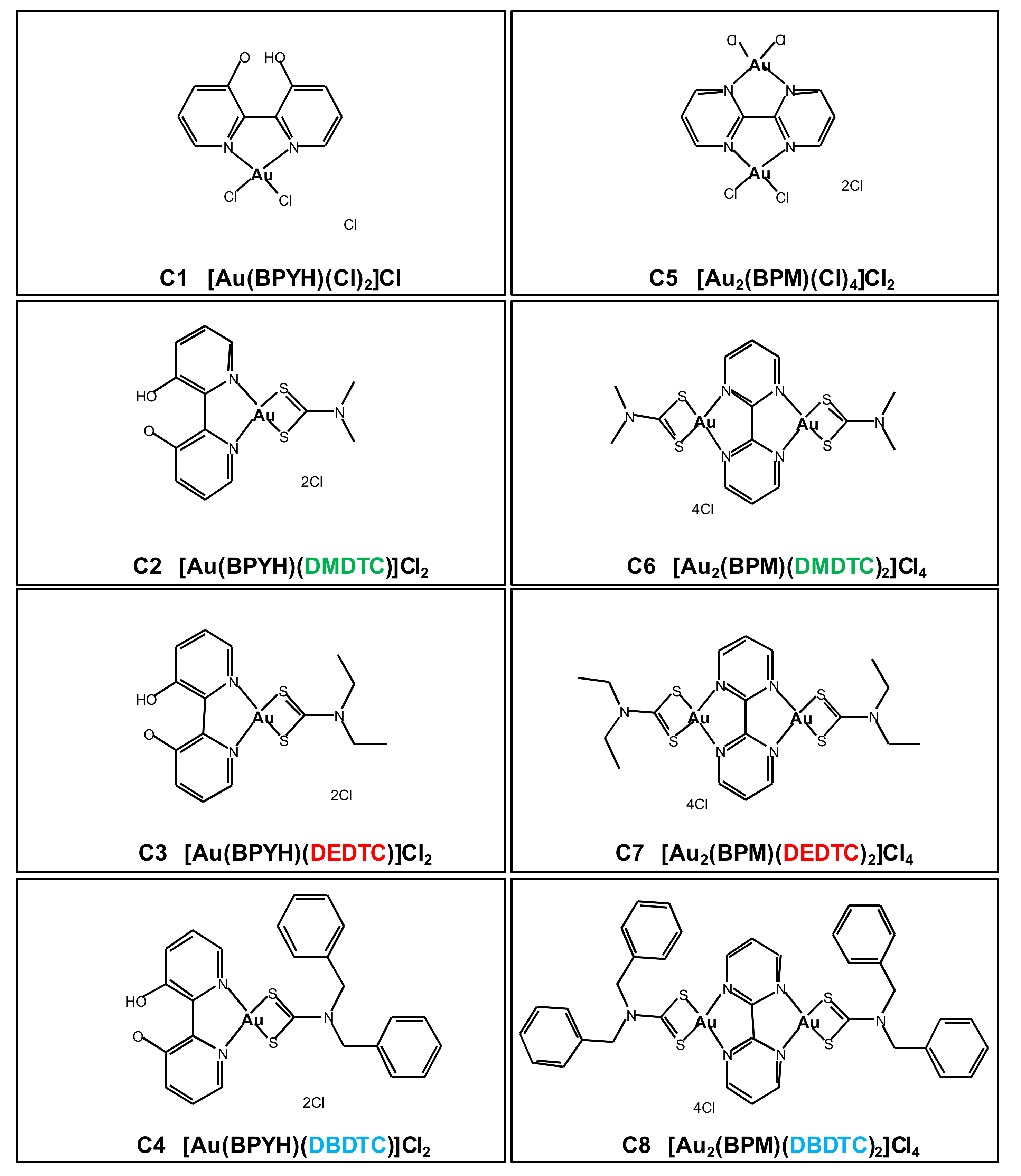
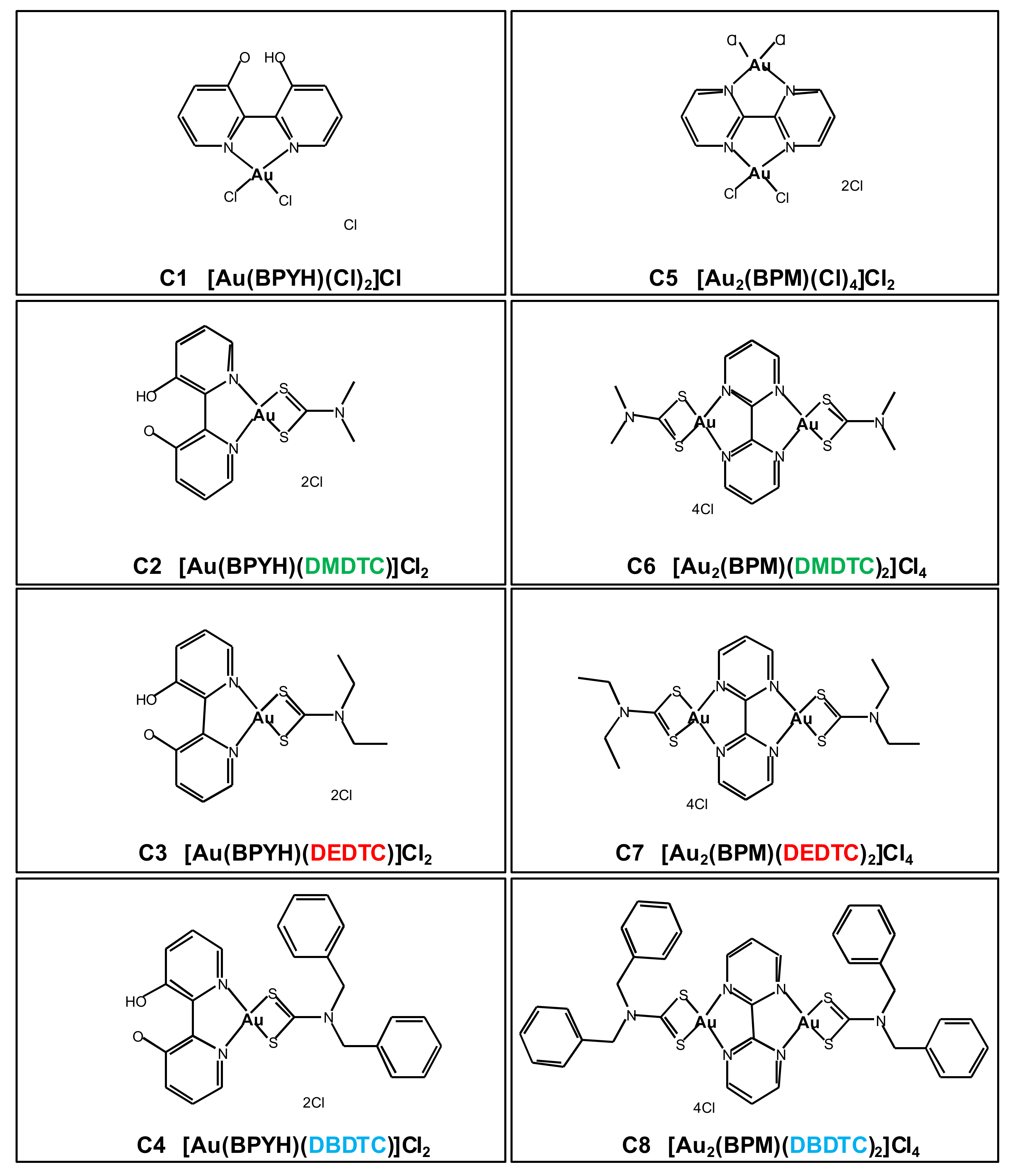
2.1. In Vitro Cytotoxicity of Gold (III) Complexes
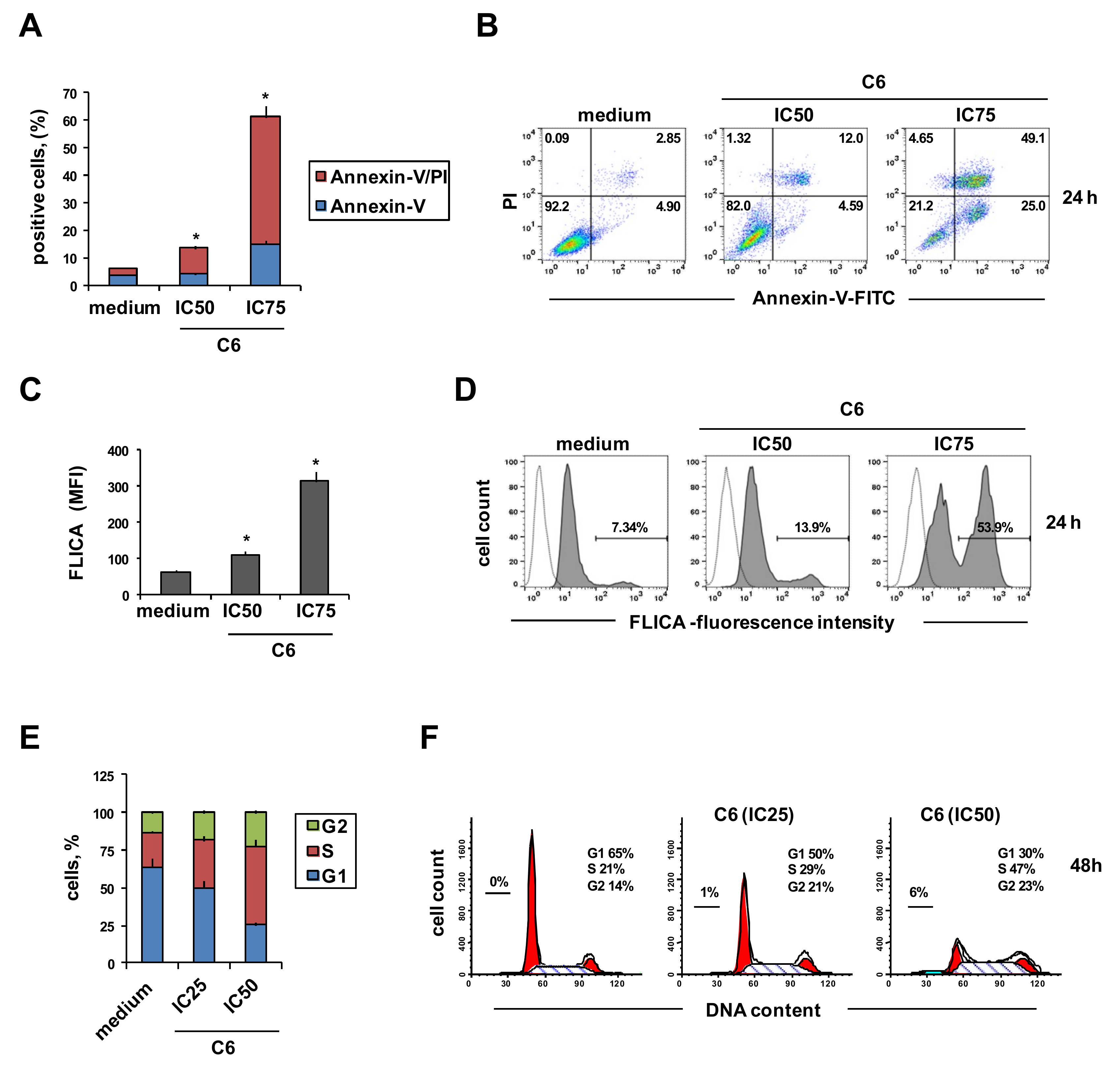
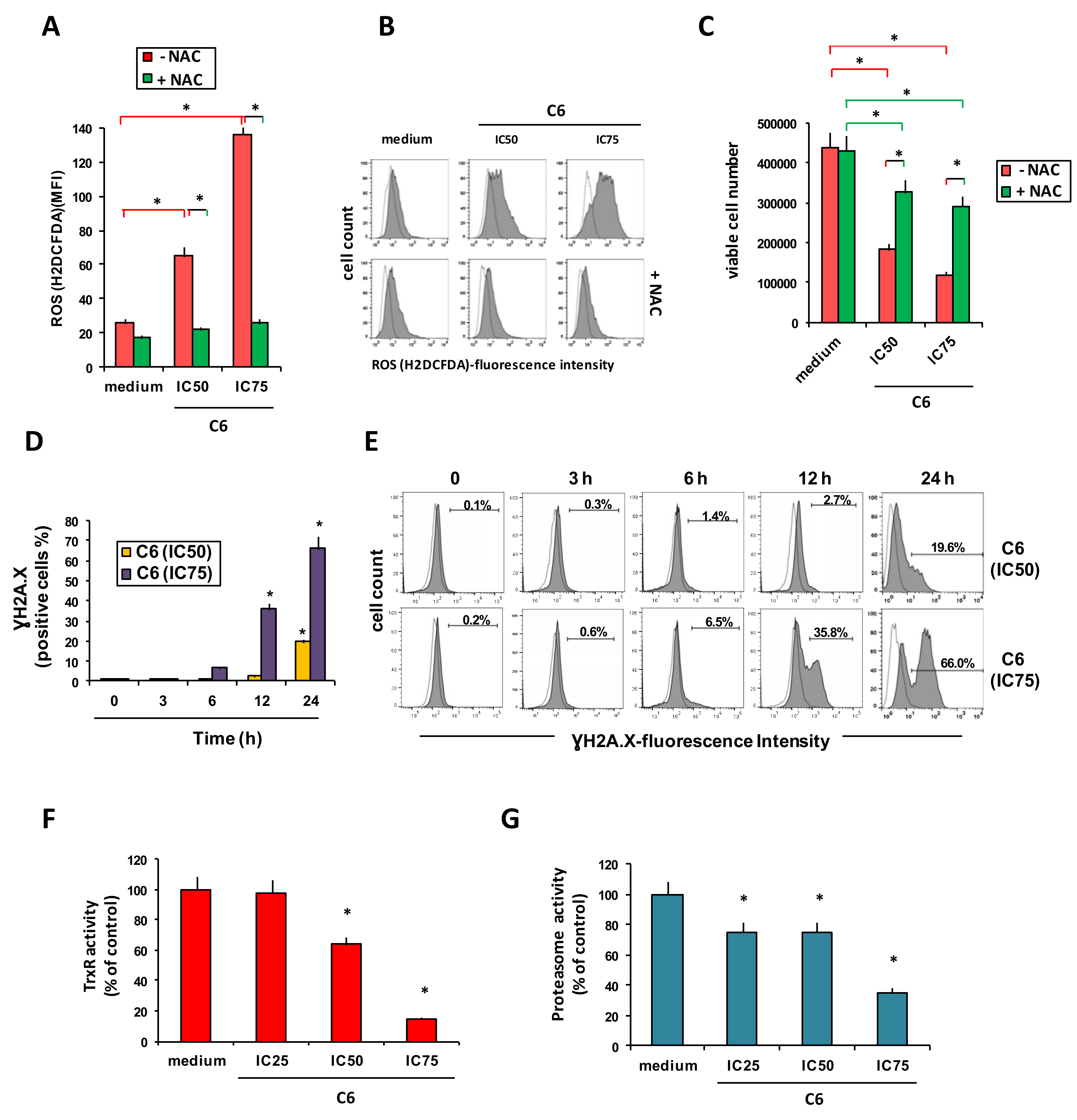
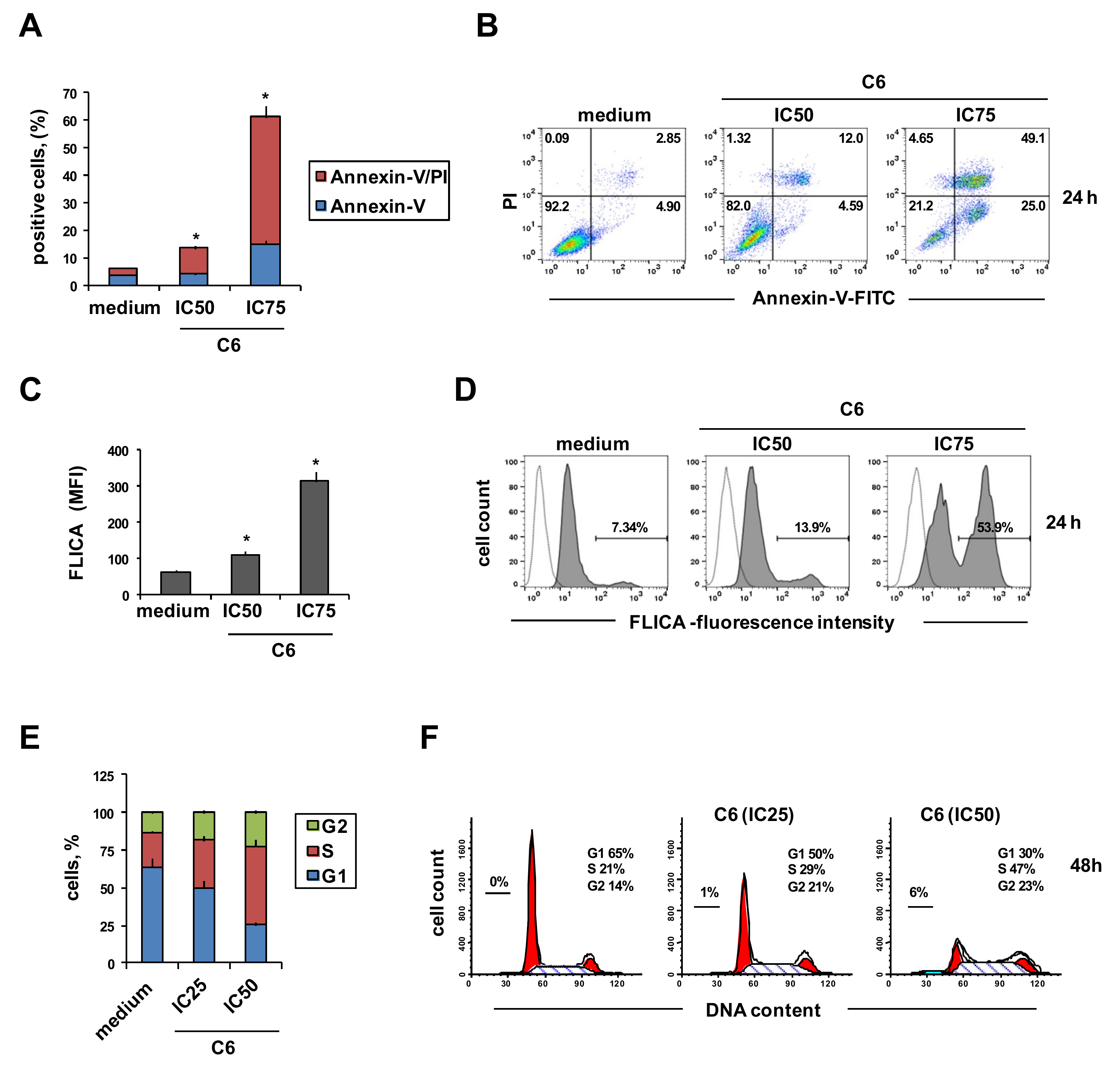
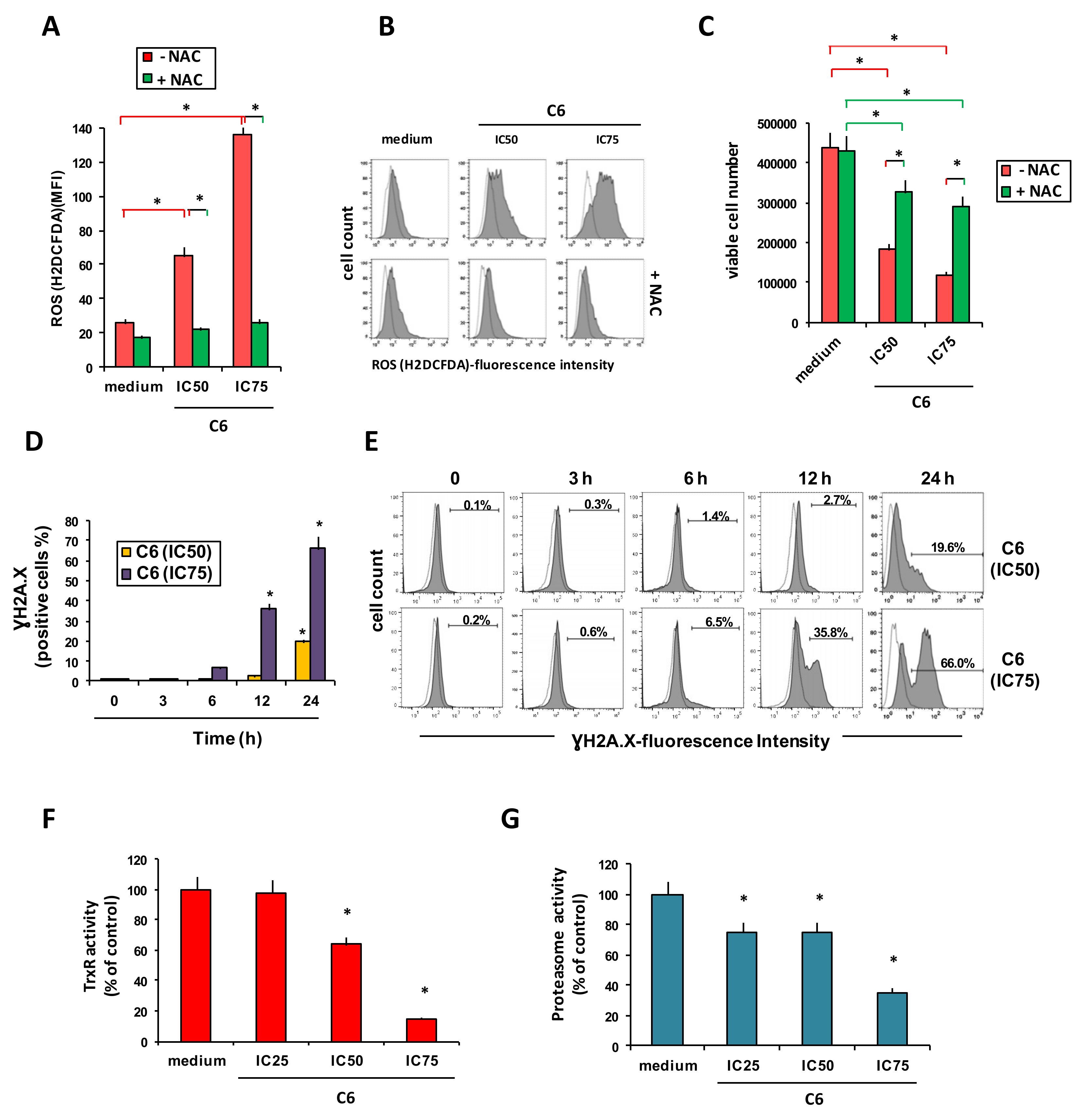
2.2. Cellular Mechanism of Action of Compound C6
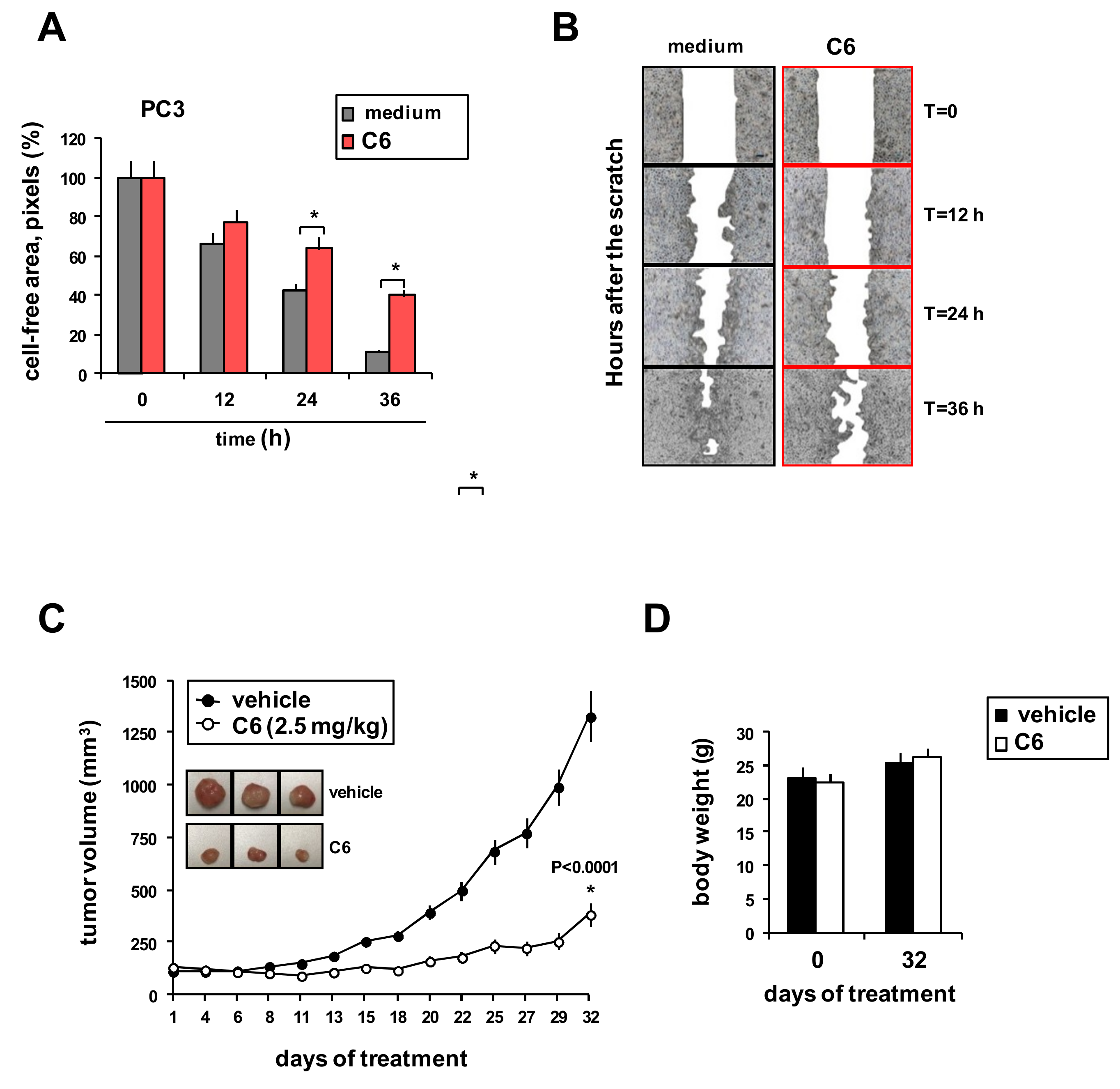
2.3. Effects of C6 on Tumor Cell Migration and Xenograft Growth
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cell Lines and Culture Conditions
4.3. Cytotoxicity Assay
4.4. Cellular Assays
4.5. Human Prostate Tumor Xenograft Experiments
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Spreckelmeyer, S.; Orvig, C.; Casini, A. Cellular transport mechanisms of cytotoxic metallodrugs: An overview beyond cisplatin. Molecules 2014, 19, 15584–15610. [Google Scholar] [CrossRef]
- Cappetta, D.; Rossi, F.; Piegari, E.; Quaini, F.; Berrino, L.; Urbanek, K.; De, A.A. Doxorubicin targets multiple players: A new view of an old problem. Pharm. Res. 2018, 127, 4–14. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharm. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Nardon, C.; Fregona, D. Editorial: Throwing Light on Recent Advances on Metallodrugs: From Deemed Poisons to a Striking Hope for the Future. Curr. Med. Chem. 2018, 25, 434–436. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, T.; Rilak, A.; Bugarcic, Z.D. Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives. Eur. J. Med. Chem. 2017, 142, 8–31. [Google Scholar] [CrossRef] [PubMed]
