Peptide Mediated In Vivo Tumor Targeting of Nanoparticles through Optimization in Single and Multilayer In Vitro Cell Models


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
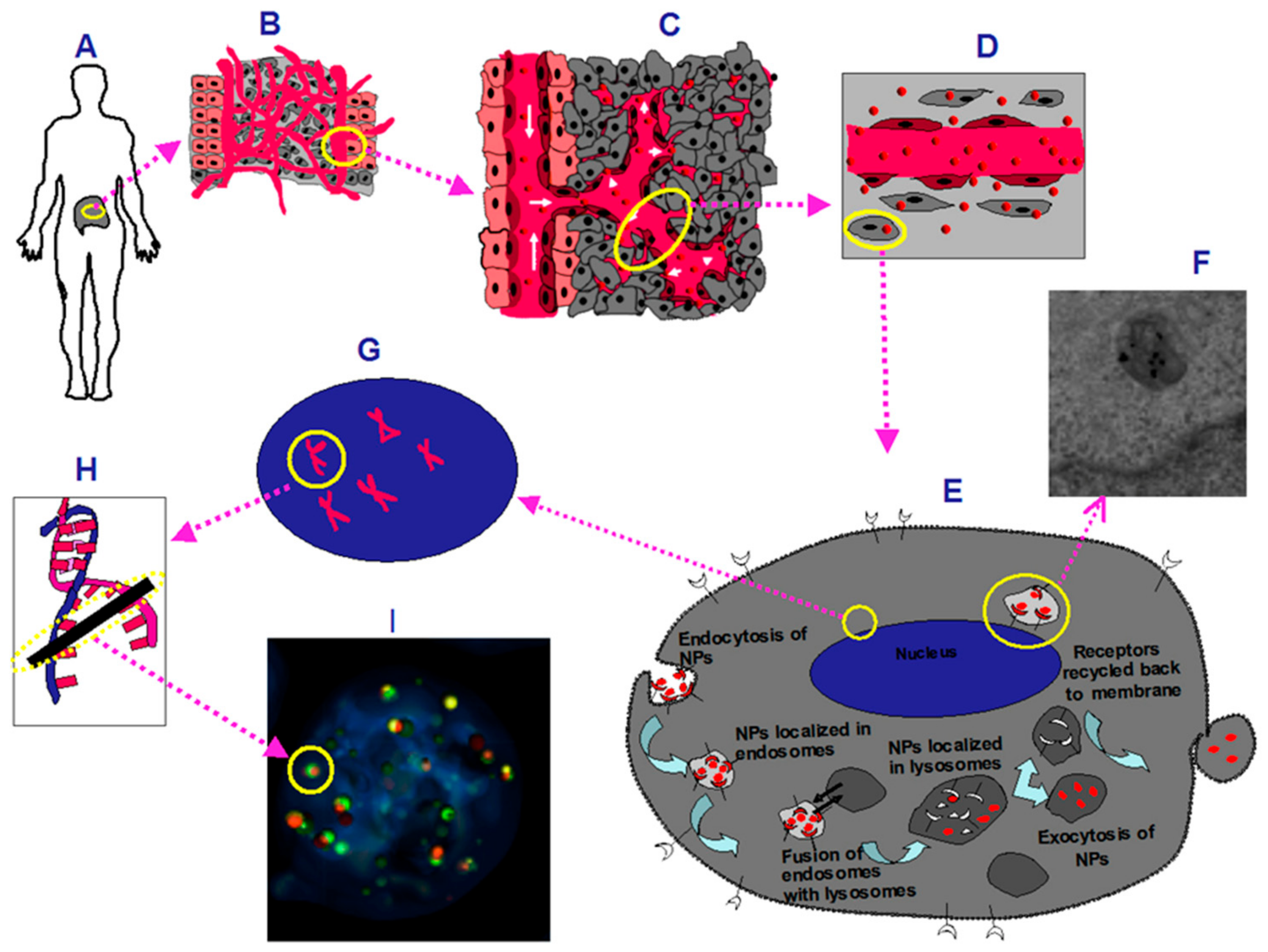
1. Introduction
2. Results
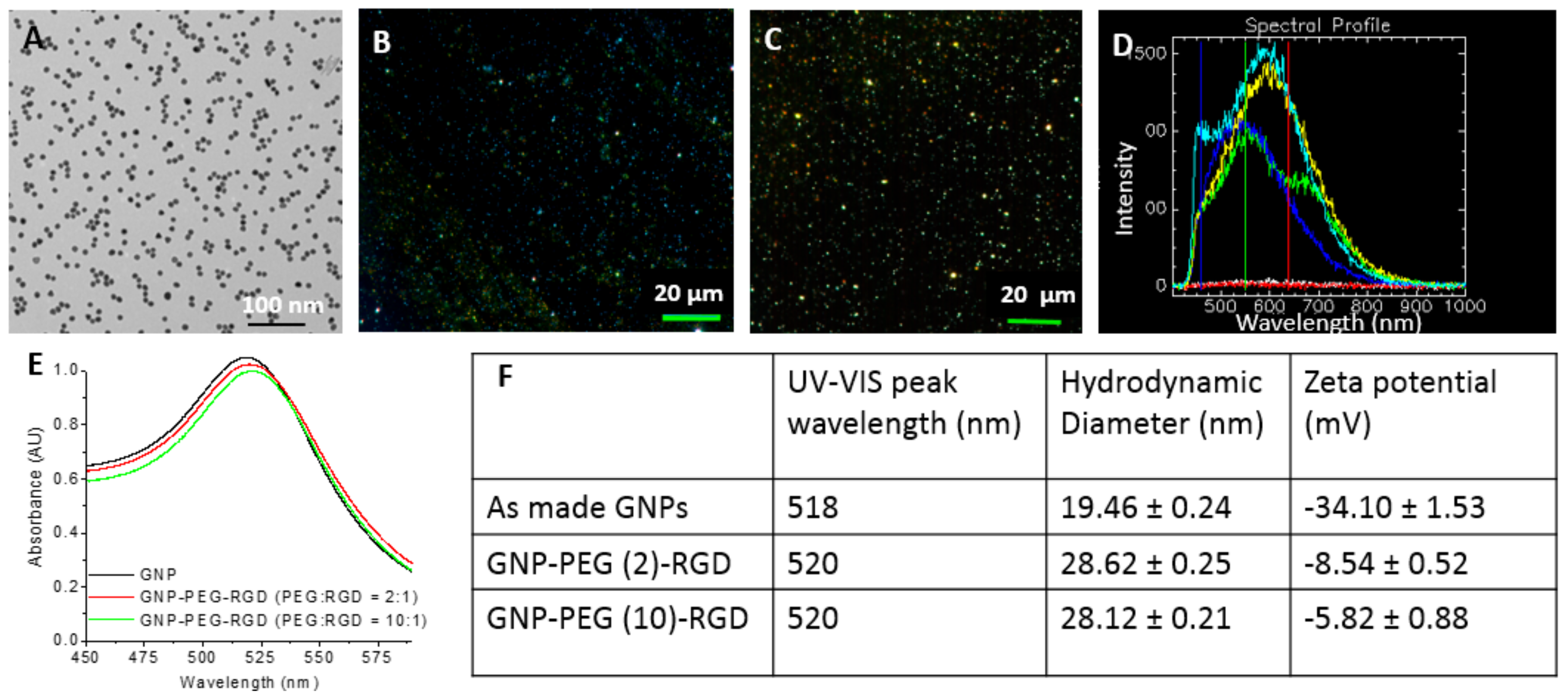
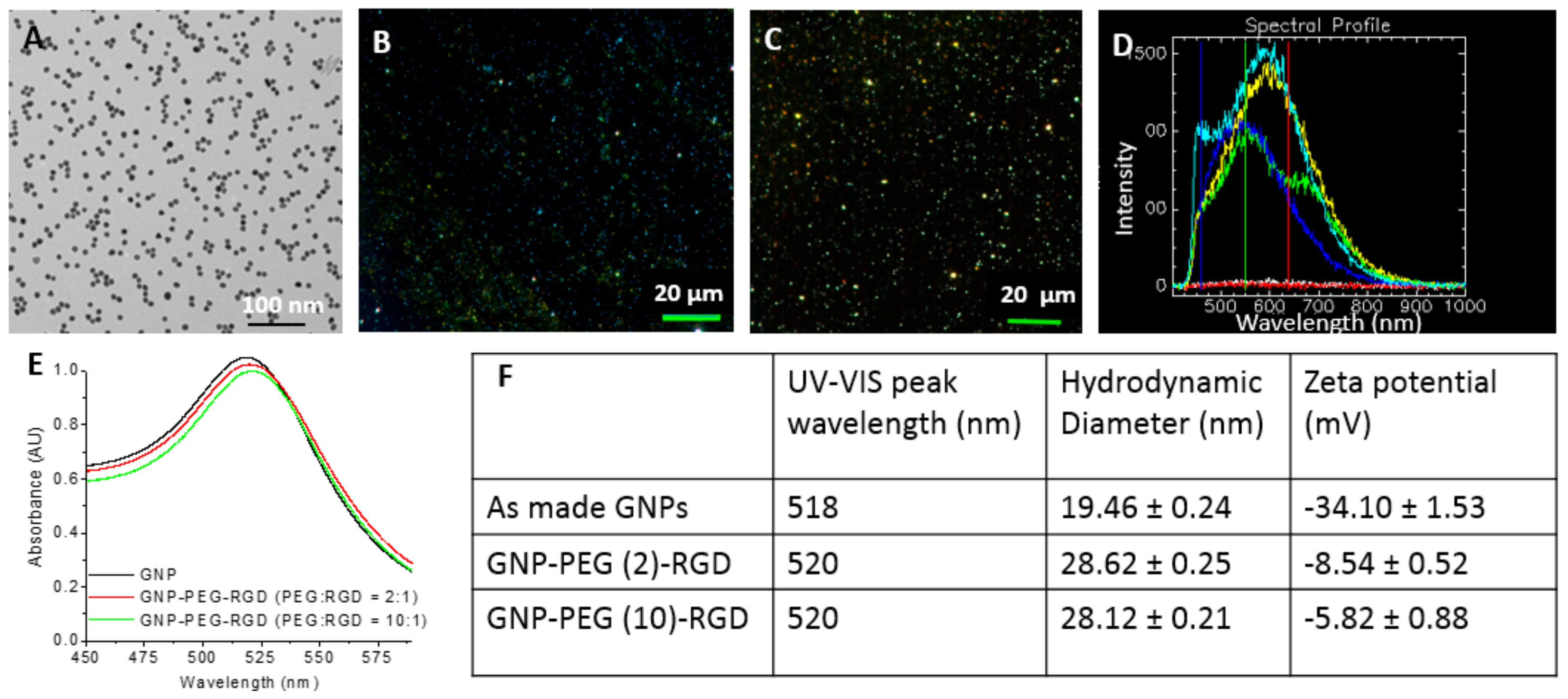
2.1. Characterization of Gold Nanoparticles Functionalized with Peptide and PEG Molecules
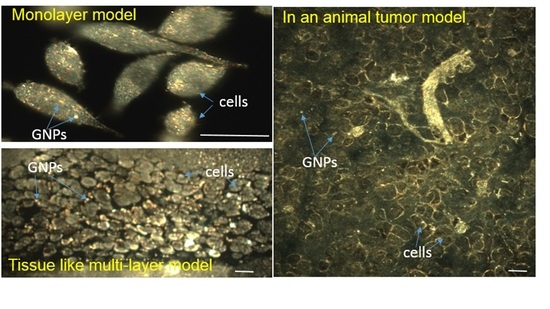
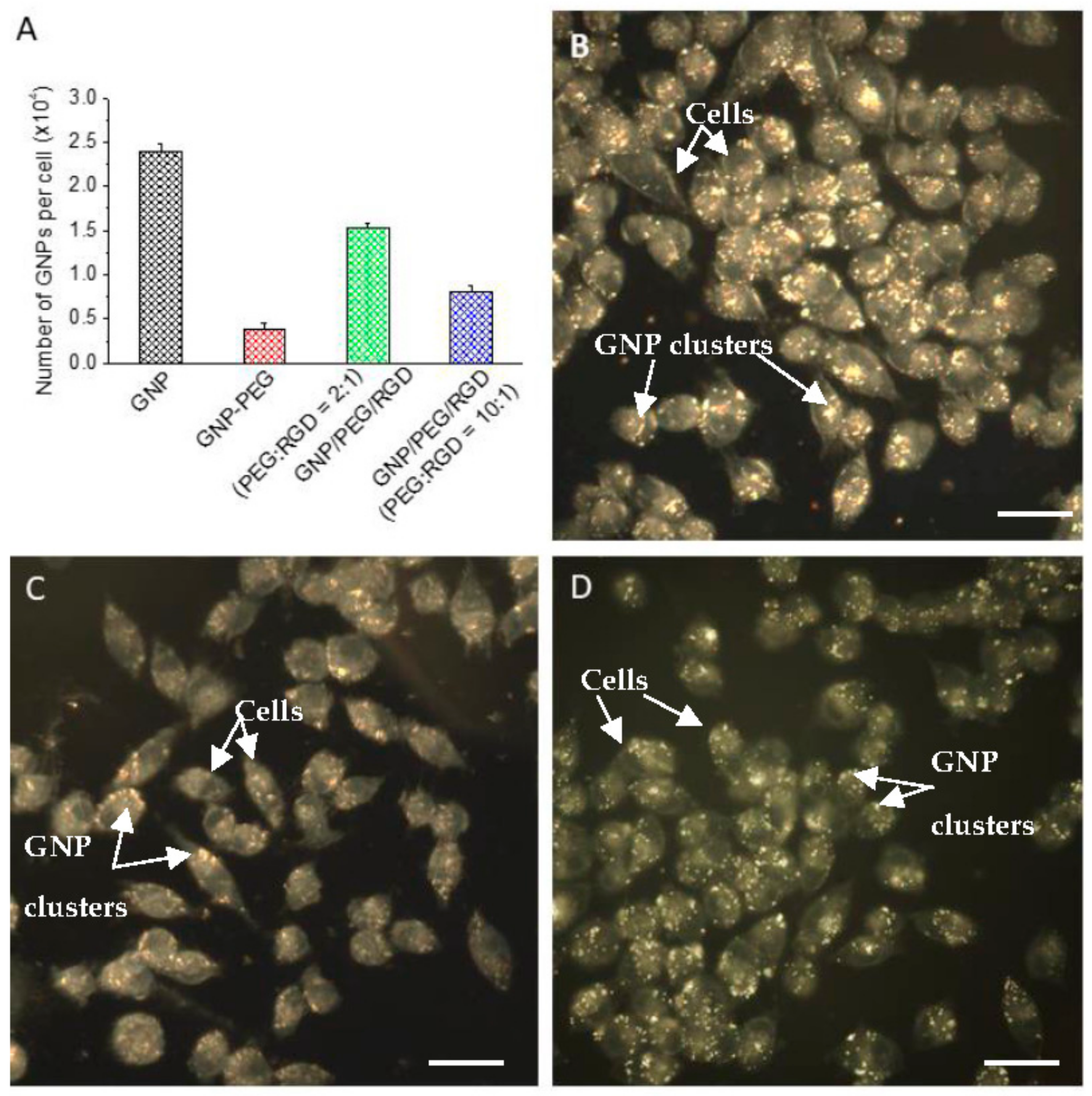
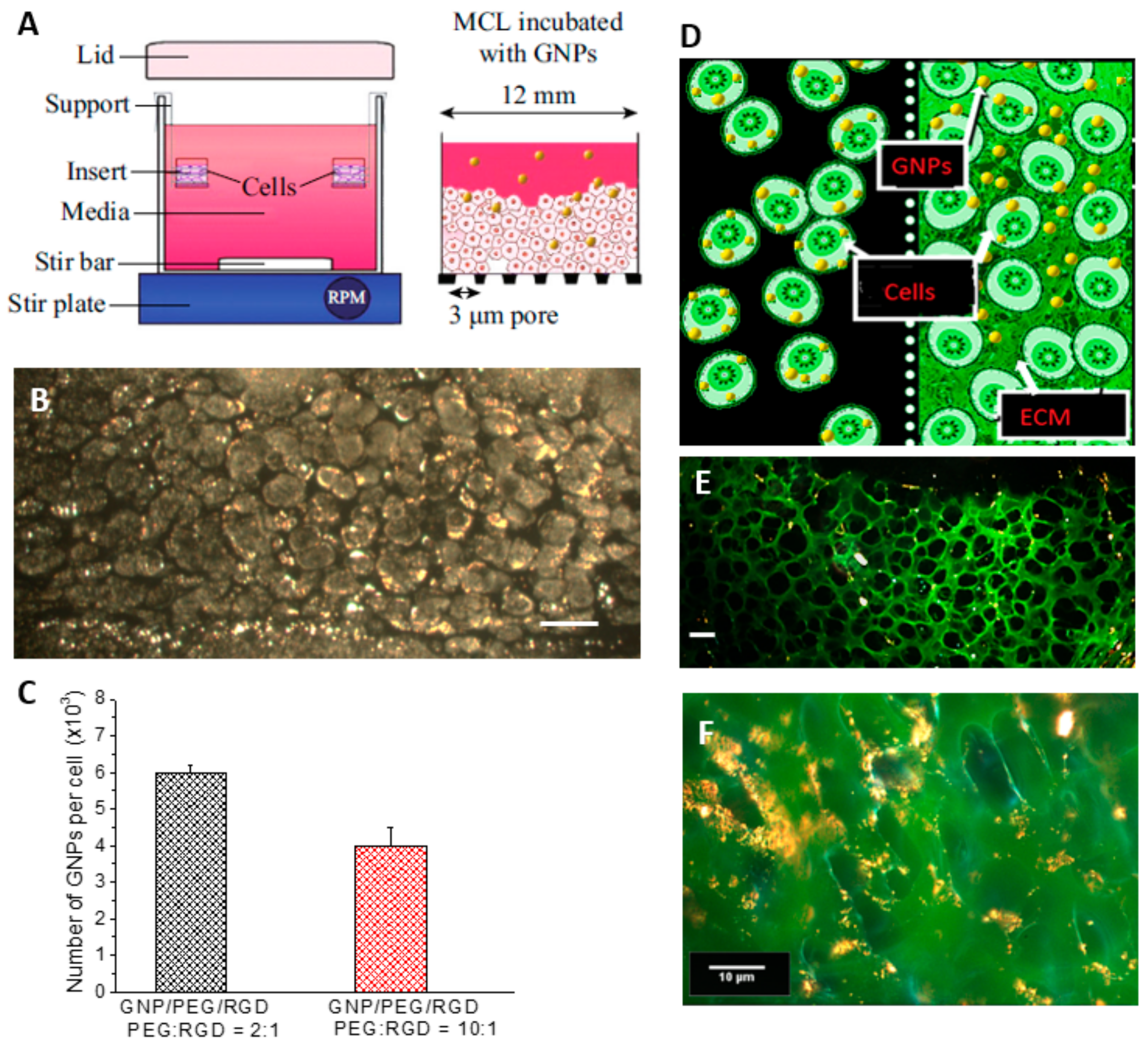
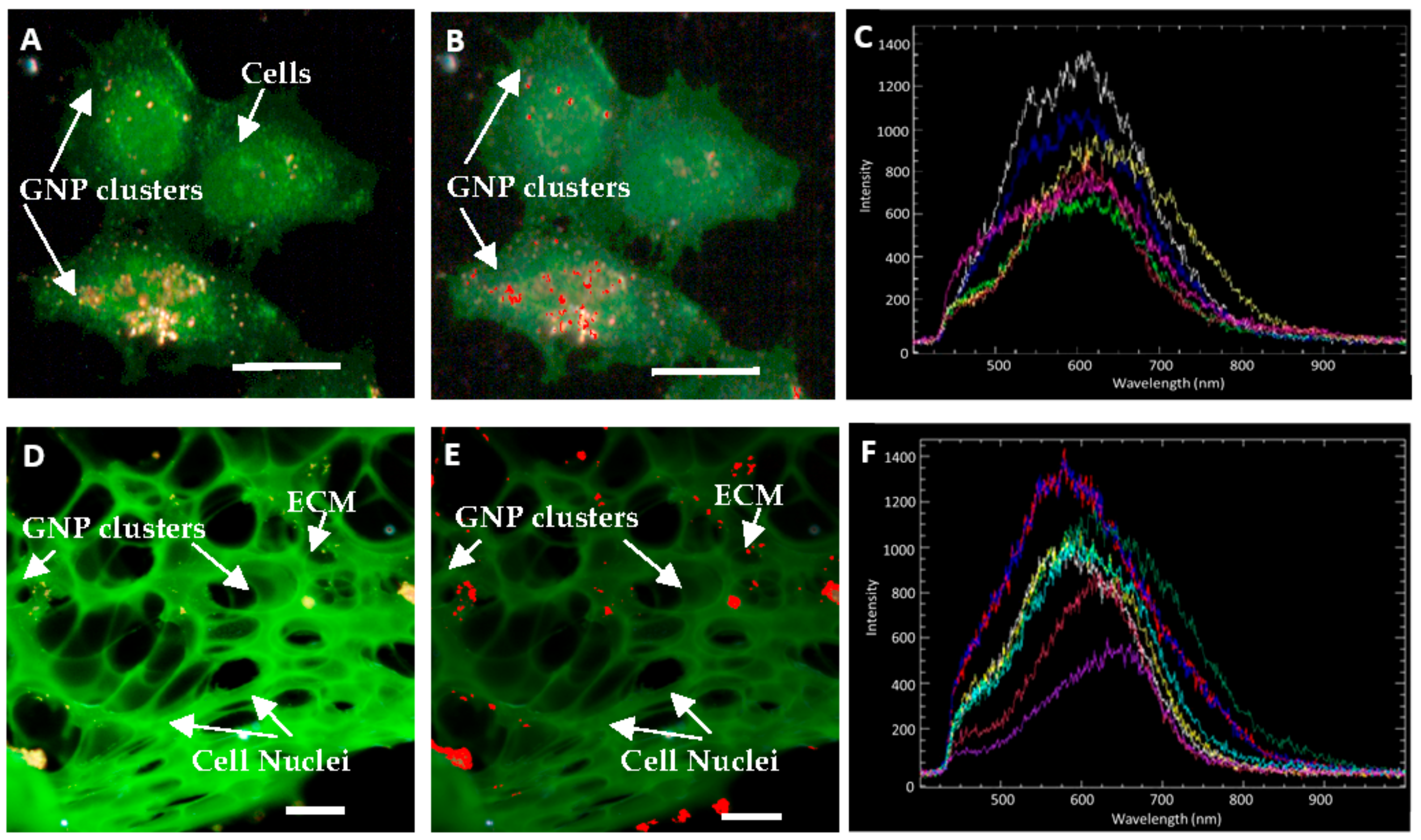
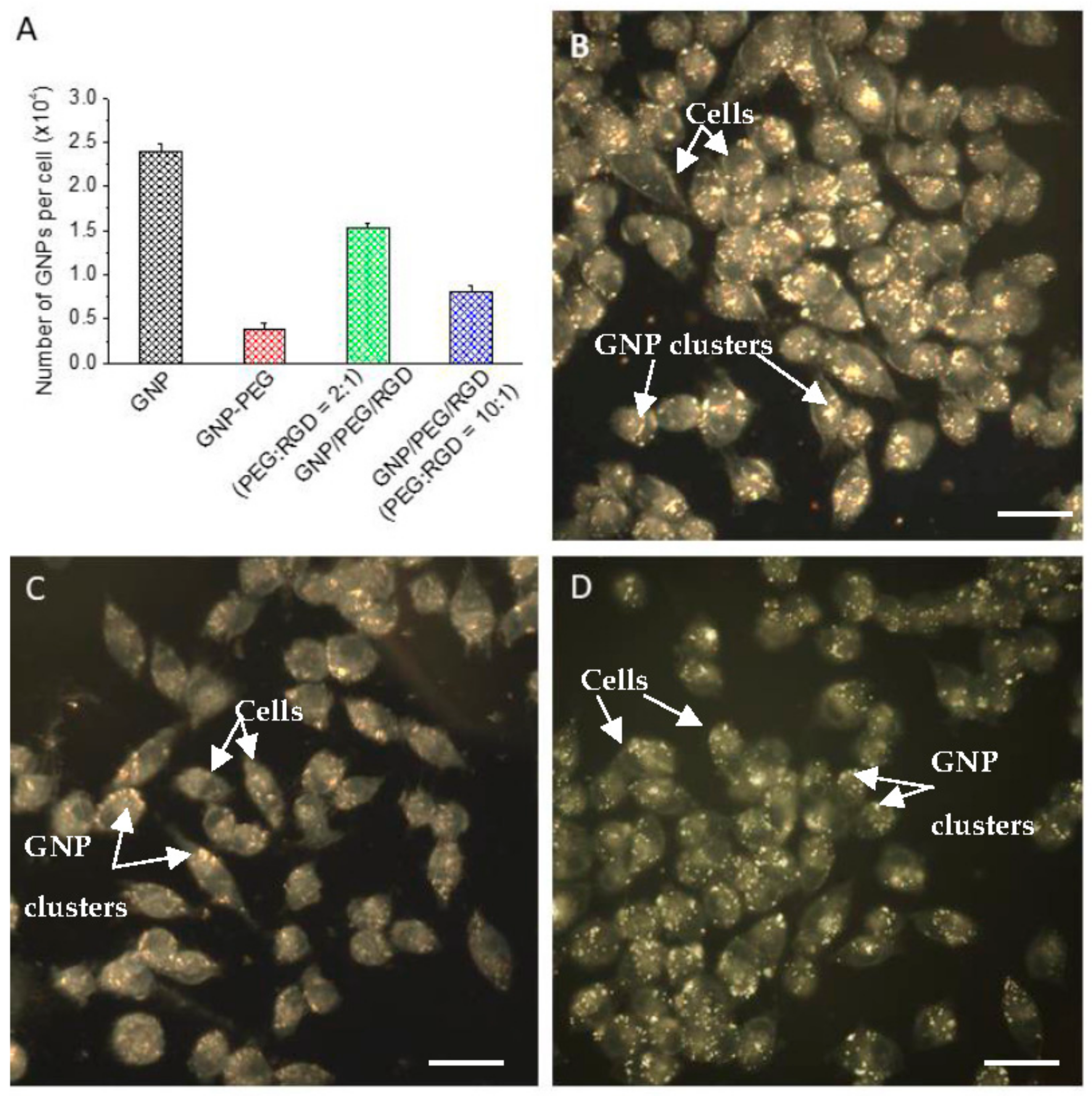
2.2. Accumulation of Gold Nanoparticles at the Monolayer Level
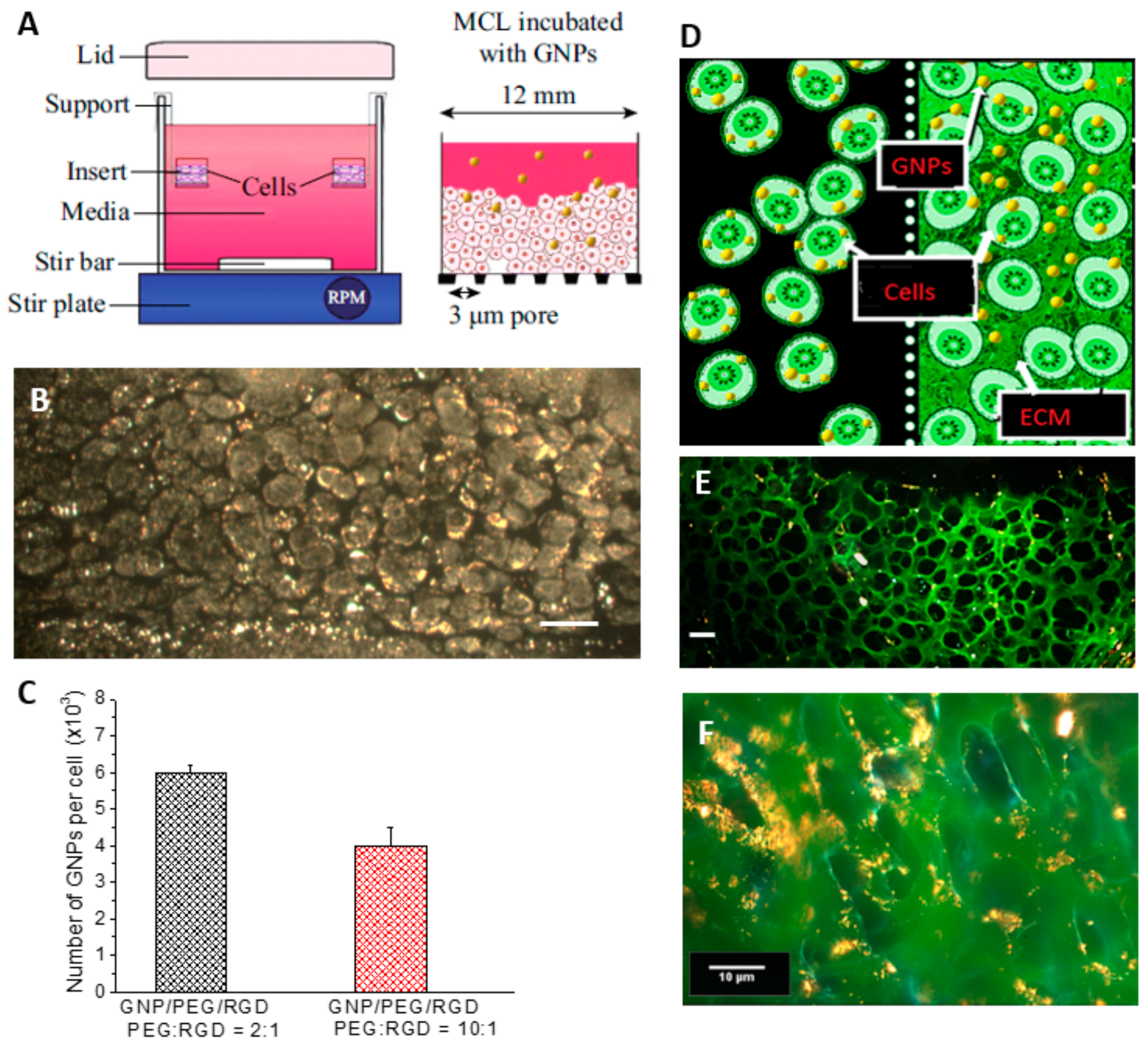
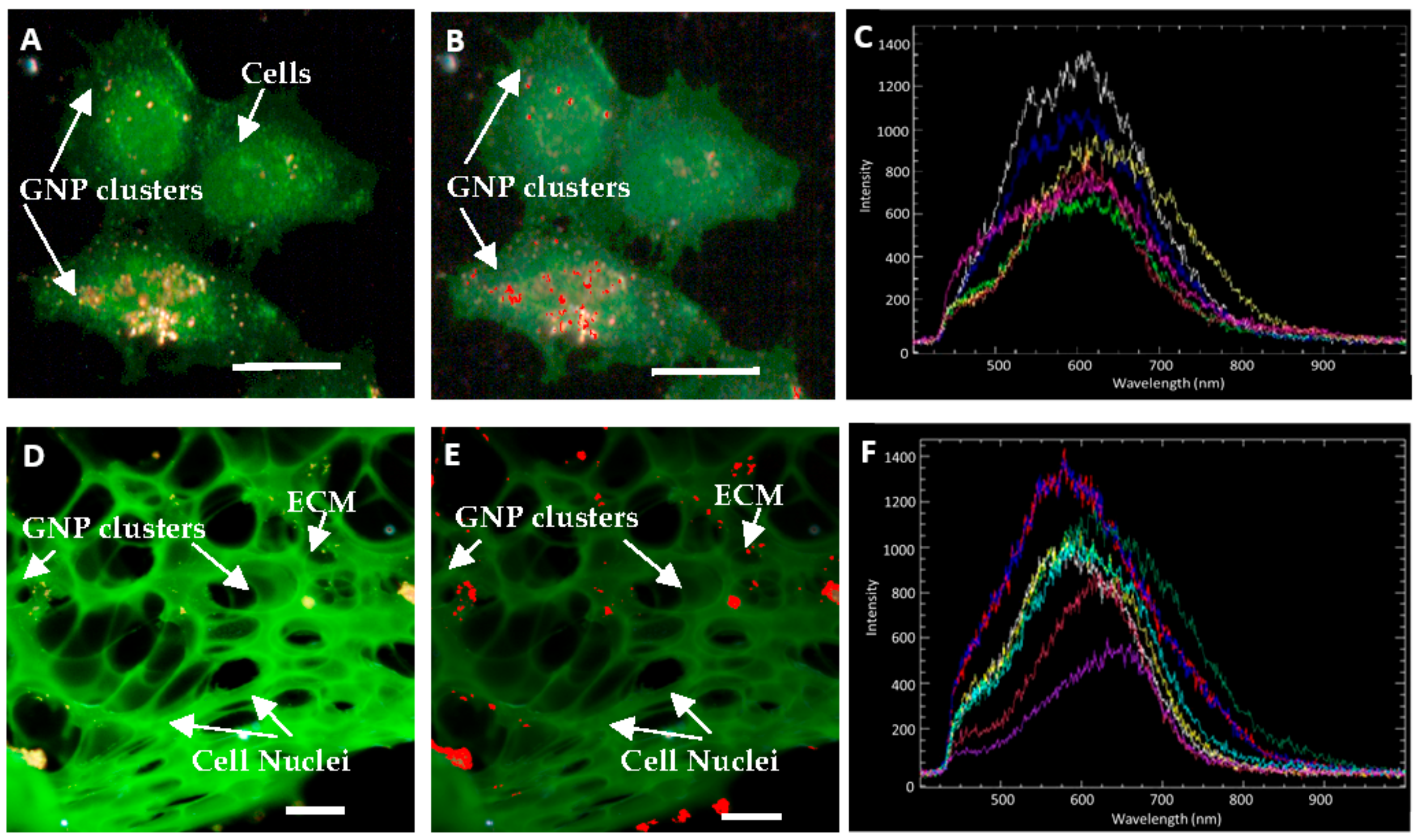
2.3. Accumulation of Gold Nanoparticles in Multilayer the Model
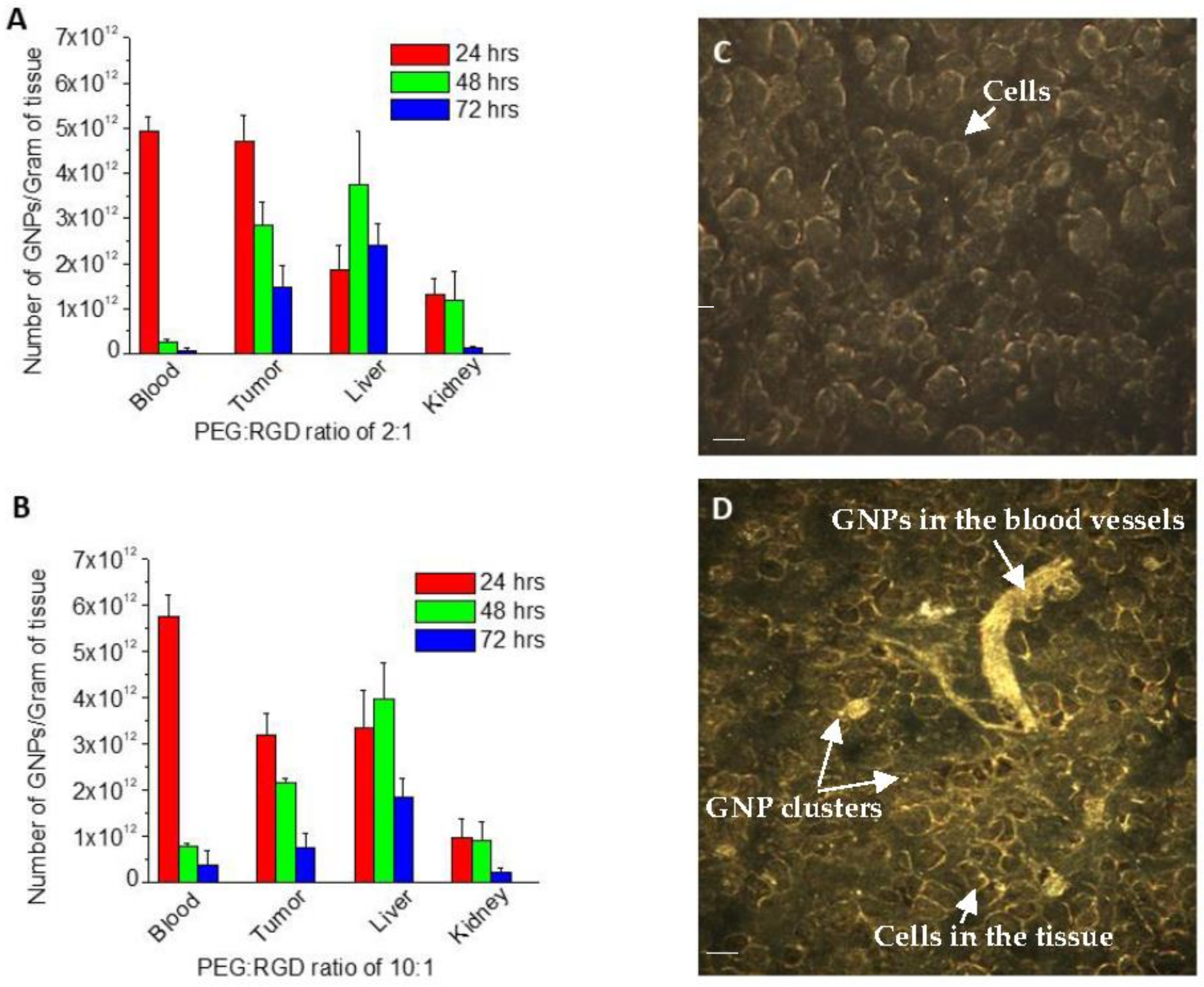
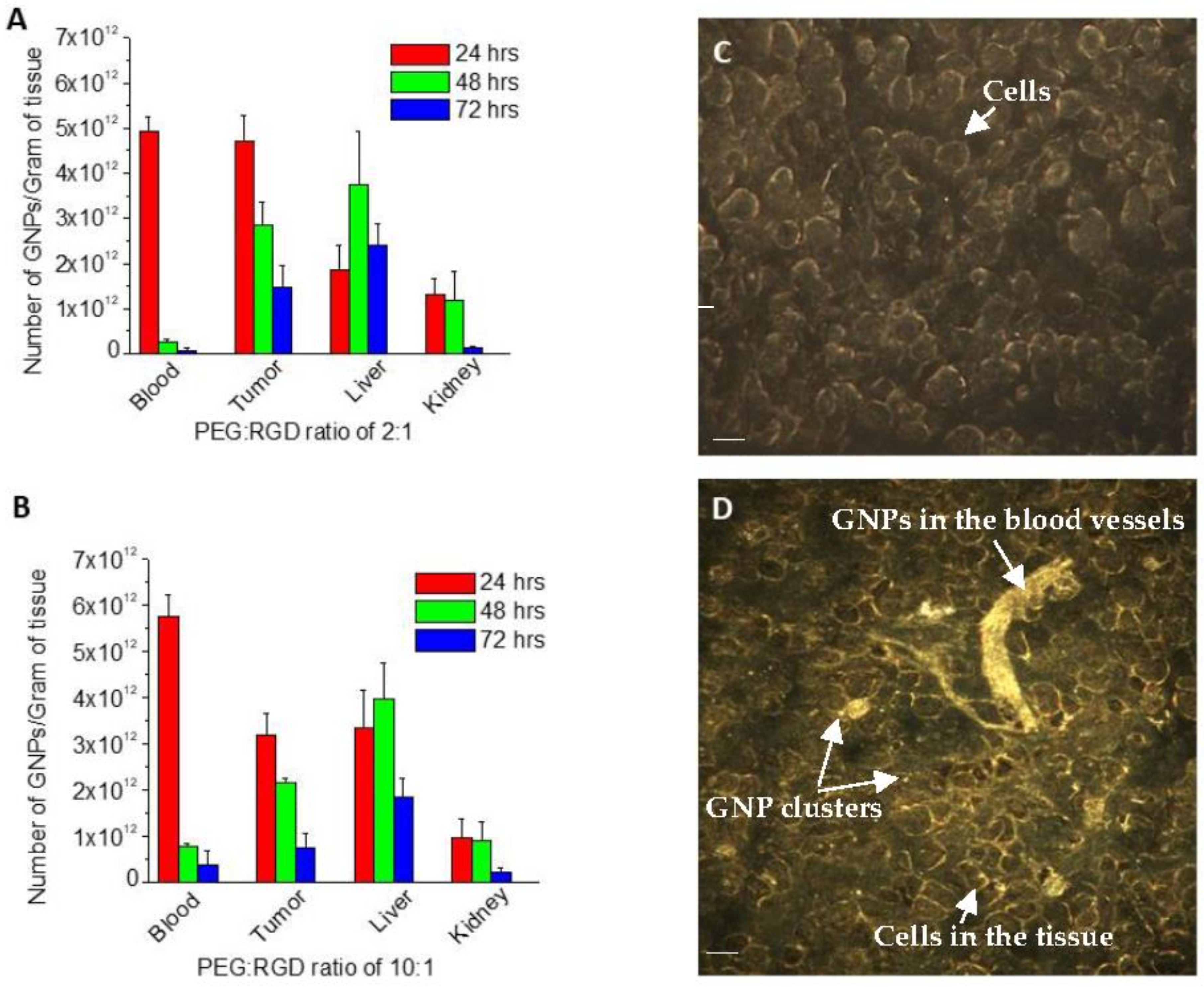
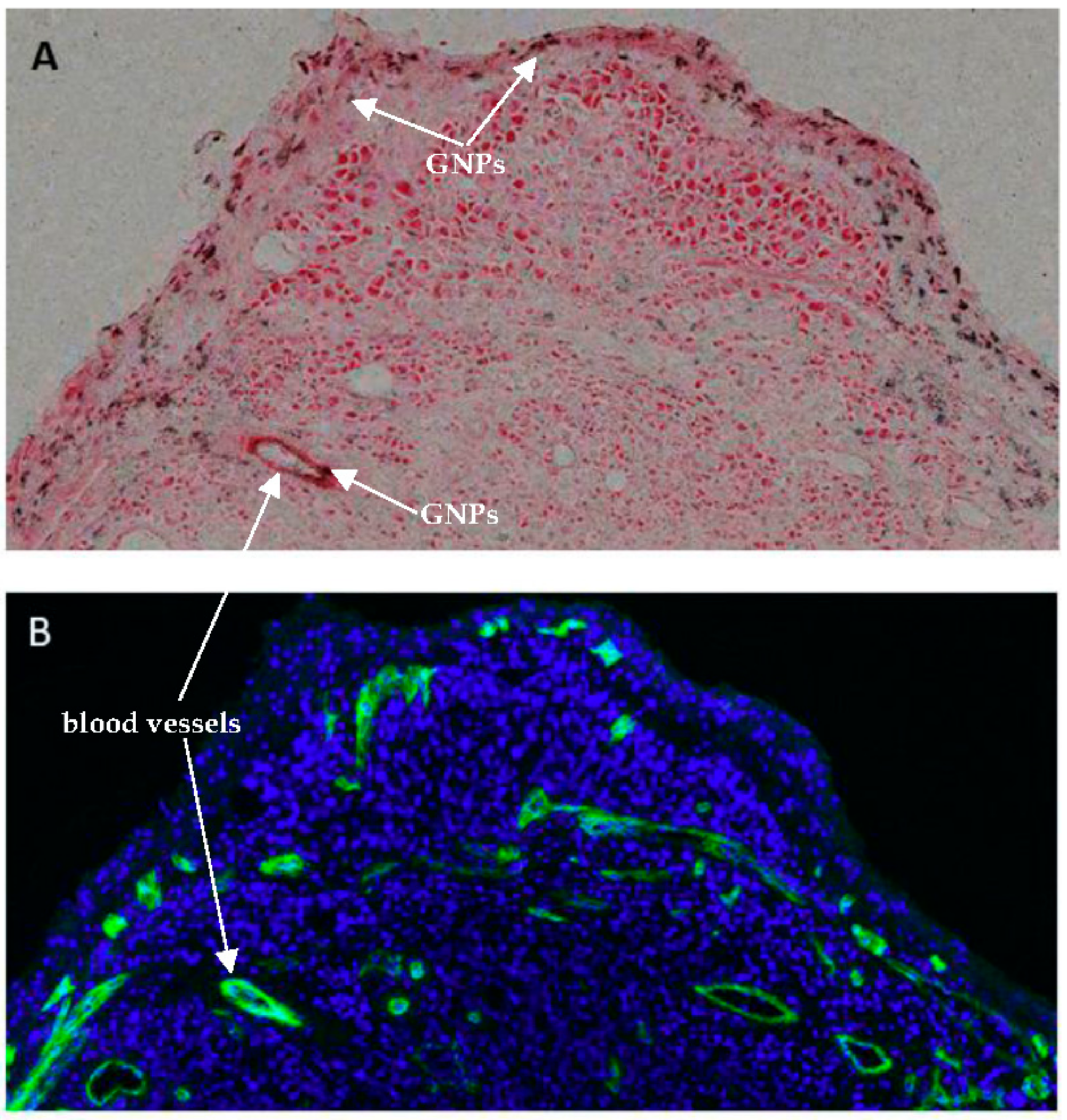
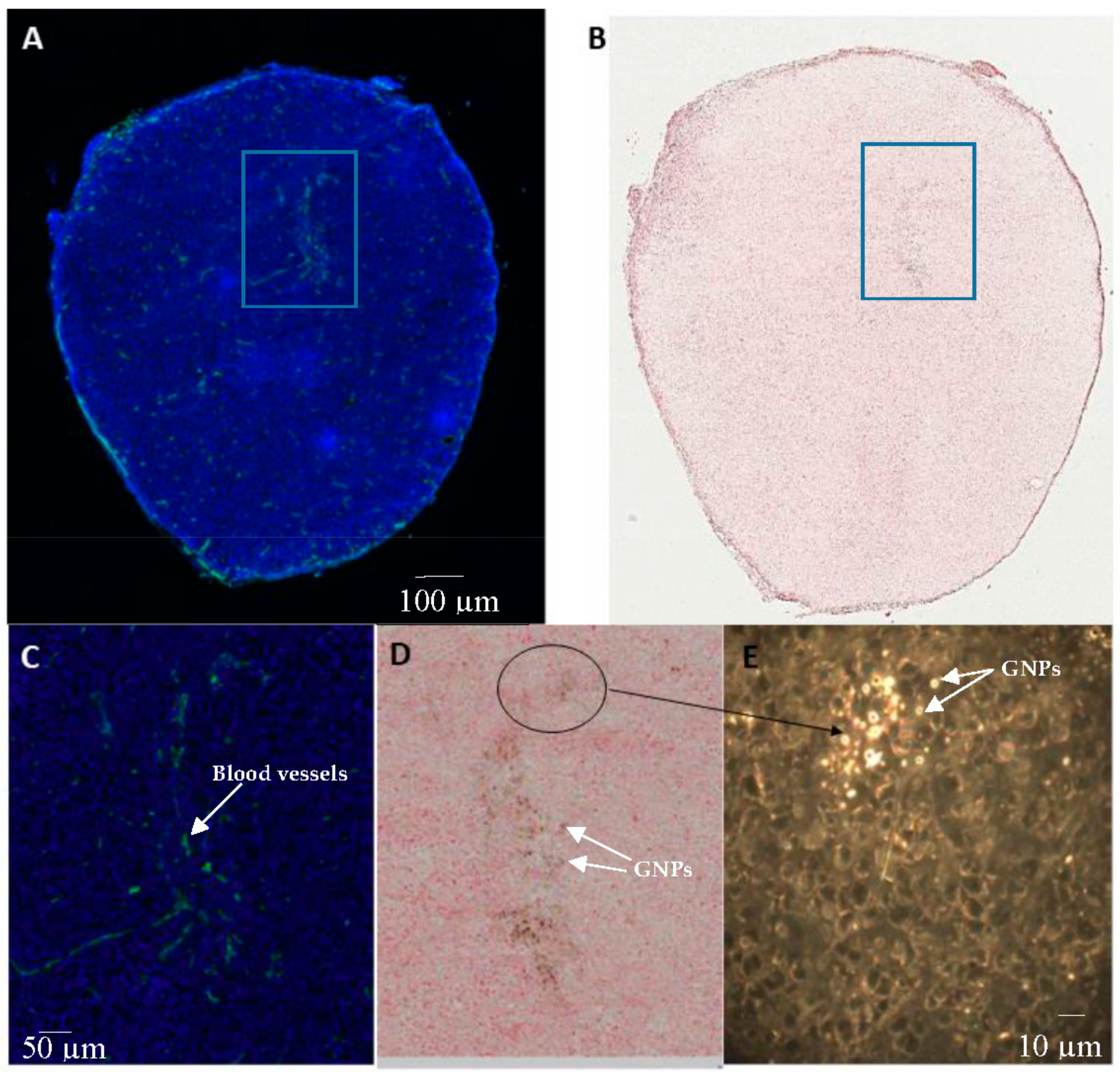
2.4. In Vivo Accumulation and Pharmacokinetics of GNPs in a Pancreatic Cancer Model
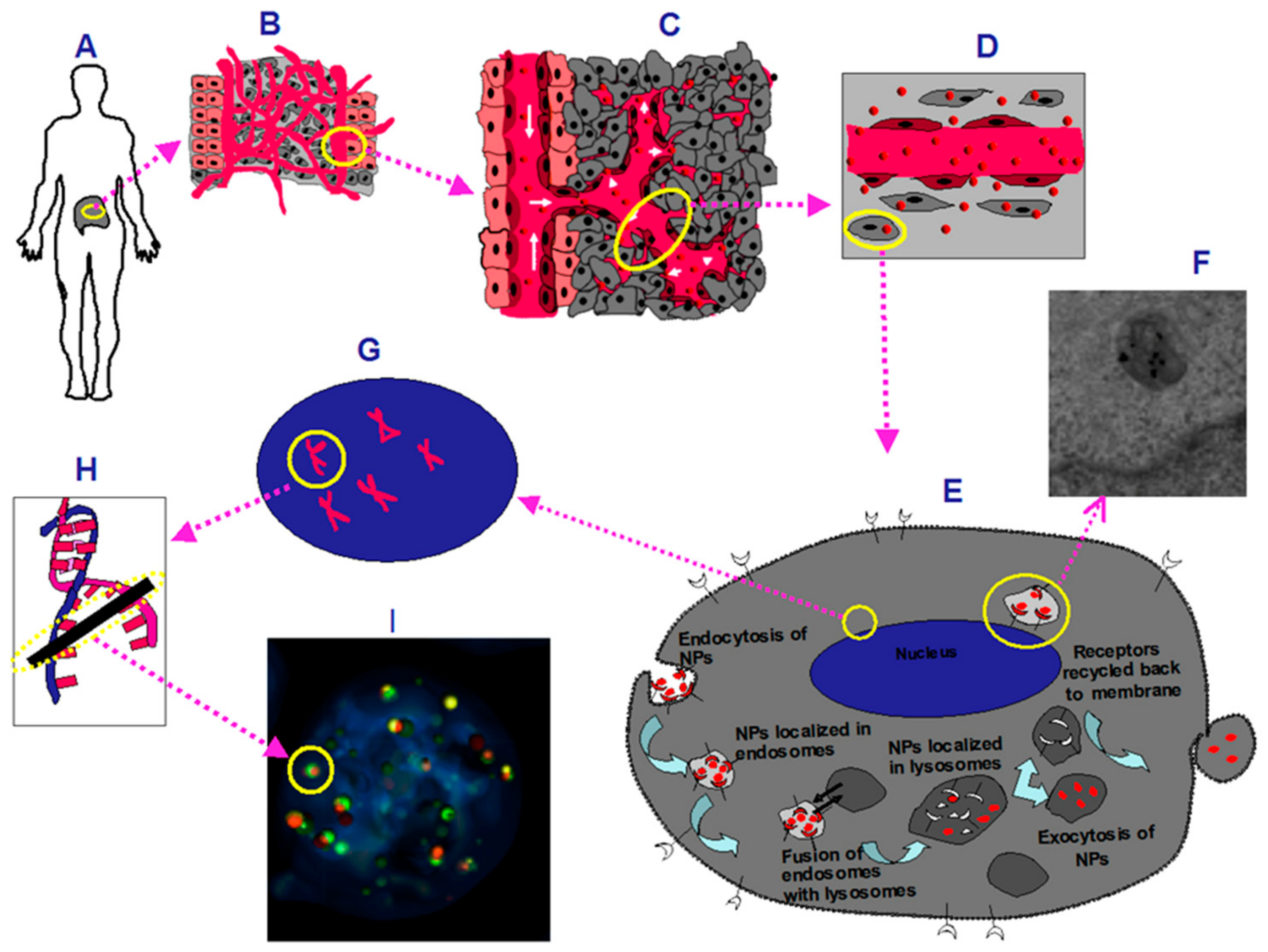
3. Discussion
4. Materials and Methods
4.1. Synthesis and Characterization of NPs
4.2. Conjugation of Peptides and PEG onto GNPs
4.3. Cellular Accumulation Studies
4.4. Growth of Multi-Cellular Layers (MCLs)
4.5. Pancreatic Xenograft Model
4.6. In Vivo Comprehensive Acute and Physical Toxicity Assay
4.7. In Vivo Biodiversity Assay
4.8. Immunohistochemistry
4.9. Quantification of GNPs
4.10. Hyperspectral Imaging
