The Complex Interplay between Chronic Inflammation, the Microbiome, and Cancer: Understanding Disease Progression and What We Can Do to Prevent It



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Inflammation and Disease
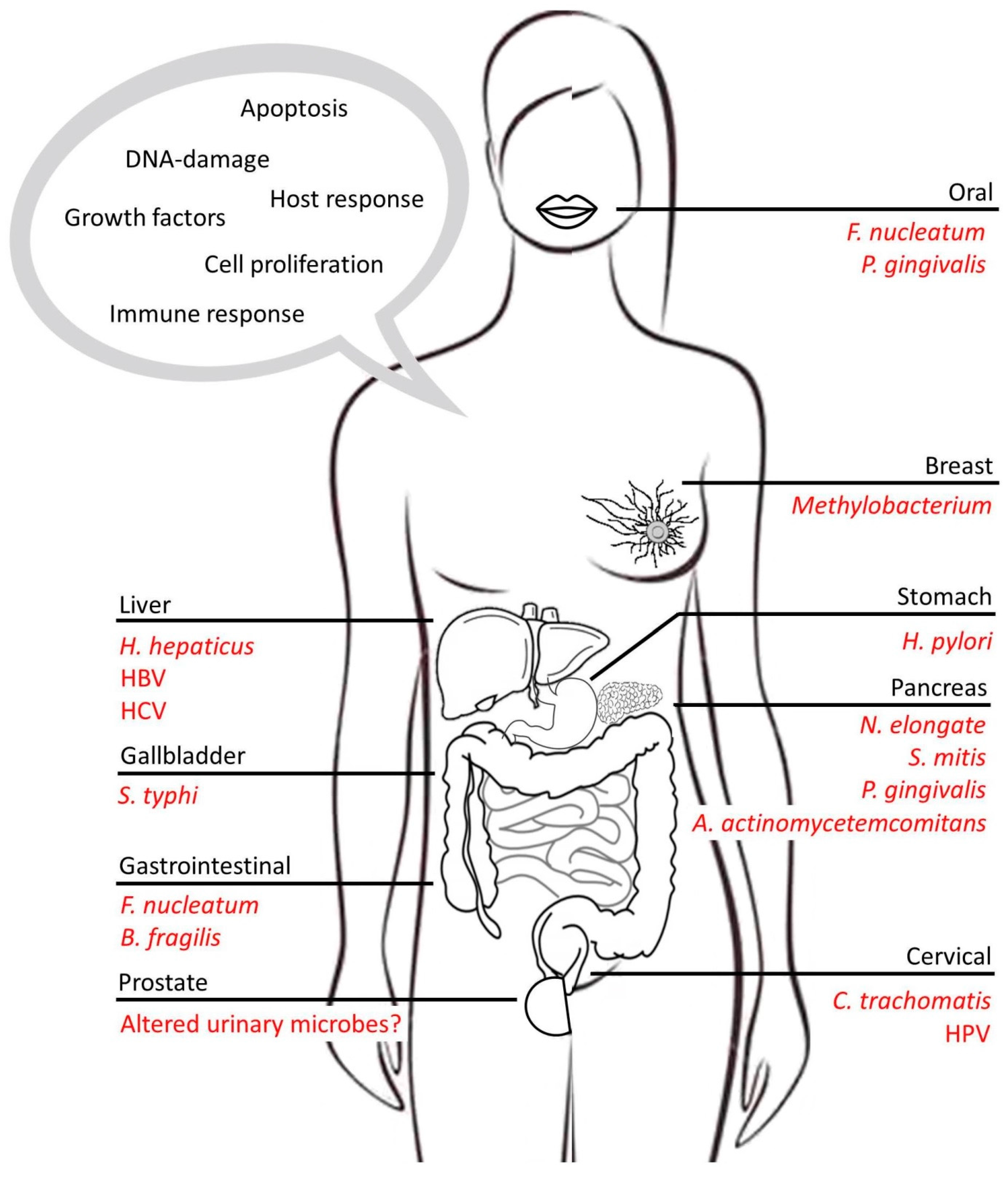
3. Microbes as Drivers of Chronic Inflammation: The Link to Cancer
3.1. Gastric Cancers
3.2. Liver Cancers
3.3. Pancreatic Cancers
3.4. Colorectal Cancer
3.5. Breast and Prostate Cancer
4. Microbes and Inflammation
5. Mechanism of Action of Microbes Associated with the Development of Cancer
5.1. Regulation of Immune Cells
5.2. Growth Factors
5.3. Promoting the Hallmarks of Cancer
6. Microbes and Inflammation in Cancer: Means of Treatment and Prevention
6.1. Response to Chemotherapy
6.2. Immunotherapy
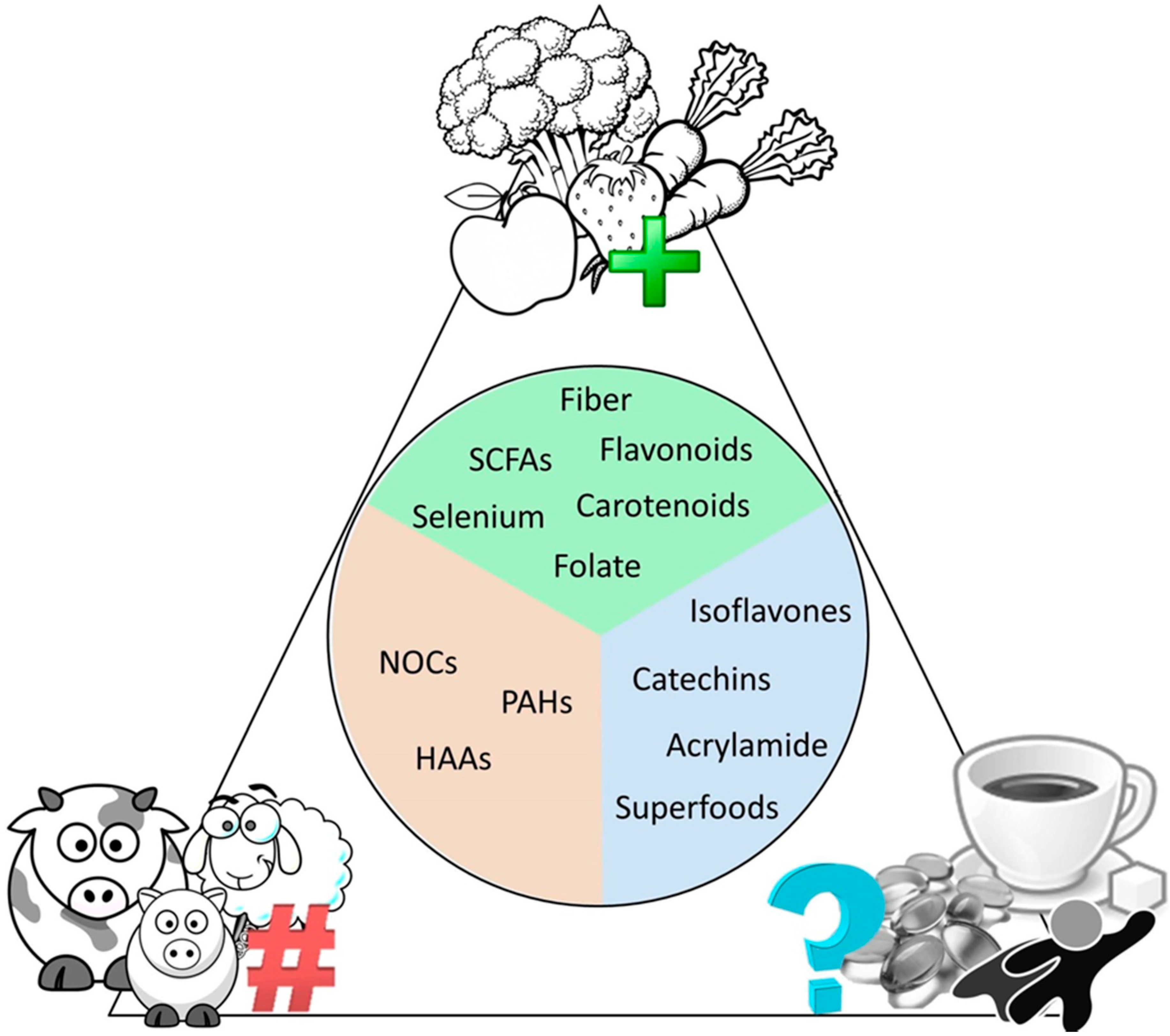
6.3. Diet as a Bridge between Microbes and Cancer
6.4. Common Dietary Misconceptions
7. The Take Home Message
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BFT | B. fragilis toxin |
| Cag | cytotoxin-associated gene |
| CDDL | cytidine deaminase |
| CDT | cytolethal distending toxin |
| CRC | colorectal cancer |
| CPT-11 | tissue carboxylesterase transforms Irinotecan |
| CTLA4 | anti–cytotoxic T-lymphocyte-associated protein 4 |
| CTX | cyclophosphamide |
| DII® | epidermal growth factor receptor |
| EFAs | essential fatty acids |
| HAAs | heterocyclic aromatic amines |
| HB-EGF | heparin-binding EGF-like growth factor |
| HBV | Hepatitis B |
| HCC | hepatocellular carcinoma |
| HCV | Hepatitis C |
| IBD | inflammatory bowel diseases |
| IL | interleukin |
| MALT | mucosa-associated lymphoid tissue |
| MAPK | mitogen-activated protein kinase |
| NF-κB | nuclear factor-κB |
| NK | natural killer |
| NOCs | N-nitroso compounds |
| NOX2 | NADPH oxidase 2 |
| PAHs | polycyclic aromatic hydrocarbons |
| PD-1 | programmed cell death protein 1 |
| PSA | polysaccharide A |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| SCFAs | short chain fatty acids |
| STAT3 | signal transducer and activator of transcription 3 |
| TGFβ | Transforming growth factor beta |
| Th | T helper |
| TAMs | tumor-associated macrophages |
| TNF | tumor necrosis factor |
References
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Lashinger, L.M.; Ford, N.A.; Hursting, S.D. Interacting inflammatory and growth factor signals underlie the obesity-cancer link. J. Nutr. 2014, 144, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kidane, D.; Chae, W.J.; Czochor, J.; Eckert, K.A.; Glazer, P.M.; Bothwell, A.L.; Sweasy, J.B. Interplay between DNA repair and inflammation, and the link to cancer. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 116–139. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Murata, M. Crosstalk between DNA damage and inflammation in the multiple steps of carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1808. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir. J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Grivennikov, S.I. Inflammation and colorectal cancer: Colitis-associated neoplasia. Semin. Immunopathol. 2013, 35, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer. Oncology 2002, 16, 217–226. [Google Scholar] [PubMed]
- Rubin, D.C.; Shaker, A.; Levin, M.S. Chronic intestinal inflammation: Inflammatory bowel disease and colitis-associated colon cancer. Front. Immunol. 2012, 3, 107. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef] [PubMed]
- Homey, B.; Muller, A.; Zlotnik, A. Chemokines: Agents for the immunotherapy of cancer? Nat. Rev. Immunol. 2002, 2, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A. Smad signalling network. J. Cell Sci. 2002, 115, 3355–3356. [Google Scholar] [PubMed]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Houghteling, P.D.; Walker, W.A. Why is initial bacterial colonization of the intestine important to infants’ and children’s health? J. Pediatr. Gastroenterol. Nutr. 2015, 60, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Walker, A. Intestinal colonization and programming of the intestinal immune response. J. Clin. Gastroenterol. 2014, 48 (Suppl. 1), S8–S11. [Google Scholar] [CrossRef] [PubMed]