- Soldevila-Barreda, J.J.; Sadler, P.J. Approaches to the design of catalytic metallodrugs. Curr. Opin. Chem. Biol. 2015, 25, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casini, A.; Sun, R.W.; Ott, I. Medicinal Chemistry of Gold Anticancer Metallodrugs. Met. Ions Life Sci. 2018, 18. [Google Scholar] [CrossRef]
- Bertrand, B.; Williams, M.R.M.; Bochmann, M. Gold(III) Complexes for Antitumor Applications: An Overview. Chemistry 2018, 24, 11840–11851. [Google Scholar] [CrossRef] [PubMed]
- Celegato, M.; Borghese, C.; Casagrande, N.; Mongiat, M.; Kahle, X.U.; Paulitti, A.; Spina, M.; Colombatti, A.; Aldinucci, D. Preclinical activity of the repurposed drug Auranofin in classical Hodgkin lymphoma. Blood 2015, 126, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Lorenzon, D.; Stefani, L.; Giovagnini, L.; Colombatti, A.; Fregona, D. Antiproliferative and apoptotic effects of two new gold(III) methylsarcosinedithiocarbamate derivatives on human acute myeloid leukemia cells in vitro. Anticancer Drugs 2007, 18, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Milacic, V.; Chen, D.; Ronconi, L.; Landis-Piwowar, K.R.; Fregona, D.; Dou, Q.P. A novel anticancer gold(III) dithiocarbamate compound inhibits the activity of a purified 20S proteasome and 26S proteasome in human breast cancer cell cultures and xenografts. Cancer Res. 2006, 66, 10478–10486. [Google Scholar] [CrossRef]
