Upregulation of Complement Factor H by SOCS-1/3–STAT4 in Lung Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
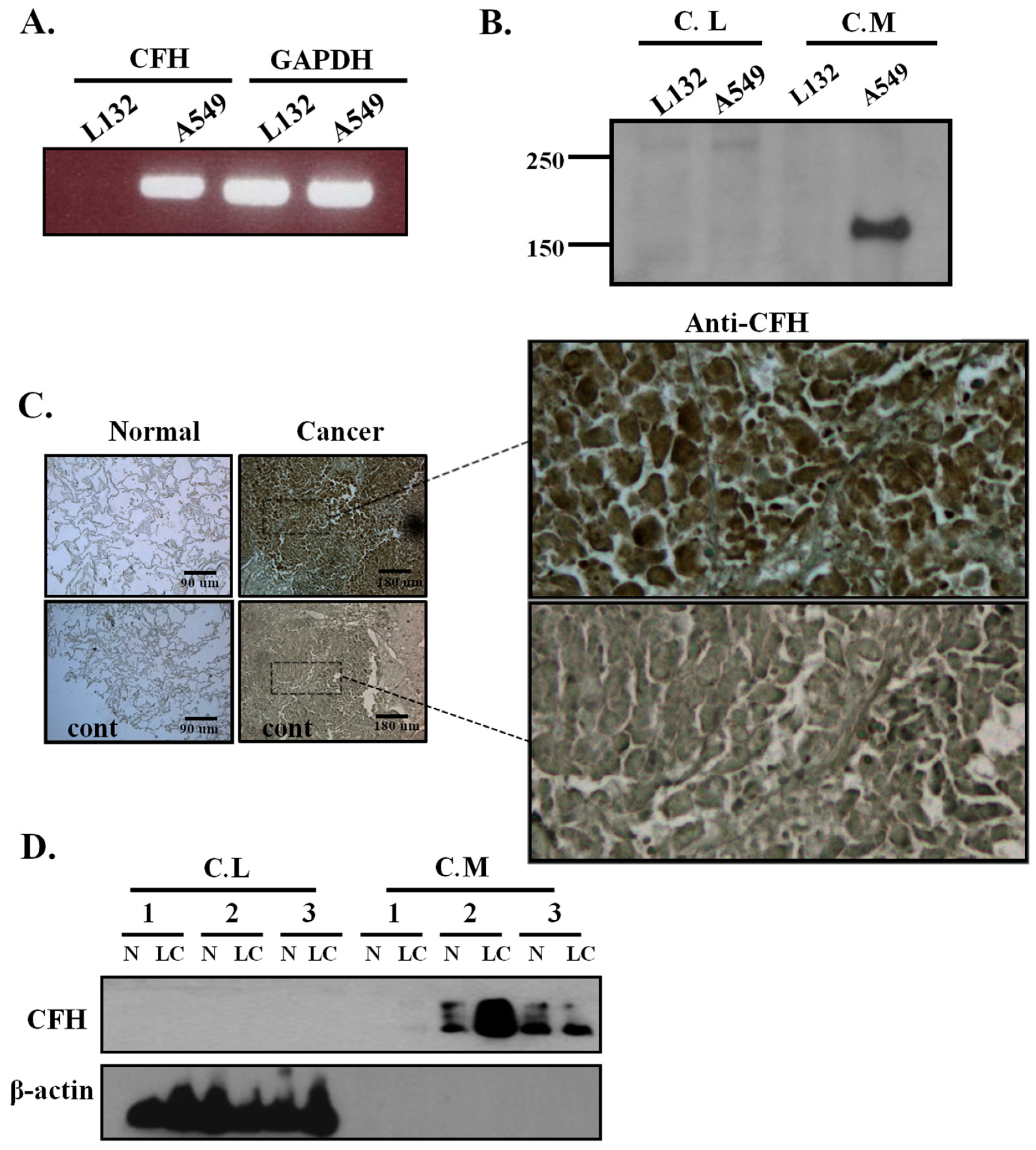
2.1. CFH Expression in Lung Cell Lines, Human Lung Tissues, and Primary Cultured Lung Cells