- Fiebiger, U.; Bereswill, S.; Heimesaat, M.M. Dissecting the interplay between intestinal microbiota and host immunity in health and disease: Lessons learned from germfree and gnotobiotic animal models. Eur. J. Microbiol. Immunol. 2016, 6, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.S.; Yim, H.C.H. Microbial factors in inflammatory diseases and cancers. Adv. Exp. Med. Biol. 2017, 1024, 153–174. [Google Scholar] [PubMed]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Takeda, K. Role of gut microbiota in rheumatoid arthritis. J. Clin. Med. 2017, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Mellemkjaer, L.; Linet, M.S.; Gridley, G.; Frisch, M.; Moller, H.; Olsen, J.H. Rheumatoid arthritis and cancer risk. Eur. J. Cancer 1996, 32A, 1753–1757. [Google Scholar] [CrossRef]
- Sussman, D.A.; Santaolalla, R.; Strobel, S.; Dheer, R.; Abreu, M.T. Cancer in inflammatory bowel disease: Lessons from animal models. Curr. Opin. Gastroenterol. 2012, 28, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yu, Y.; Yue, Y.; Zhang, Z.; Su, K. Microbial infection and rheumatoid arthritis. J. Clin. Cell. Immunol. 2013, 4, 174. [Google Scholar] [PubMed]
- Fox, J.G.; Wang, T.C. Inflammation, atrophy, and gastric cancer. J. Clin. Investig. 2007, 117, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Meng, W.; Wang, B.; Qiao, L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014, 345, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Tegtmeyer, N.; Brandt, S.; Yamaoka, Y.; De Poire, E.; Sgouras, D.; Wessler, S.; Torres, J.; Smolka, A.; Backert, S. C-src and c-abl kinases control hierarchic phosphorylation and function of the caga effector protein in western and east asian helicobacter pylori strains. J. Clin. Investig. 2012, 122, 1553–1566. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Kwok, T.; Hartig, R.; Konig, W.; Backert, S. Nf-kappab activation and potentiation of proinflammatory responses by the helicobacter pylori caga protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9300–9305. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Israel, D.A.; Washington, M.K.; Krishna, U.; Fox, J.G.; Rogers, A.B.; Neish, A.S.; Collier-Hyams, L.; Perez-Perez, G.I.; Hatakeyama, M.; et al. Activation of beta-catenin by carcinogenic helicobacter pylori. Proc. Natl. Acad. Sci. USA 2005, 102, 10646–10651. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, A.M.; Held, M.; Hansson, L.E.; Engstrand, L.; Nyren, O. Helicobacter pylori in gastric cancer established by caga immunoblot as a marker of past infection. Gastroenterology 2001, 121, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Peek, R.M., Jr.; Crabtree, J.E. Helicobacter infection and gastric neoplasia. J. Pathol. 2006, 208, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, J.L.; Whary, M.T.; Ge, Z.; Muthupalani, S.; Taylor, N.S.; Mobley, M.; Potter, A.; Varro, A.; Eibach, D.; Suerbaum, S.; et al. Lack of commensal flora in helicobacter pylori-infected ins-gas mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 2011, 140, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Rickman, B.; Rogers, A.B.; Ge, Z.; Wang, T.C.; Fox, J.G. Helicobacter pylori eradication prevents progression of gastric cancer in hypergastrinemic ins-gas mice. Cancer Res. 2008, 68, 3540–3548. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.J.; Lin, J.C.; Tu, S.P. Etiology and prevention of gastric cancer. Gastrointest. Tumors 2016, 3, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Dang, S.; Hou, P. Gene methylation in gastric cancer. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 424, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Sharma, P.C. Next generation sequencing-based emerging trends in molecular biology of gastric cancer. Am. J. Cancer Res. 2018, 8, 207–225. [Google Scholar] [PubMed]