- Gratteri, P.; Massai, L.; Michelucci, E.; Rigo, R.; Messori, L.; Cinellu, M.A.; Musetti, C.; Sissi, C.; Bazzicalupi, C. Interactions of selected gold(III) complexes with DNA G quadruplexes. Dalton Trans. 2015, 44, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Coronnello, M.; Marcon, G.; Carotti, S.; Caciagli, B.; Mini, E.; Mazzei, T.; Orioli, P.; Messori, L. Cytotoxicity, DNA damage, and cell cycle perturbations induced by two representative gold(III) complexes in human leukemic cells with different cisplatin sensitivity. Oncol Res. 2000, 12, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Saggioro, D.; Rigobello, M.P.; Paloschi, L.; Folda, A.; Moggach, S.A.; Parsons, S.; Ronconi, L.; Fregona, D.; Bindoli, A. Gold(III)-dithiocarbamato complexes induce cancer cell death triggered by thioredoxin redox system inhibition and activation of ERK pathway. Chem. Biol. 2007, 14, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Marzano, C.; Ronconi, L.; Chiara, F.; Giron, M.C.; Faustinelli, I.; Cristofori, P.; Trevisan, A.; Fregona, D. Gold(III)-dithiocarbamato anticancer agents: Activity, toxicology and histopathological studies in rodents. Int. J. Cancer 2011, 129, 487–496. [Google Scholar] [CrossRef]
- Cattaruzza, L.; Fregona, D.; Mongiat, M.; Ronconi, L.; Fassina, A.; Colombatti, A.; Aldinucci, D. Antitumor activity of gold(III)-dithiocarbamato derivatives on prostate cancer cells and xenografts. Int. J. Cancer 2011, 128, 206–215. [Google Scholar] [CrossRef]