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2016, 7, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.B.; Bristow, R.G.; Kim, J.; Siu, L.L. Combined-modality treatment of solid tumors using radiotherapy and molecular targeted agents. J. Clin. Oncol. 2003, 21, 2760–2776. [Google Scholar] [CrossRef] [PubMed]
- Herscher, L.L.; Cook, J.A.; Pacelli, R.; Pass, H.; Russo, A.; Mitchell, J. Principles of chemoradiation: Theoretical and practical considerations. Oncology (Williston Park NY) 1999, 13, 11–22. [Google Scholar]
- Chithrani, B.D. Optimization of bio-nano interface using gold nanostructures as a model nanoparticle system. Insci. J. 2011, 1, 136–156. [Google Scholar] [CrossRef]
- Cruje, C.; Chithrani, B. Integration of peptides for enhanced uptake of pegylayed gold nanoparticles. J. Nanosci. Nanotechnol. 2015, 15, 2125–2131. [Google Scholar] [CrossRef] [PubMed]
- Cruje, C.; Chithrani, D.B. Polyethylene glycol functionalized nanoparticles for improved cancer treatment. Rev. Nanosci. Nanotechnol. 2014, 3, 20–30. [Google Scholar] [CrossRef]
- Khawar, I.A.; Kim, J.H.; Kuh, H.-J. Improving drug delivery to solid tumors: Priming the tumor microenvironment. J. Control. Release 2015, 201, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Patra, C.R.; Bhattacharya, R.; Wang, E.; Katarya, A.; Lau, J.S.; Dutta, S.; Muders, M.; Wang, S.; Buhrow, S.A.; Safgren, S.L.; et al. Targeted delivery of gemcitabine to pancreatic adenocarcinoma using cetuximab as a targeting agent. Cancer Res. 2008, 68, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Paunesku, T.; Woloschak, G. Peptide mediated cancer targeting of nanoconjugates. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011, 3, 269–281. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.E.; Ginsberg, M.H.; Plow, E.F. Arginyl-glycyl-aspartic acid (rgd): A cell adhesion motif. Trends Biochem. Sci. 1991, 16, 246–250. [Google Scholar] [CrossRef]