- Camargo, M.C.; Mera, R.; Correa, P.; Peek, R.M., Jr.; Fontham, E.T.; Goodman, K.J.; Piazuelo, M.B.; Sicinschi, L.; Zabaleta, J.; Schneider, B.G. Interleukin-1beta and interleukin-1 receptor antagonist gene polymorphisms and gastric cancer: A meta-analysis. Cancer Epidemiol. Biomark. 2006, 15, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- El-Omar, E.M.; Carrington, M.; Chow, W.H.; McColl, K.E.; Bream, J.H.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.C.; Rothman, N.; et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000, 404, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Puls, J.; Buhrdorf, R.; Gebert, B.; Odenbreit, S.; Haas, R. Systematic mutagenesis of the helicobacter pylori cag pathogenicity island: Essential genes for caga translocation in host cells and induction of interleukin-8. Mol. Microbiol. 2001, 42, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Krisch, L.M.; Posselt, G.; Hammerl, P.; Wessler, S. Caga phosphorylation in helicobacter pylori-infected b cells is mediated by the nonreceptor tyrosine kinases of the src and abl families. Infect. Immun. 2016, 84, 2671–2680. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: Globocan 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A. The role of inflammation and liver cancer. Adv. Exp. Med. Biol. 2014, 816, 401–435. [Google Scholar] [PubMed]
- Buchmann, P.; Dembek, C.; Kuklick, L.; Jager, C.; Tedjokusumo, R.; von Freyend, M.J.; Drebber, U.; Janowicz, Z.; Melber, K.; Protzer, U. A novel therapeutic hepatitis b vaccine induces cellular and humoral immune responses and breaks tolerance in hepatitis b virus (hbv) transgenic mice. Vaccine 2013, 31, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, Y.; Guidotti, L.G.; Kuhlen, C.V.; Fowler, P.; Chisari, F.V. Immune pathogenesis of hepatocellular carcinoma. J. Exp. Med. 1998, 188, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Feng, Y.; Theve, E.J.; Raczynski, A.R.; Fiala, J.L.; Doernte, A.L.; Williams, M.; McFaline, J.L.; Essigmann, J.M.; Schauer, D.B.; et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut 2010, 59, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Algul, H.; Treiber, M.; Lesina, M.; Schmid, R.M. Mechanisms of disease: Chronic inflammation and cancer in the pancreas—A potential role for pancreatic stellate cells? Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Whitcomb, D.C.; Shimosegawa, T.; Esposito, I.; Lerch, M.M.; Gress, T.; Mayerle, J.; Drewes, A.M.; Rebours, V.; Akisik, F.; et al. Chronic pancreatitis. Nat. Rev. Dis. Prim. 2017, 3, 17060. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Nakagawa, S.; Sawayama, H.; Ishimoto, T.; Imai, K.; Iwatsuki, M.; Hashimoto, D.; Baba, Y.; Yamashita, Y.I.; Yoshida, N.; et al. The microbiome and hepatobiliary-pancreatic cancers. Cancer Lett. 2017, 402, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2016. [Google Scholar] [CrossRef] [PubMed]
- Tenesa, A.; Dunlop, M.G. New insights into the aetiology of colorectal cancer from genome-wide association studies. Nat. Rev. Genet. 2009, 10, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Szigethy, E.; McLafferty, L.; Goyal, A. Inflammatory bowel disease. Child Adolesc. Psychiatr. Clin. N. Am. 2010, 19, 301–318. [Google Scholar] [CrossRef] [PubMed]
- De Souza, H.S.; Fiocchi, C. Immunopathogenesis of ibd: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Stokkers, P.C.; Hommes, D.W. New cytokine therapeutics for inflammatory bowel disease. Cytokine 2004, 28, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. Ikkbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Hou, V.; Grivennikov, S.I. Cytokines, ibd, and colitis-associated cancer. Inflamm. Bowel Dis. 2015, 21, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking tnf-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar] [PubMed]