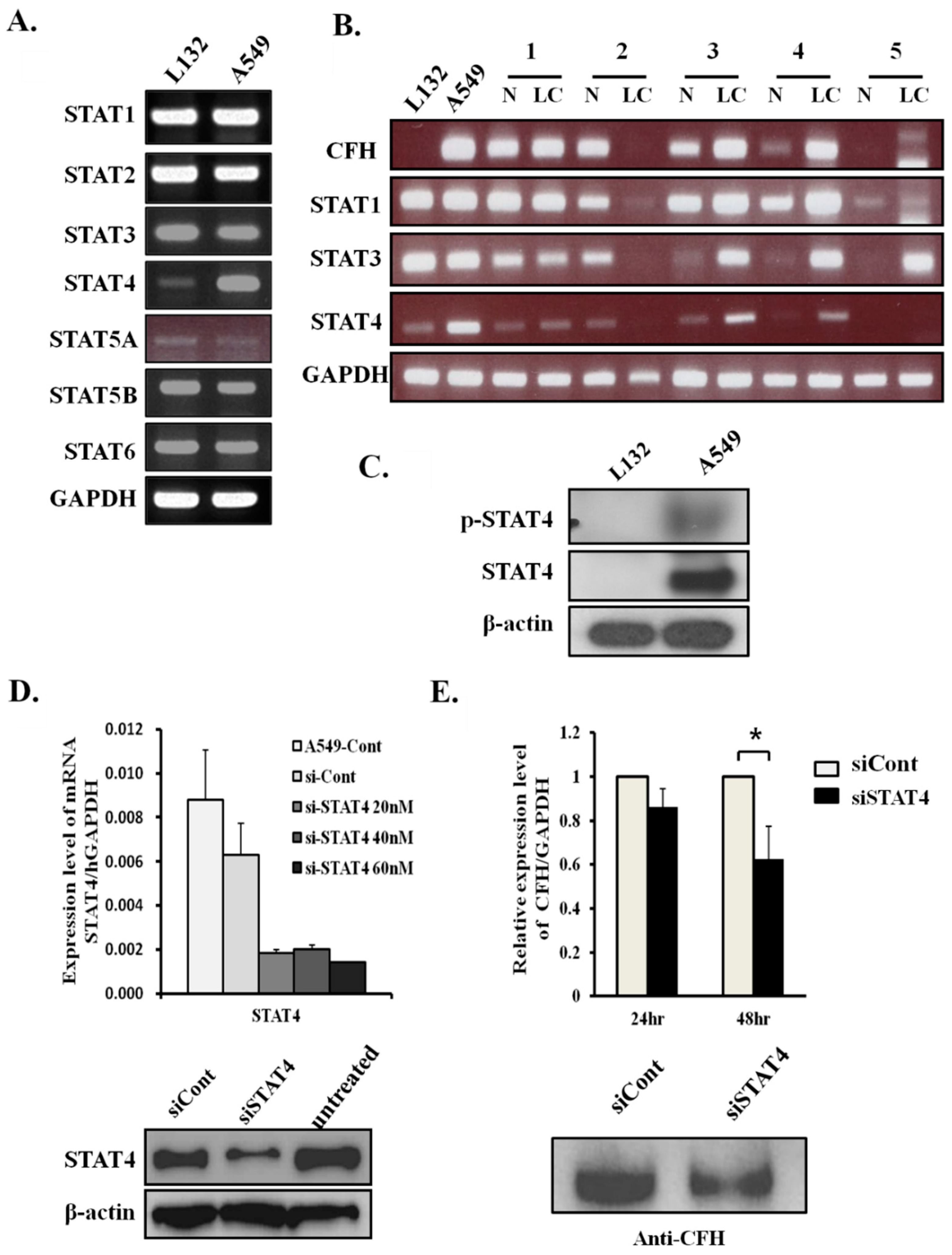
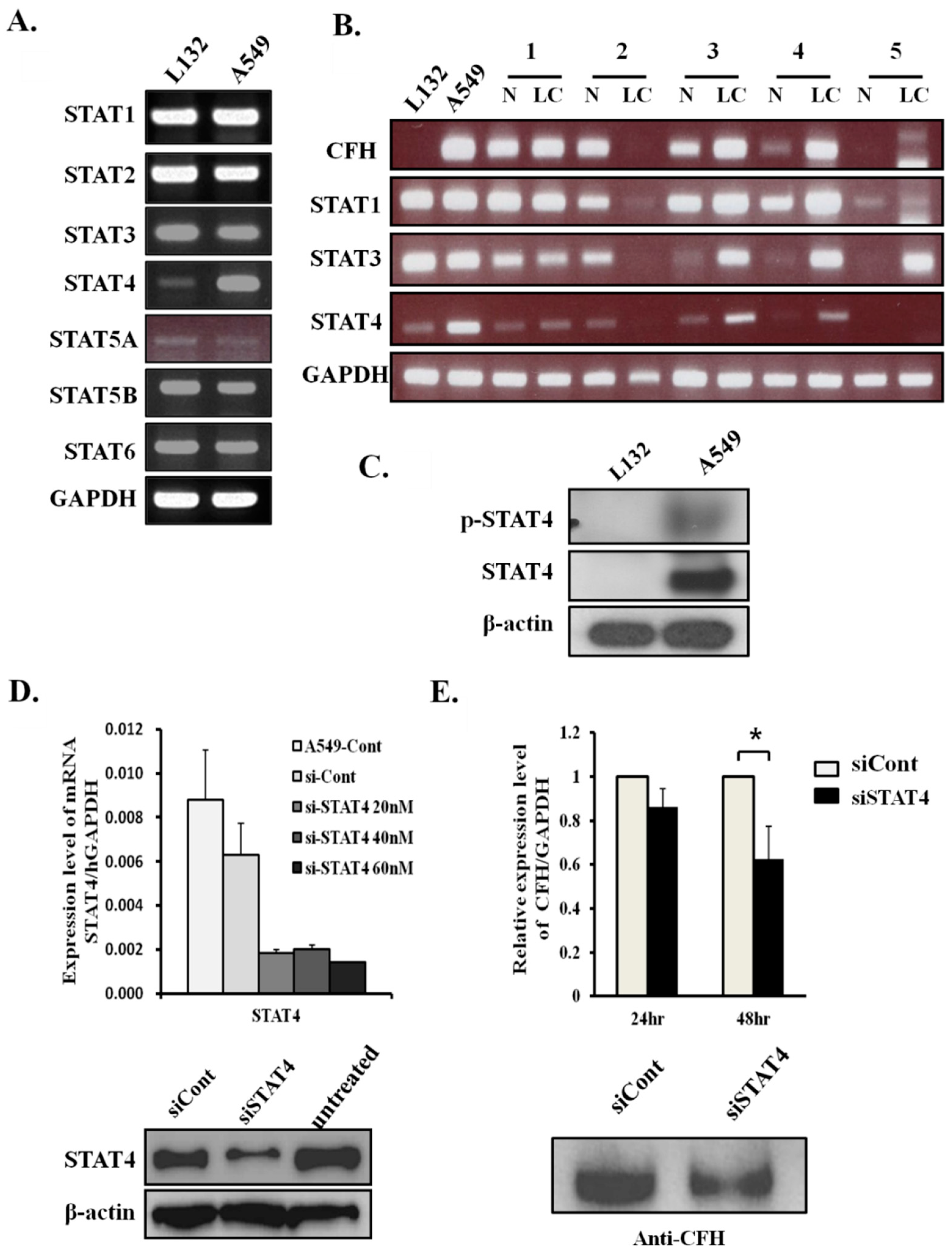
2.2. CFH Expression Levels Correlate with STAT4 Expression Levels in Lung Cancers
2.3. STAT4 Regulates CFH Expression Levels