- Gehlsen, K.R.; Argraves, W.S.; Pierschbacher, M.D.; Ruoslahti, E. Inhibition of in vitro tumor cell invasion by arg-gly-asp-containing synthetic peptides. J. Cell Biol. 1988, 106, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Uertz, J.; Yohan, D.; Chithrani, B. Peptide modified gold nanoparticles for improved cellular uptake, nuclear transport, and intracellular retention. Nanoscale 2014, 6, 12026–12033. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.; Patel, D.; Chuttani, K.; Mishra, A.K.; Misra, A. In vitro mechanistic study of cell death and in vivo performance evaluation of rgd grafted pegylated docetaxel liposomes in breast cancer. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, Y.; Su, S.; Li, W.; Chen, C.; Wu, Y. Anti-tumor activity of paclitaxel through dual-targeting carrier of cyclic rgd and transferrin conjugated hyperbranched copolymer nanoparticles. Biomaterials 2012, 33, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, S.; Qian, L.; Pei, Y.; Qiu, Y.; Jiang, Y. Rgd-modified peg–pamam–dox conjugates: In vitro and in vivo studies for glioma. Eur. J. Pharm. Biopharm. 2011, 79, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-F.; Wang, J.-C.; Bian, D.-Y.; Zhang, X.; Zhang, Q. Targeted delivery of rgd-modified liposomes encapsulating both combretastatin a-4 and doxorubicin for tumor therapy: In vitro and in vivo studies. Eur. J. Pharm. Biopharm. 