- Atreya, R.; Mudter, J.; Finotto, S.; Mullberg, J.; Jostock, T.; Wirtz, S.; Schutz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of interleukin 6 trans signaling suppresses t-cell resistance against apoptosis in chronic intestinal inflammation: Evidence in crohn disease and experimental colitis in vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Kai, Y.; Takahashi, I.; Ishikawa, H.; Hiroi, T.; Mizushima, T.; Matsuda, C.; Kishi, D.; Hamada, H.; Tamagawa, H.; Ito, T.; et al. Colitis in mice lacking the common cytokine receptor gamma chain is mediated by il-6-producing CD4+ T cells. Gastroenterology 2005, 128, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Benjamin, J.L.; McCarthy, N.E.; Hedin, C.R.; Koutsoumpas, A.; Plamondon, S.; Price, C.L.; Hart, A.L.; Kamm, M.A.; Forbes, A.; et al. Relationship between human intestinal dendritic cells, gut microbiota, and disease activity in crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Izcue, A.; Hue, S.; Buonocore, S.; Arancibia-Carcamo, C.V.; Ahern, P.P.; Iwakura, Y.; Maloy, K.J.; Powrie, F. Interleukin-23 restrains regulatory t cell activity to drive t cell-dependent colitis. Immunity 2008, 28, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.H.; Kljavin, N.M.; Ota, N.; Leonard, J.; Roose-Girma, M.; Diehl, L.; Ouyang, W.; Ghilardi, N. Opposing consequences of il-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol. 2012, 5, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. Il-23 is essential for t cell-mediated colitis and promotes inflammation via il-17 and il-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Seiderer, J.; Elben, I.; Diegelmann, J.; Glas, J.; Stallhofer, J.; Tillack, C.; Pfennig, S.; Jurgens, M.; Schmechel, S.; Konrad, A.; et al. Role of the novel th17 cytokine il-17f in inflammatory bowel disease (ibd): Upregulated colonic il-17f expression in active crohn’s disease and analysis of the il17f p.His161arg polymorphism in ibd. Inflamm. Bowel Dis. 2008, 14, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. Il-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Altemus, J.; Niazi, F.; Green, H.; Calhoun, B.C.; Sturgis, C.; Grobmyer, S.R.; Eng, C. Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget 2017, 8, 88122–88138. [Google Scholar] [CrossRef] [PubMed]
- Mani, S. Microbiota and breast cancer. Prog. Mol. Biol. Transl. Sci. 2017, 151, 217–229. [Google Scholar] [PubMed]
- Sfanos, K.S.; Yegnasubramanian, S.; Nelson, W.G.; De Marzo, A.M. The inflammatory microenvironment and microbiome in prostate cancer development. Nat. Rev. Urol. 2018, 15, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Bhudia, R.; Ahmad, A.; Akpenyi, O.; Whiley, A.; Wilks, M.; Oliver, T. Identification of low oxygen-tolerating bacteria in prostate secretions of cancer patients and discussion of possible aetiological significance. Sci. Rep. 2017, 7, 15164. [Google Scholar] [CrossRef] [PubMed]
- Arora, H.C.; Eng, C.; Shoskes, D.A. Gut microbiome and chronic prostatitis/chronic pelvic pain syndrome. Ann. Transl. Med. 2017, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, E.; White, J.R.; Yu, S.H.; Kulac, I.; Ertunc, O.; De Marzo, A.M.; Yegnasubramanian, S.; Mangold, L.A.; Partin, A.W.; Sfanos, K.S. Profiling the urinary microbiome in men with positive versus negative biopsies for prostate cancer. J. Urol. 2018, 199, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Hou, V.; Grivennikov, S.I. Microbiome, inflammation, and cancer. Cancer J. 2014, 20, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Miquel, S.; Ulmer, J.; Kechaou, N.; Langella, P.; Bermudez-Humaran, L.G. Role of commensal and probiotic bacteria in human health: A focus on inflammatory bowel disease. Microb. Cell Fact. 2013, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.J.; Carneiro, M.B.; dos Anjos Pultz, B.; Pereira Silva, D.; Lopes, M.E.; dos Santos, L.M. The multifaceted role of commensal microbiota in homeostasis and gastrointestinal diseases. J. Immunol. Res. 2015, 2015, 321241. [Google Scholar] [CrossRef] [PubMed]
- Engering, A.; Hogerwerf, L.; Slingenbergh, J. Pathogen-host-environment interplay and disease emergence. Emerg. Microb. Infect. 2013, 2, e5. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Burcharth, J.; Pommergaard, H.C. Linking gut microbiota to colorectal cancer. J. Cancer 2017, 8, 3378–3395. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Zaidi, D.; Valcheva, R.; Jovel, J.; Martinez, I.; Sergi, C.; Walter, J.; Mason, A.L.; Wong, G.K.; Dieleman, L.A.; et al. Mucosal barrier depletion and loss of bacterial diversity are primary abnormalities in paediatric ulcerative colitis. J. Crohn’s Colitis 2016, 10, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.; Al-Hebshi, N.N.; Speicher, D.J.; Perera, I.; Johnson, N.W. Emerging role of bacteria in oral carcinogenesis: A review with special reference to perio-pathogenic bacteria. J. Oral Microbiol. 