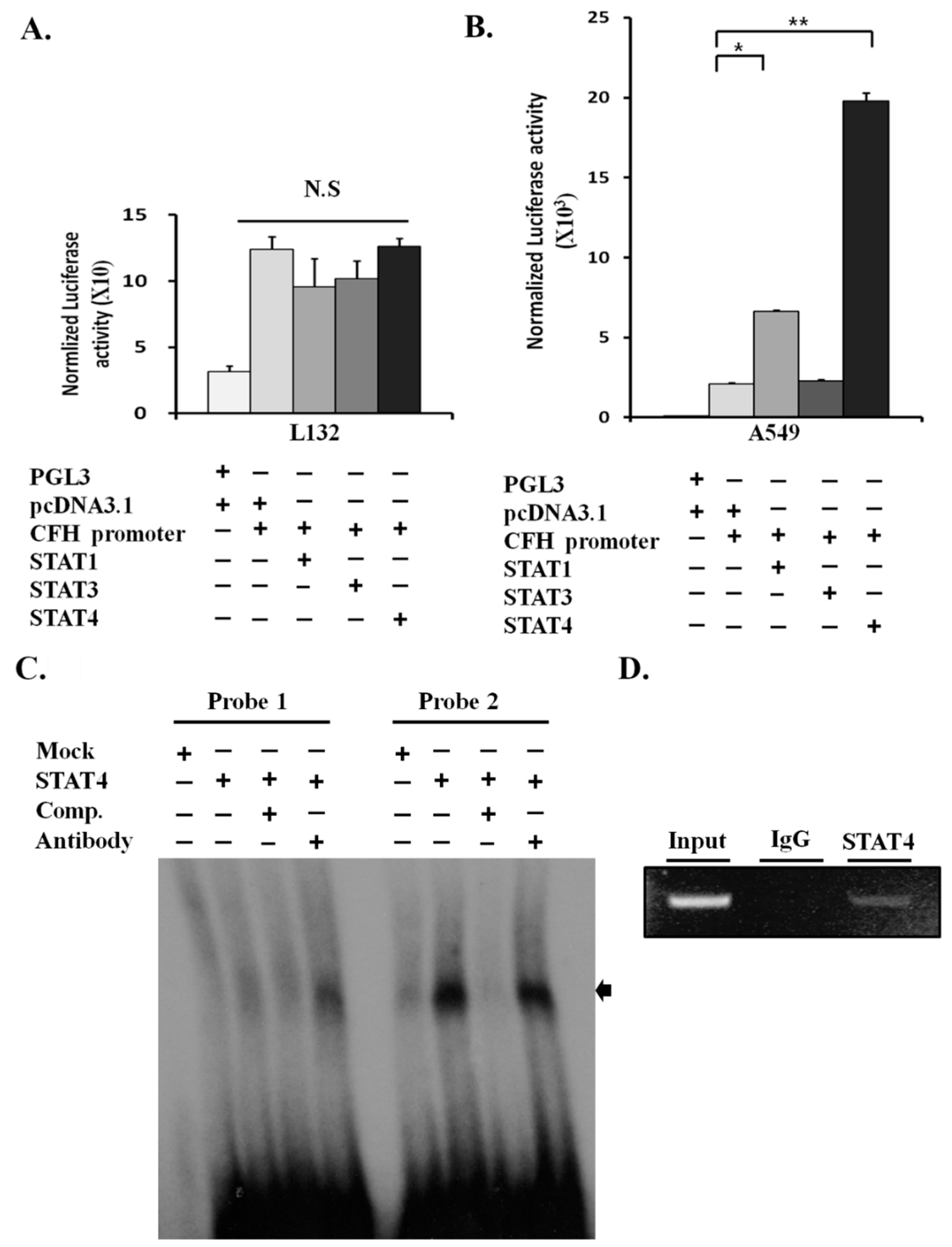
2.4. STAT4 Upregulates the Activity of the CFH Promoter
2.5. STAT4 Directly Binds and Activates the CFH Promoter
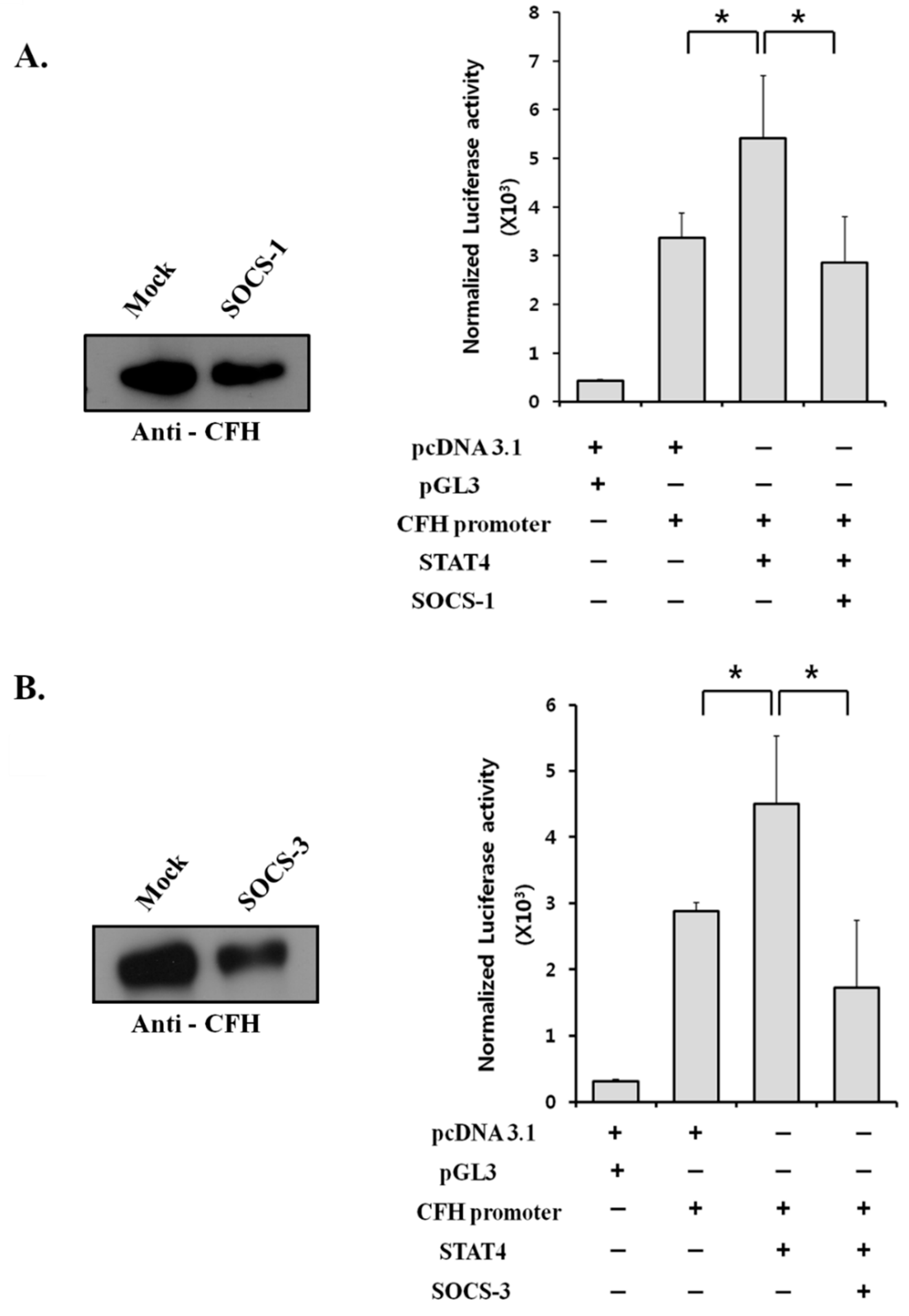