2010, 74, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Shi, W.; Freund, L.B. Mechanics of receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 2005, 102, 9469–9474. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Curry, T.; Che, Y.; Kopelman, R. Targeted Delivery of Peptide-Conjugated Biocompatible Gold Nanoparticles into Cancer Cell Nucleus; SPIE BiOS, 2013; International Society for Optics and Photonics: Bellingham, WA, USA, 2013; p. 85951D. [Google Scholar]
- Kang, B.; Mackey, M.; El-Sayed, M. Nuclear targeting of gold nanoparticles in cancer cells induces DNA damage, causing cytokinesis arrest and apoptosis. J. Am. Chem. Soc. 2010, 132, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Park, R.; Shahinian, A.H.; Tohme, M.; Khankaldyyan, V.; Bozorgzadeh, M.H.; Bading, J.R.; Moats, R.; Laug, W.E.; Conti, P.S. 18 f-labeled rgd peptide: Initial evaluation for imaging brain tumor angiogenesis. Nucl. Med. Biol. 2004, 31, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Yohan, D.; Cruje, C.; Lu, X.; Chithrani, D. Elucidating the uptake and distribution of nanoparticles in solid tumors via a multilayered cell culture model. Nano-Micro Lett. 2015, 7, 127–137. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Zuber, A.; Purdey, M.; Schartner, E.; Forbes, C.; van der Hoek, B.; Giles, D.; Abell, A.; Monro, T.; Ebendorff-Heidepriem, H. Detection of gold nanoparticles with different sizes using absorption and fluorescence based method. Sens. Actuators B Chem. 2016, 227, 117–127. [Google Scholar] [CrossRef]
- Link, S.; El-Sayed, M.A. Spectral properties and relaxation dynamics of surface plasmon electronic oscillations in gold and silver nanodots and nanorods. J. Phys. Chem. B 1999, 103, 8410–8426. [Google Scholar] [CrossRef]