- Kouodom, M.N.; Ronconi, L.; Celegato, M.; Nardon, C.; Marchio, L.; Dou, Q.P.; Aldinucci, D.; Formaggio, F.; Fregona, D. Toward the selective delivery of chemotherapeutics into tumor cells by targeting peptide transporters: Tailored gold-based anticancer peptidomimetics. J. Med. Chem. 2012, 55, 2212–2226. [Google Scholar] [CrossRef] [PubMed]
- Nardon, C.; Schmitt, S.M.; Yang, H.; Zuo, J.; Fregona, D.; Dou, Q.P. Gold(III)-dithiocarbamato peptidomimetics in the forefront of the targeted anticancer therapy: Preclinical studies against human breast neoplasia. PLoS ONE 2014, 9, e84248. [Google Scholar] [CrossRef]
- Celegato, M.; Fregona, D.; Mongiat, M.; Ronconi, L.; Borghese, C.; Canzonieri, V.; Casagrande, N.; Nardon, C.; Colombatti, A.; Aldinucci, D. Preclinical activity of multiple-target gold(III)-dithiocarbamato peptidomimetics in prostate cancer cells and xenografts. Future Med. Chem. 2014, 6, 1249–1263. [Google Scholar] [CrossRef]
- Altaf, M.; Monim-ul-Mehboob, M.; Seliman, A.A.; Sohail, M.; Wazeer, M.I.; Isab, A.A.; Li, L.; Dhuna, V.; Bhatia, G.; Dhuna, K. Synthesis, characterization and anticancer activity of gold(I) complexes that contain tri-tert-butylphosphine and dialkyl dithiocarbamate ligands. Eur. J. Med. Chem. 2015, 95, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altaf, M.; Monom-ul-Mehboob, M.; Selimam, A.A.; Isab, A.A.; Dhuna, V.; Bhatia, G.; Dhuna, K. Synthesis, X–ray Structures, Spectroscopic Analysis and Anticancer Activity of Novel Gold(I) Carbene Complexes. J. Organomet. Chem. 2014, 765, 68–79. [Google Scholar] [CrossRef]