2.6. Suppressor of Cytokine Signaling (SOCS) Proteins Negatively Regulate CFH Expression by Suppressing STAT4 Activity
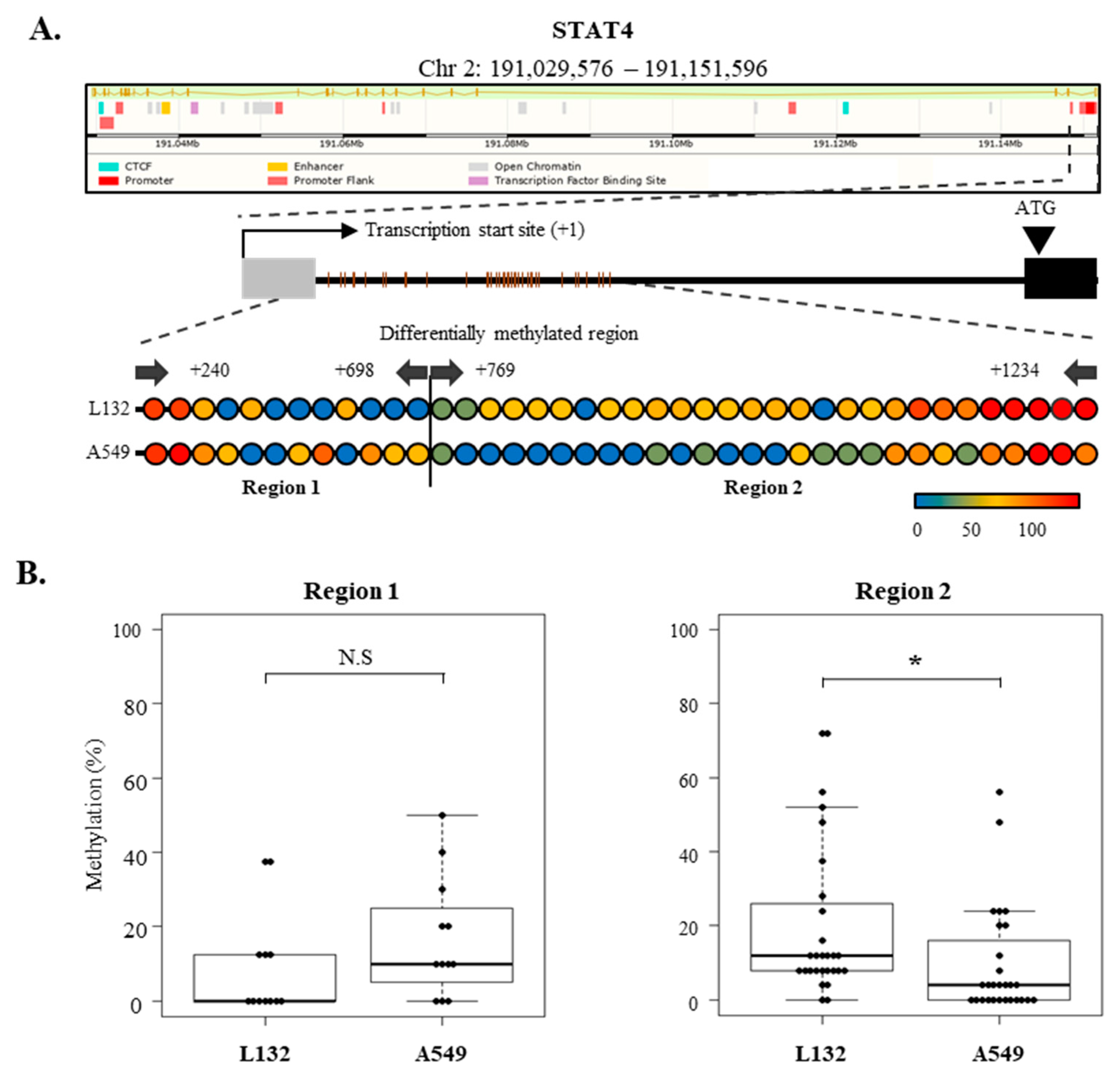
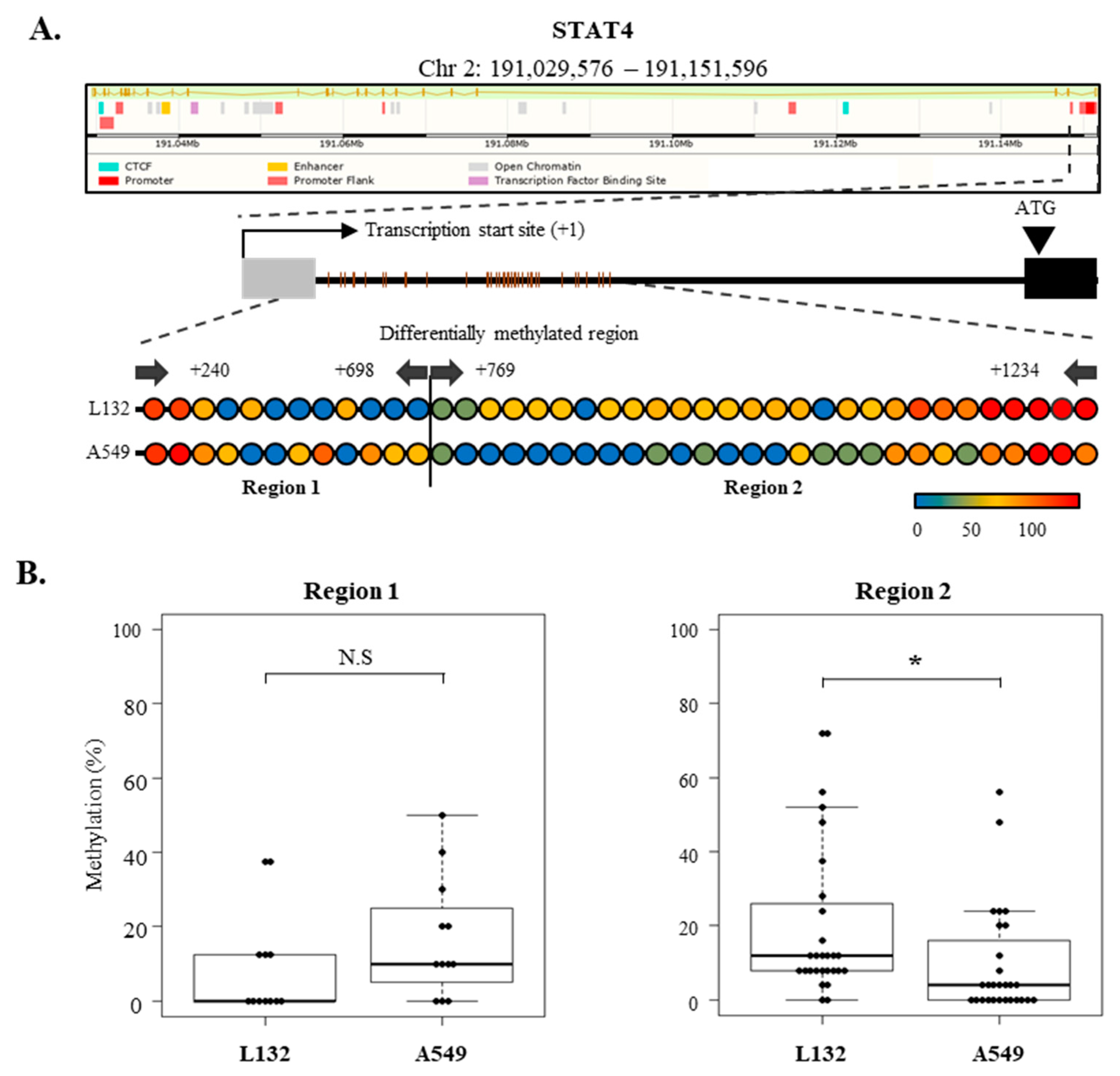
2.7. Epigenetic Regulation of SOCS-1, SOCS-3, and STAT4 Expression in Lung Cancer Cells