2016, 8, 32762. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Totsika, M.; Morrison, M.; Punyadeera, C. Oral microbiome: A new biomarker reservoir for oral and oropharyngeal cancers. Theranostics 2017, 7, 4313–4321. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.L. Bacteria and cancer: Cause, coincidence or cure? A review. J. Transl. Med. 2006, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Toprak, N.U.; Yagci, A.; Gulluoglu, B.M.; Akin, M.L.; Demirkalem, P.; Celenk, T.; Soyletir, G. A possible role of bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin. Microbiol. Infect. 2006, 12, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Hinuma, Y.; Nagata, K.; Hanaoka, M.; Nakai, M.; Matsumoto, T.; Kinoshita, K.I.; Shirakawa, S.; Miyoshi, I. Adult t-cell leukemia: Antigen in an atl cell line and detection of antibodies to the antigen in human sera. Proc. Natl. Acad. Sci. USA 1981, 78, 6476–6480. [Google Scholar] [CrossRef] [PubMed]
- Satou, Y.; Yasunaga, J.; Yoshida, M.; Matsuoka, M. Htlv-i basic leucine zipper factor gene mrna supports proliferation of adult T cell leukemia cells. Proc. Natl. Acad. Sci. USA 2006, 103, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Markowska, J.; Fischer, N.; Markowski, M.; Nalewaj, J. The role of chlamydia trachomatis infection in the development of cervical neoplasia and carcinoma. Medycyna Wieku Rozwojowego 2005, 9, 83–86. [Google Scholar] [PubMed]
- Burd, E.M. Human papillomavirus and cervical cancer. Clin. Microbiol. Rev. 2003, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Raab-Traub, N. Novel mechanisms of ebv-induced oncogenesis. Curr. Opin. Virol. 2012, 2, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, V.; Eslick, G.D. Systematic review with meta-analysis: The relationship between chronic salmonella typhi carrier status and gall-bladder cancer. Aliment. Pharmacol. Ther. 2014, 39, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.M.; Luo, T.; Kamarajan, P.; Fenno, J.C.; Rickard, A.H.; Kapila, Y.L. Microbial communities associated with primary and metastatic head and neck squamous cell carcinoma—A high fusobacterial and low streptococcal signature. Sci. Rep. 2017, 7, 9934. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.; Boshoff, C.; Lagos, D. Kaposi sarcoma as a model of oncogenesis and cancer treatment. Expert Rev. Anticancer Ther. 2007, 7, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Yakoob, J.; Abbas, Z.; Ahmad, Z.; Tariq, K.; Awan, S.; Mustafa, K.; Khan, R. Gastric lymphoma: Association with helicobacter pylori outer membrane protein q (hopq) and cytotoxic-pathogenicity activity island (cpai) genes. Epidemiol. Infect. 2017, 145, 3468–3476. [Google Scholar] [CrossRef] [PubMed]
- Goubran, H.A.; Kotb, R.R.; Stakiw, J.; Emara, M.E.; Burnouf, T. Regulation of tumor growth and metastasis: The role of tumor microenvironment. Cancer Growth Metastasis 2014, 7, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Bremnes, R.M.; Al-Shibli, K.; Donnem, T.; Sirera, R.; Al-Saad, S.; Andersen, S.; Stenvold, H.; Camps, C.; Busund, L.T. The role of tumor-infiltrating immune cells and chronic inflammation at the tumor site on cancer development, progression, and prognosis: Emphasis on non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Yang, K.; Wu, B.; Chen, H.; Chen, X.; Chen, X.; Jiang, L.; Ye, F.; He, D.; Lu, Z.; et al. Tumor-infiltrating immune cells are associated with prognosis of gastric cancer. Medicine 2015, 94, e1631. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Cao, Y.; Chan, A.T.; Qian, Z.R.; Nowak, J.A.; Masugi, Y.; Shi, Y.; Song, M.; da Silva, A.; Gu, M.; et al. Fusobacterium nucleatum in colorectal carcinoma tissue according to tumor location. Clin. Transl. Gastroenterol. 2016, 7, e200. [Google Scholar] [CrossRef] [PubMed]