- Cruje, C.; Yang, C.; Uertz, J.; van Prooijen, M.; Chithrani, B.D. Optimization of peg coated nanoscale gold particles for enhanced radiation therapy. RSC Adv. 2015, 5, 101525–101532. [Google Scholar] [CrossRef]
- Kirchhausen, T. Three ways to make a vesicle. Nat. Rev. Mol. Cell Biol. 2000, 1, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Kam, N.W.; Liu, Z.; Dai, H. Carbon nanotubes as intracellular transporters for proteins and DNA: An investigation of the uptake mechanism and pathway. Angew. Chem. 2006, 45, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Ghosh, R.N.; Maxfield, F.R. Endocytosis. Physiol. Rev. 1997, 77, 759–803. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Heller, D.A.; Sharma, R.; Strano, M.S. Size-dependent cellular uptake and expulsion of single-walled carbon nanotubes: Single particle tracking and a generic uptake model for nanoparticles. ACS Nano 2009, 3, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, B.D.; Chan, W.C. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Schroter, C.J.; Braun, M.; Englert, J.; Beck, H.; Schmid, H.; Kalbacher, H. A rapid method to separate endosomes from lysosomal contents using differential centrifugation and hypotonic lysis of lysosomes. J. Immunol. Methods 1999, 227, 161–168. [Google Scholar] [CrossRef]