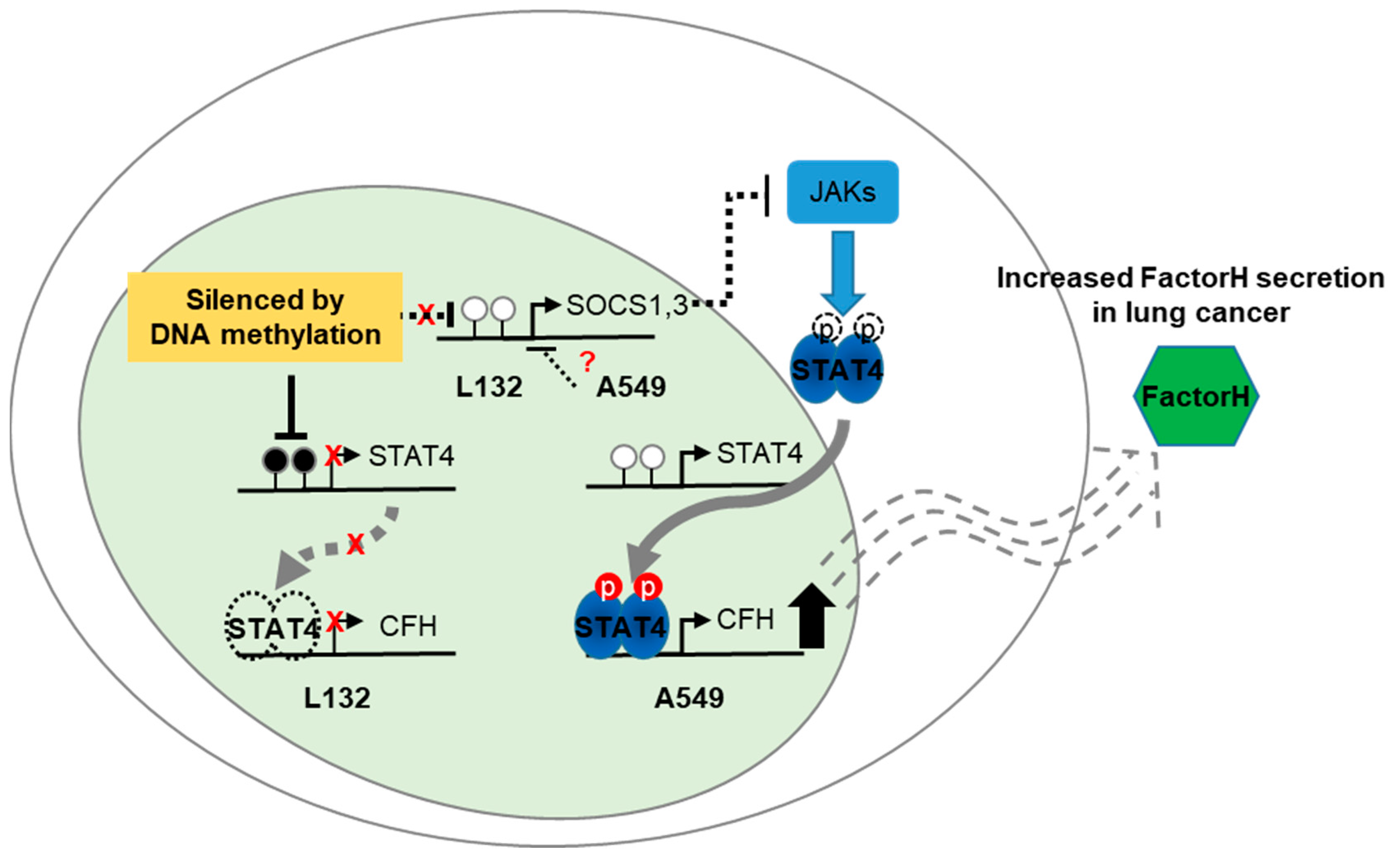
3. Discussion
4. Materials and Methods
4.1. Lung Cell Lines and Human Lung Tissues
4.2. Antibodies and Western Blot Analysis
4.3. RNA Extraction and RT-PCR
4.4. Immunohistochemistry
4.5. DNA Constructs
4.6. Luciferase Assay
4.7. RNA Interference Knockdown
4.8. EMSA
4.9. ChIP Assays
- F:5′-AAACTCGAGCCAAATTCATCAAGCACTGCATTCTTGGCA-3′;
- R:5′-AAAAAGCTTGGATCTTTTAAGAGGACATTTACCAGCTAA-3′.