- Sagiv-Barfi, I.; Czerwinski, D.K.; Levy, S.; Alam, I.S.; Mayer, A.T.; Gambhir, S.S.; Levy, R. Eradication of spontaneous malignancy by local immunotherapy. Sci. Transl. Med. 2018, 10, eaan4488. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, K.A.; Schulz, H.M.; Regner, E.H.; Severs, E.L.; Hendrickson, J.D.; Mehta, G.; Whitney, A.K.; Ir, D.; Ohri, N.; Robertson, C.E.; et al. Bacteroidales recruit il-6-producing intraepithelial lymphocytes in the colon to promote barrier integrity. Mucosal Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Falasca, K.; Ucciferri, C.; Dalessandro, M.; Zingariello, P.; Mancino, P.; Petrarca, C.; Pizzigallo, E.; Conti, P.; Vecchiet, J. Cytokine patterns correlate with liver damage in patients with chronic hepatitis b and c. Ann. Clin. Lab. Sci. 2006, 36, 144–150. [Google Scholar] [PubMed]
- Holzerlandt, R.; Orengo, C.; Kellam, P.; Alba, M.M. Identification of new herpesvirus gene homologs in the human genome. Genome Res. 2002, 12, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Shahanavaj, K.; Gil-Bazo, I.; Castiglia, M.; Bronte, G.; Passiglia, F.; Carreca, A.P.; del Pozo, J.L.; Russo, A.; Peeters, M.; Rolfo, C. Cancer and the microbiome: Potential applications as new tumor biomarker. Expert Rev. Anticancer Ther. 2015, 15, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Dechelotte, P.; Bonnet, M.; et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014, 63, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, G.; Cougnoux, A.; Delmas, J.; Darfeuille-Michaud, A.; Bonnet, R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2014, 5, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, N.; Lee, C. Mysteries of tgf-beta paradox in benign and malignant cells. Front. Oncol. 2014, 4, 94. [Google Scholar] [CrossRef] [PubMed]
- Menzies, B.E. The role of fibronectin binding proteins in the pathogenesis of staphylococcus aureus infections. Curr. Opin. Infect. Dis. 2003, 16, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ren, A.; Wang, X.; Fan, X.; Zhao, Y.; Gao, G.F.; Cleary, P.; Wang, B. Influenza viral neuraminidase primes bacterial coinfection through tgf-beta-mediated expression of host cell receptors. Proc. Natl. Acad. Sci. USA 2015, 112, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Bailey-Bucktrout, S.; Xi, Y.; Xu, D.; Du, D.; Zhang, Q.; Xiang, W.; Liu, J.; Melton, A.; Sheppard, D.; et al. Innate antiviral host defense attenuates tgf-beta function through irf3-mediated suppression of smad signaling. Mol. Cell 2014, 56, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. Emt, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.O.; Fang, Y.; Li, Z.; Chen, Z.; Xiang, J. Role of cellular cytoskeleton in epithelial-mesenchymal transition process during cancer progression. Biomed. Rep. 2015, 3, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Duncan, R. Prostate cancer cell migration induced by myopodin isoforms is associated with formation of morphologically and biochemically distinct actin networks. FASEB J. 2013, 27, 5046–5058. [Google Scholar] [CrossRef] [PubMed]
- Haynes, J.; Srivastava, J.; Madson, N.; Wittmann, T.; Barber, D.L. Dynamic actin remodeling during epithelial-mesenchymal transition depends on increased moesin expression. Mol. Biol. Cell 2011, 22, 4750–4764. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Buret, A.; Gall, D.G.; Olson, M.E.; Hardin, J.A. The role of the epidermal growth factor receptor in microbial infections of the gastrointestinal tract. Microbes Infect. 1999, 1, 1139–1144. [Google Scholar] [CrossRef]
- Gunawardhana, N.; Jang, S.; Choi, Y.H.; Hong, Y.A.; Jeon, Y.E.; Kim, A.; Su, H.; Kim, J.H.; Yoo, Y.J.; Merrell, D.S.; et al. Helicobacter pylori-induced hb-egf upregulates gastrin expression via the egf receptor, c-raf, mek1, and erk2 in the mapk pathway. Front. Cell. Infect. Microbiol. 2017, 7, 541. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. Shp-2 tyrosine phosphatase as an intracellular target of helicobacter pylori caga protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Kwok, T.; Zabler, D.; Urman, S.; Rohde, M.; Hartig, R.; Wessler, S.; Misselwitz, R.; Berger, J.; Sewald, N.; Konig, W.; et al. Helicobacter exploits integrin for type iv secretion and kinase activation. Nature 2007, 449, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Ahn, J.H.; Park, S.H.; Do, K.H.; Kim, J.; Moon, Y. Enhanced wound healing by recombinant escherichia coli nissle 1917 via human epidermal growth factor receptor in human intestinal epithelial cells: Therapeutic implication using recombinant probiotics. Infect. Immun. 