- Silverstein, S.C.; Steinman, R.M.; Cohn, Z.A. Endocytosis. Annu. Rev. Biochem. 1977, 46, 669–722. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, Q.; Liang, L.; Li, J.; Wang, K.; Li, J.; Lv, M.; Chen, N.; Song, H.; Lee, J. Real-time visualization of clustering and intracellular transport of gold nanoparticles by correlative imaging. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Transport into the cell from the plasma membrane: Endocytosis. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Schulz, F.; Lutz, D.; Rusche, N.; Bastús, N.G.; Stieben, M.; Höltig, M.; Grüner, F.; Weller, H.; Schachner, M.; Vossmeyer, T. Gold nanoparticles functionalized with a fragment of the neural cell adhesion molecule L1 stimulate L1-mediated functions. Nanoscale 2013, 5, 10605–10617. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Chan, W.C. Effect of gold nanoparticle aggregation on cell uptake and toxicity. ACS Nano 2011, 5, 5478–5489. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.P.B.; Robert, B. The scientific basis of radiotherapy. In The Basic Science of Oncology; Tannock, I.F.H., Richard, P., Robert, G.B., Harrington, L., Eds.; McGraw-Hill: Toronto, ON, Canada, 2008; pp. 289–321. [Google Scholar]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198. [Google Scholar] [CrossRef] [PubMed]
- Chouikrat, R.; Seve, A.; Vanderesse, R.; Benachour, H.; Barberi-Heyob, M.; Richeter, S.; Raehm, L.; Durand, J.O.; Verelst, M.; Frochot, C. Non polymeric nanoparticles for photodynamic therapy applications: Recent developments. Curr. Med. Chem. 2012, 19, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Alkilany, A.M.; Murphy, C.J. Toxicity and cellular uptake of gold nanoparticles: What we have learned so far? J. Nanopart. Res. Interdiscip. Forum Nanoscale Sci. Technol. 2010, 12, 2313–2333. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.M.; Loadman, P.M.; Cronin, B.P. Evaluation of a novel in vitro assay for assessing drug penetration into avascular regions of tumours. Br. J. Cancer 1998, 77, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Wendt, K.R.; Clow, K.A.; Fryer, K.H. Multilayers of cells growing on a permeable support. An in vitro tumour model. Acta Oncol. 1997, 36, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.; Bradley, D.A.; Winlove, C.P. Effects of ionizing radiation on extracellular matrix. Nucl. Instrum. Methods Phys. Res. Sect. A 2007, 580, 566–569. [Google Scholar] [CrossRef]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of extracellular matrix assembly in interstitial transport in sold tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar] [PubMed]
- Ji, T.; Lang, J.; Wang, J.; Cai, R.; Zhang, Y.; Qi, F.; Zhang, L.; Zhao, X.; Wu, W.; Hao, J.; et al. Designing liposomes to suppress extraceullular matrix expression to enhance drug penetration and pancreatic tumor therapy. ACS Nano 2017, 11, 8668–8678. [Google Scholar] [CrossRef] [PubMed]
- Sindhwani, S.; Syed, A.M.; Wilhelm, S.; Glancy, D.R.; Chen, Y.Y.; Dobosz, M.; Chan, W.C. Three-dimensional optical mapping of nanoparticle distribution in intact tissues. ACS Nano 2016, 10, 5468–5478. [Google Scholar] [CrossRef] [PubMed]
- Lunardi, S.; Muschel, R.J.; Brunner, T.B. The stromal compartments in pancreatic cancer: Are there any therapeutic targets? Cancer Lett. 2014, 343, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Ware, M.J.; Curtis, L.T.; Wu, M.; Ho, J.C.; Corr, S.J.; Curley, S.A.; Godin, B.; Frieboes, H.B. Pancreatic adenocarcinoma response to chemotherapy enhanced with non-invasive radio frequency evaluated via an integrated experimental/computational approach. Sci. Rep. 2017, 7, 3437. [Google Scholar] [CrossRef] [PubMed]