- Al-Jaroudi, S.S.; Altaf, M.; Al-Saadi, A.A.; Kawde, A.N.; Altuwaijri, S.; Ahmad, S.; Isab, A.A. Synthesis, characterization and theoretical calculations of (1,2-diaminocyclohexane)(1,3-diaminopropane)gold(III) chloride complexes: In Vitro cytotoxic evaluations against human cancer cell lines. Biometals 2015, 28, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Altaf, M.; Monim-ul-Mehboob, M.; Kawde, A.N.; Corona, G.; Larcher, R.; Ogasawara, M.; Casagrande, N.; Celegato, M.; Borghese, C.; Siddik, Z.H.; et al. New bipyridine gold(III) dithiocarbamate-containing complexes exerted a potent anticancer activity against cisplatin-resistant cancer cells independent of p53 status. Oncotarget 2017, 8, 490–505. [Google Scholar] [CrossRef] [PubMed]
- Scalcon, V.; Bindoli, A.; Rigobello, M.P. Significance of the mitochondrial thioredoxin reductase in cancer cells: An update on role, targets and inhibitors. Free Radic. Biol. Med. 2018, 127, 62–79. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharm. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Shan, W.; Zhong, W.; Zhao, R.; Oberley, T.D. Thioredoxin 1 as a subcellular biomarker of redox imbalance in human prostate cancer progression. Free Radic. Biol. Med. 2010, 49, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Tomida, A.; Yoshikawa, H.; Taketani, Y.; Tsuruo, T. Increased expression of thioredoxin/adult T-cell leukemia-derived factor in cisplatin-resistant human cancer cell lines. Clin. Cancer Res. 1996, 2, 427–432. [Google Scholar]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K. Protein metabolism: How the proteasome