2012, 80, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Dzutsev, A.; Badger, J.H.; Perez-Chanona, E.; Roy, S.; Salcedo, R.; Smith, C.K.; Trinchieri, G. Microbes and cancer. Annu. Rev. Immunol. 2017, 35, 199–228. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of t helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.I.; Yatsunenko, T.; Manary, M.J.; Trehan, I.; Mkakosya, R.; Cheng, J.; Kau, A.L.; Rich, S.S.; Concannon, P.; Mychaleckyj, J.C.; et al. Gut microbiomes of malawian twin pairs discordant for kwashiorkor. Science 2013, 339, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Carbonero, F.; Benefiel, A.C.; Alizadeh-Ghamsari, A.H.; Gaskins, H.R. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 2012, 3, 448. [Google Scholar] [CrossRef] [PubMed]
- Huycke, M.M.; Gaskins, H.R. Commensal bacteria, redox stress, and colorectal cancer: Mechanisms and models. Exp. Biol. Med. 2004, 229, 586–597. [Google Scholar] [CrossRef]
- Wang, X.; Huycke, M.M. Extracellular superoxide production by enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology 2007, 132, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Moore, D.R.; Nimmo, S.L.; Lightfoot, S.A.; Huycke, M.M. 4-hydroxy-2-nonenal mediates genotoxicity and bystander effects caused by enterococcus faecalis-infected macrophages. Gastroenterology 2012, 142, 543–551.e7. [Google Scholar] [CrossRef] [PubMed]
- Nesic, D.; Hsu, Y.; Stebbins, C.E. Assembly and function of a bacterial genotoxin. Nature 2004, 429, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Simanski, M.; Rademacher, F.; Schroder, L.; Glaser, R.; Harder, J. The inflammasome and the epidermal growth factor receptor (egfr) are involved in the staphylococcus aureus-mediated induction of il-1alpha and il-1beta in human keratinocytes. PLoS ONE 2016, 11, e0147118. [Google Scholar] [CrossRef] [PubMed]
- Breshears, L.M.; Schlievert, P.M.; Peterson, M.L. A disintegrin and metalloproteinase 17 (adam17) and epidermal growth factor receptor (egfr) signaling drive the epithelial response to staphylococcus aureus toxic shock syndrome toxin-1 (tsst-1). J. Biol. Chem. 2012, 287, 32578–32587. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.I.; Seaghdha, M.O.; Prince, A.S. Staphylococcus aureus protein a activates tace through egfr-dependent signaling. EMBO J. 2007, 26, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, K.; Schwichtenberg, L.; Schroder, J.M.; Harder, J. Pseudomonas aeruginosa- and il-1beta-mediated induction of human beta-defensin-2 in keratinocytes is controlled by nf-kappab and ap-1. J. Investig. Dermatol. 2006, 126, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and atp. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.S.; Pietras, E.M.; Uricchio, L.H.; Hirano, K.; Rao, S.; Lin, H.; O’Connell, R.M.; Iwakura, Y.; Cheung, A.L.; Cheng, G.; et al. Inflammasome-mediated production of il-1beta is required for neutrophil recruitment against staphylococcus aureus in vivo. J. Immunol. 2007, 179, 6933–6942. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Franchi, L.; Miller, L.S.; Nunez, G. A critical role for hemolysins and bacterial lipoproteins in staphylococcus aureus-induced activation of the nlrp3 inflammasome. J. Immunol. 2009, 183, 3942–3948. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Park, B.G.; Wolf, A.J.; Brikos, C.; Goodridge, H.S.; Becker, C.A.; Reyes, C.N.; Miao, E.A.; Aderem, A.; Gotz, F.; et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and il-1beta secretion. Cell Host Microbe 2010, 7, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberger, B.M.; Gerber, P.A.; Holcmann, M.; Buhren, B.A.; Amberg, N.; Smolle, V.; Schrumpf, H.; Boelke, E.; Ansari, P.; Mackenzie, C.; et al. Epidermal egfr controls cutaneous host defense and prevents inflammation. Sci. Transl. Med. 2013, 5, 199ra111. [Google Scholar] [CrossRef] [PubMed]