4.10. Bisulfite Conversion PCR and Sequencing
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ajona, D.; Castano, Z.; Garayoa, M.; Zudaire, E.; Pajares, M.J.; Martinez, A.; Cuttitta, F.; Montuenga, L.M.; Pio, R. Expression of complement factor H by lung cancer cells: effects on the activation of the alternative pathway of complement. Cancer Res. 2004, 64, 6310–6318. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.J.; Ahn, J.M.; Yoon, Y.H.; Rhim, T.Y.; Park, C.S.; Park, J.Y.; Lee, S.Y.; Kim, J.W.; Cho, J.Y. Identification and validation of SAA as a potential lung cancer biomarker and its involvement in metastatic pathogenesis of lung cancer. J. Proteome Res. 2011, 10, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Bjorge, L.; Hakulinen, J.; Vintermyr, O.K.; Jarva, H.; Jensen, T.S.; Iversen, O.E.; Meri, S. Ascitic complement system in ovarian cancer. Br. J. Cancer 2005, 92, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Wilczek, E.; Rzepko, R.; Nowis, D.; Legat, M.; Golab, J.; Glab, M.; Gorlewicz, A.; Konopacki, F.; Mazurkiewicz, M.; Sladowski, D.; et al. The possible role of factor H in colon cancer resistance to complement attack. Int. J. Cancer 2008, 122, 2030–2037. [Google Scholar] [CrossRef]
- Hsu, Y.F.; Ajona, D.; Corrales, L.; Lopez-Picazo, J.M.; Gurpide, A.; Montuenga, L.M.; Pio, R. Complement activation mediates cetuximab inhibition of non-small cell lung cancer tumor growth in vivo. Mol. Cancer 2010, 9, 139–146. [Google Scholar] [CrossRef]
- Larbouret, C.; Gaborit, N.; Chardes, T.; Coelho, M.; Campigna, E.; Bascoul-Mollevi, C.; Mach, J.P.; Azria, D.; Robert, B.; Pelegrin, A. In pancreatic carcinoma, dual EGFR/HER2 targeting with cetuximab/trastuzumab is more effective than treatment with trastuzumab/erlotinib or lapatinib alone: implication of receptors’ down-regulation and dimers’ disruption. Neoplasia 2012, 14, 121–130. [Google Scholar] [CrossRef]
- Plosker, G.L.; Figgitt, D.P. Rituximab: A review of its use in non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Drugs 2003, 63, 803–843. [Google Scholar] [CrossRef]
- Jurianz, K.; Ziegler, S.; Garcia-Schuler, H.; Kraus, S.; Bohana-Kashtan, O.; Fishelson, Z.; Kirschfink, M. Complement resistance of tumor cells: Basal and induced mechanisms. Mol. Immunol. 1999, 36, 929–939. [Google Scholar] [CrossRef]
- Pangburn, M.K. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunopharmacology 2000, 49, 149–157. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Fenaille, F.; Le Mignon, M.; Groseil, C.; Ramon, C.; Riande, S.; Siret, L.; Bihoreau, N. Site-specific N-glycan characterization of human complement factor H. Glycobiology 2007, 17, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Timar, K.K.; Pasch, M.C.; van den Bosch, N.H.; Jarva, H.; Junnikkala, S.; Meri, S.; Bos, J.D.; Asghar, S.S. Human keratinocytes produce the complement inhibitor factor H: synthesis is regulated by interferon-gamma. Mol. Immunol. 2006, 43, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.P.; Daha, M.R. Complement in glomerular injury. Semin. Immunopathol. 2007, 29, 375–384. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Zhao, Y.; Cui, J.G. An NF-kappaB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 2008, 283, 31315–31322. [Google Scholar] [CrossRef]
- Nan, R.; Farabella, I.; Schumacher, F.F.; Miller, A.; Gor, J.; Martin, A.C.; Jones, D.T.; Lengyel, I.; Perkins, S.J. Zinc binding to the Tyr402 and His402 allotypes of complement factor H: possible implications for age-related macular degeneration. J. Mol. Biol. 2011, 408, 714–735. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Asmat, T.M.; Luo, S.; Jensch, I.; Zipfel, P.F.; Hammerschmidt, S. Complement regulator Factor H mediates a two-step uptake of Streptococcus pneumoniae by human cells. J. Biol. Chem. 2010, 285, 23486–23495. [Google Scholar] [CrossRef] [PubMed]
- Blom, A.M.; Hallstrom, T.; Riesbeck, K. Complement evasion strategies of pathogens-acquisition of inhibitors and beyond. Mol. Immunol. 2009, 46, 2808–2817. [Google Scholar] [CrossRef]
- Cui, T.; Chen, Y.; Knosel, T.; Yang, L.; Zoller, K.; Galler, K.; Berndt, A.; Mihlan, M.; Zipfel, P.F.; Petersen, I. Human complement factor H is a novel diagnostic marker for lung adenocarcinoma. Int. J. Oncol. 2011, 39, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Ajona, D.; Hsu, Y.F.; Corrales, L.; Montuenga, L.M.; Pio, R. Down-regulation of human complement factor H sensitizes non-small cell lung cancer cells to complement attack and reduces in vivo tumor growth. J. Immunol. 2007, 178, 5991–5998. [Google Scholar] [CrossRef]