adapts to stress. Nat. Rev. Mol. Cell Biol. 2014, 15, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Tomasello, M.F.; Nardon, C.; Lanza, V.; Di, N.G.; Pettenuzzo, N.; Salmaso, S.; Milardi, D.; Caliceti, P.; Pappalardo, G.; Fregona, D. New comprehensive studies of a gold(III) Dithiocarbamate complex with proven anticancer properties: Aqueous dissolution with cyclodextrins, pharmacokinetics and upstream inhibition of the ubiquitin-proteasome pathway. Eur. J. Med. Chem. 2017, 138, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Quero, J.; Cabello, S.; Fuertes, T.; Marmol, I.; Laplaza, R.; Polo, V.; Gimeno, M.C.; Rodriguez-Yoldi, M.J.; Cerrada, E. Proteasome versus Thioredoxin Reductase Competition as Possible Biological Targets in Antitumor Mixed Thiolate-Dithiocarbamate Gold(III) Complexes. Inorg. Chem. 2018, 57, 10832–10845. [Google Scholar] [CrossRef] [PubMed]
- Ritch, C.; Cookson, M. Recent trends in the management of advanced prostate cancer. F1000Reseach 2018, 7. [Google Scholar] [CrossRef]
- Nobili, S.; Mini, E.; Landini, I.; Gabbiani, C.; Casini, A.; Messori, L. Gold compounds as anticancer agents: Chemistry, cellular pharmacology, and preclinical studies. Med. Res. Rev. 2010, 30, 550–580. [Google Scholar] [CrossRef]
- Micale, N.; Schirmeister, T.; Ettari, R.; Cinellu, M.A.; Maiore, L.; Serratrice, M.; Gabbiani, C.; Massai, L.; Messori, L. Selected cytotoxic gold compounds cause significant inhibition of 20S proteasome catalytic activities. J. Inorg. Biochem. 2014, 141, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Line | ||||