- Ware, M.J.; Ware, M.J.; Keshishian, V.; Law, J.J.; Ho, J.C. Generation of an in vitro 3d pdac stroma rich spheroid model. Biomaterials 2016, 108, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Nagy, J.; Chang, S.; Dvorak, A.; Dvorak, H. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865. [Google Scholar] [CrossRef] [PubMed]
- Frens, G. Controlled nucleation for the particle size in monodisperse gold suspensions. Nature 1973, 241, 20–22. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Bromma, K.; Chithrani, D. Peptide Mediated In Vivo Tumor Targeting of Nanoparticles through Optimization in Single and Multilayer In Vitro Cell Models. Cancers 2018, 10, 84. https://doi.org/10.3390/cancers10030084
Yang C, Bromma K, Chithrani D. Peptide Mediated In Vivo Tumor Targeting of Nanoparticles through Optimization in Single and Multilayer In Vitro Cell Models. Cancers. 2018; 10(3):84. https://doi.org/10.3390/cancers10030084
Chicago/Turabian StyleYang, Celina, Kyle Bromma, and Devika Chithrani. 2018. "Peptide Mediated In Vivo Tumor Targeting of Nanoparticles through Optimization in Single and Multilayer In Vitro Cell Models" Cancers 10, no. 3: 84. https://doi.org/10.3390/cancers10030084
APA StyleYang, C., Bromma, K., & Chithrani, D. (2018). Peptide Mediated In Vivo Tumor Targeting of Nanoparticles through Optimization in Single and Multilayer In Vitro Cell Models. Cancers, 10(3), 84. https://doi.org/10.3390/cancers10030084