- Ihle, J.N. STATs: signal transducers and activators of transcription. Cell 1996, 84, 331–334. [Google Scholar] [CrossRef]
- Wu, Z.; Lauer, T.W.; Sick, A.; Hackett, S.F.; Campochiaro, P.A. Oxidative stress modulates complement factor H expression in retinal pigmented epithelial cells by acetylation of FOXO3. J. Biol. Chem. 2007, 282, 22414–22425. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef]
- Eyles, J.L.; Metcalf, D.; Grusby, M.J.; Hilton, D.J.; Starr, R. Negative regulation of interleukin-12 signaling by suppressor of cytokine signaling-1. J. Biol. Chem. 2002, 277, 43735–43740. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 14133–14138. [Google Scholar] [CrossRef]
- Lee, T.L.; Yeh, J.; Van Waes, C.; Chen, Z. Epigenetic modification of SOCS-1 differentially regulates STAT3 activation in response to interleukin-6 receptor and epidermal growth factor receptor signaling through JAK and/or MEK in head and neck squamous cell carcinomas. Mol. Cancer Ther. 2006, 5, 8–19. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Matsubara, K.; Qian, G.S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar] [CrossRef]
- Zhang, S.; Guo, D.; Jiang, L.; Zhang, Q.; Qiu, X.; Wang, E. SOCS3 inhibiting migration of A549 cells correlates with PYK2 signaling in vitro. BMC Cancer 2008, 8, 150. [Google Scholar] [CrossRef]
- Shin, H.J.; Park, H.Y.; Jeong, S.J.; Park, H.W.; Kim, Y.K.; Cho, S.H.; Kim, Y.Y.; Cho, M.L.; Kim, H.Y.; Min, K.U.; et al. STAT4 expression in human T cells is regulated by DNA methylation but not by promoter polymorphism. J. Immunol. 2005, 175, 7143–7150. [Google Scholar] [CrossRef]
- Varsano, S.; Rashkovsky, L.; Shapiro, H.; Ophir, D.; Mark-Bentankur, T. Human lung cancer cell lines express cell membrane complement inhibitory proteins and are extremely resistant to complement-mediated lysis; a comparison with normal human respiratory epithelium in vitro, and an insight into mechanism(s) of resistance. Clin. Exp. Immunol. 1998, 113, 173–182. [Google Scholar] [CrossRef]
- Barnes, J.; Agarwal, S.K. Targeting STAT4 in systemic sclerosis: A promising new direction. Expert Rev. Clin. Immunol. 2011, 7, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Korman, B.D.; Kastner, D.L.; Gregersen, P.K.; Remmers, E.F. STAT4: genetics, mechanisms, and implications for autoimmunity. Curr. Allergy Asthma Rep. 2008, 8, 398–403. [Google Scholar] [CrossRef]
- Slattery, M.L.; Lundgreen, A.; Kadlubar, S.A.; Bondurant, K.L.; Wolff, R.K. JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol. Carcinog. 2013, 52, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, L.; Zhang, Y.; Zhang, Y.; Zhao, Y.; You, X.; Lin, Z.; Zhang, X.; Ye, L. The oncoprotein HBXIP uses two pathways to up-regulate S100A4 in promotion of growth and migration of breast cancer cells. J. Biol. Chem. 2012, 287, 30228–30239. [Google Scholar] [CrossRef] [PubMed]
- Mallette, F.A.; Calabrese, V.; Ilangumaran, S.; Ferbeyre, G. SOCS1, a novel interaction partner of p53 controlling oncogene-induced senescence. Aging (Albany NY) 2010, 2, 445–452. [Google Scholar] [CrossRef]
- Yoon, K.A.; Chae, Y.M.; Cho, J.Y. FGF2 stimulates SDF-1 expression through the Erm transcription factor in Sertoli cells. J. Cell Physiol. 2009, 220, 245–256. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, B.G.; Lee, S.J.; Heo, S.H.; Kim, J.Y.; Kwon, T.H.; Lee, E.B.; Ryoo, H.M.; Cho, J.Y. Proteomic profiling of endothelial cells in human lung cancer. J. Proteome Res. 2008, 7, 1138–1150. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, Y.-H.; Hwang, H.-J.; Sung, H.-J.; Heo, S.-H.; Kim, D.-S.; Hong, S.-H.; Lee, K.-H.; Cho, J.-Y. Upregulation of Complement Factor H by SOCS-1/3–STAT4 in Lung Cancer. Cancers 2019, 11, 471. https://doi.org/10.3390/cancers11040471
Yoon Y-H, Hwang H-J, Sung H-J, Heo S-H, Kim D-S, Hong S-H, Lee K-H, Cho J-Y. Upregulation of Complement Factor H by SOCS-1/3–STAT4 in Lung Cancer. Cancers. 2019; 11(4):471. https://doi.org/10.3390/cancers11040471
Chicago/Turabian StyleYoon, Yeon-Hee, Hyeon-Ji Hwang, Hye-Jin Sung, Sun-Hee Heo, Dong-Sun Kim, Su-Hyung Hong, Kang-Hoon Lee, and Je-Yoel Cho. 2019. "Upregulation of Complement Factor H by SOCS-1/3–STAT4 in Lung Cancer" Cancers 11, no. 4: 471. https://doi.org/10.3390/cancers11040471
APA StyleYoon, Y.-H., Hwang, H.-J., Sung, H.-J., Heo, S.-H., Kim, D.-S., Hong, S.-H., Lee, K.-H., & Cho, J.-Y. (2019). Upregulation of Complement Factor H by SOCS-1/3–STAT4 in Lung Cancer. Cancers, 11(4), 471. https://doi.org/10.3390/cancers11040471