|---|---|---|---|---|---|
| A549 | DU145 | PC3 | MCF-7 | MG-63 | |
| Cisplatin | 52.0 (4.7) | 4.5 (0.4) | 3.3 (0.3) | 22.2 (0.2) | 58.0 (0.5) |
| C1 | >80 | 39.0 (3.5) | 28.3 (2.6) | 59.0 (5.3) | 43.0 (3.9) |
| C2 | 6.1 (0.6) | 2.8 (0.3) | 1.5 (0.1) | 2.3 (0.2) | 3.8 (0.3) |
| C3 | 3.8 (0.3) | 3.5 (0.3) | 1.3 (0.1) | 1.7 (0.2) | 1.2 (0.1) |
| C4 | 25.0 (2.3) | 6.4 (0.6) | 8.5 (0.8) | 13.0 (1.2) | 12.3 (1.1) |
| C5 | >80 | >80 | >80 | 65.0 (5.9) | 26.0 (2.3) |
| C6 | 0.8 (0.1) | 0.7 (0.1) | 0.6 (0.1) | 0.5 (0.1) | 0.8 (0.1) |
| C7 | 1.4 (0.1) | 0.8 (0.1) | 0.8 (0.1) | 0.6 (0.1) | 0.7 (0.1) |
| C8 | 23.0 (2.1) | 22.8 (2.1) | 19.5 (1.8) | 9.5 (0.9) | 5.8 (0.5) |
| Compound | IC50, µM a | FR (A2780cis/A2780) | |
|---|---|---|---|
| A2780 | A2780cis | ||
| Cisplatin | 1.5 (0.1) | 10.4 (0.9) | 6.9 |
| Doxorubicin | 0.02 (0.0) | 0.12 (0.0) | 9.0 |
| C1 | 23.0 (2.1) | 24.0 (2.0) | 1.0 |
| C2 | 0.9 (0.1) | 0.8 (0.1) | 0.9 |
| C3 | 0.4 (0.0) | 0.4 (0.0) | 1.1 |
| C4 | 7.3 (0.7) | 8.2 (0.7) | 1.1 |
| C5 | 15.4 (1.4) | 16.2 (5.1) | 1.1 |
| C6 | 0.2 (0.0) | 0.3 (0.0) | 1.2 |
| C7 | 0.4 (0.0) | 0.3 (0.0) | 0.9 |
| C8 | 3.8 (0.3) | 5.2 (0.5) | 1.4 |
| Compound | IC50, µM a | FR (R-ME-180/ME-180) | |
|---|---|---|---|
| ME-180 | R-ME-180 | ||
| Cisplatin | 15.0 (1.4) | 68.0 (6.1) | 4.5 |
| C1 | 70.0 (6.3) | 72.0 (6.5) | 1.0 |
| C2 | 14.0 (1.3) | 21.0 (1.9) | 1.5 |
| C3 | 3.0 (0.3) | 3.8 (0.3) | 1.3 |
| C4 | 30.0 (2.5) | 30.0 (2.7) | 1.0 |
| C5 | >80 | >80 | ND |
| C6 | 5.3 (0.5) | 4.9 (0.5) | 0.9 |
| C7 | 4.8 (0.4) | 4.1 (0.4) | 0.9 |
| C8 | 15.0 (1.4) | 16.0 (1.4) | 1.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altaf, M.; Casagrande, N.; Mariotto, E.; Baig, N.; Kawde, A.-N.; Corona, G.; Larcher, R.; Borghese, C.; Pavan, C.; Seliman, A.A.; et al. Potent In Vitro and In Vivo Anticancer Activity of New Bipyridine and Bipyrimidine Gold (III) Dithiocarbamate Derivatives. Cancers 2019, 11, 474. https://doi.org/10.3390/cancers11040474
Altaf M, Casagrande N, Mariotto E, Baig N, Kawde A-N, Corona G, Larcher R, Borghese C, Pavan C, Seliman AA, et al. Potent In Vitro and In Vivo Anticancer Activity of New Bipyridine and Bipyrimidine Gold (III) Dithiocarbamate Derivatives. Cancers. 2019; 11(4):474. https://doi.org/10.3390/cancers11040474
Chicago/Turabian StyleAltaf, Muhammad, Naike Casagrande, Elena Mariotto, Nadeem Baig, Abdel-Nasser Kawde, Giuseppe Corona, Roberto Larcher, Cinzia Borghese, Claudia Pavan, Adam A. Seliman, and et al. 2019. "Potent In Vitro and In Vivo Anticancer Activity of New Bipyridine and Bipyrimidine Gold (III) Dithiocarbamate Derivatives" Cancers 11, no. 4: 474. https://doi.org/10.3390/cancers11040474
APA StyleAltaf, M., Casagrande, N., Mariotto, E., Baig, N., Kawde, A.-N., Corona, G., Larcher, R., Borghese, C., Pavan, C., Seliman, A. A., Aldinucci, D., & Isab, A. A. (2019). Potent In Vitro and In Vivo Anticancer Activity of New Bipyridine and Bipyrimidine Gold (III) Dithiocarbamate Derivatives. Cancers, 11(4), 474. https://doi.org/10.3390/cancers11040474