- Eilers, R.E., Jr.; Gandhi, M.; Patel, J.D.; Mulcahy, M.F.; Agulnik, M.; Hensing, T.; Lacouture, M.E. Dermatologic infections in cancer patients treated with epidermal growth factor receptor inhibitor therapy. J. Natl. Cancer Inst. 2010, 102, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Adam17, shedding, tace as therapeutic targets. Pharmacol. Res. 2013, 71, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Bartok, E.; Bauernfeind, F.; Khaminets, M.G.; Jakobs, C.; Monks, B.; Fitzgerald, K.A.; Latz, E.; Hornung, V. Igluc: A luciferase-based inflammasome and protease activity reporter. Nat. Methods 2013, 10, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs—Phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar] [PubMed]
- Chen, X.; Wu, Y.; Dong, H.; Zhang, C.Y.; Zhang, Y. Platinum-based agents for individualized cancer treatment. Curr. Mol. Med. 2013, 13, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Gao, C.; Liu, Q.; Yu, C.; Zhang, Z.; Cai, L.; Yang, B.; Qian, Y.; Yang, J.; Liao, X. Research progress in modern structure of platinum complexes. Eur. J. Med. Chem. 2017, 140, 349–382. [Google Scholar] [CrossRef] [PubMed]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Ryals, M.M.; Van den Bruele, A.B.; Fitzgerald, T.S.; Cunningham, L.L. Sound preconditioning therapy inhibits ototoxic hearing loss in mice. J. Clin. Investig. 2013, 123, 4945–4949. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Pabla, N.; Tang, C.; He, L.; Dong, Z. DNA damage response in cisplatin-induced nephrotoxicity. Arch. Toxicol. 2015, 89, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Goldstein, D.; Krishnan, A.V.; Lin, C.S.; Friedlander, M.L.; Cassidy, J.; Koltzenburg, M.; Kiernan, M.C. Chemotherapy-induced peripheral neurotoxicity: A critical analysis. CA Cancer J. Clin. 2013, 63, 419–437. [Google Scholar] [CrossRef] [PubMed]
- Gui, Q.F.; Lu, H.F.; Zhang, C.X.; Xu, Z.R.; Yang, Y.H. Well-balanced commensal microbiota contributes to anti-cancer response in a lung cancer mouse model. Genet. Mol. Res. 2015, 14, 5642–5651. [Google Scholar] [CrossRef] [PubMed]
- Chitapanarux, I.; Chitapanarux, T.; Traisathit, P.; Kudumpee, S.; Tharavichitkul, E.; Lorvidhaya, V. Randomized controlled trial of live lactobacillus acidophilus plus bifidobacterium bifidum in prophylaxis of diarrhea during radiotherapy in cervical cancer patients. Radiat. Oncol. 2010, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, R.K. Gut microbiota and hepatic encephalopathy. Metab. Brain Dis. 2013, 28, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Spanogiannopoulos, P.; Bess, E.N.; Carmody, R.N.; Turnbaugh, P.J. The microbial pharmacists within us: A metagenomic view of xenobiotic metabolism. Nat. Rev. Microbiol. 2016, 14, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Sparreboom, A. Pharmacogenetics of irinotecan disposition and toxicity: A review. Curr. Clin. Pharmacol. 2010, 5, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Stringer, A.M.; Gibson, R.J.; Logan, R.M.; Bowen, J.M.; Yeoh, A.S.; Keefe, D.M. Faecal microflora and beta-glucuronidase expression are altered in an irinotecan-induced diarrhea model in rats. Cancer Biol. Ther. 2008, 7, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, F.M.; Maison, N.; Holtrop, G.; Young, P.; Stevens, V.J.; Ince, J.; Johnstone, A.M.; Lobley, G.E.; Flint, H.J.; Louis, P. Phylogenetic distribution of genes encoding beta-glucuronidase activity in human colonic bacteria and the impact of diet on faecal glycosidase activities. Environ. Microbiol. 2012, 14, 1876–1887. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.D.; Wang, H.; Lane, K.T.; Scott, J.E.; Orans, J.; Koo, J.S.; Venkatesh, M.; Jobin, C.; Yeh, L.A.; Mani, S.; et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 2010, 330, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jia, W. Cometabolism of microbes and host: Implications for drug metabolism and drug-induced toxicity. Clin. Pharmacol. Ther. 2013, 94, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Maurice, C.F.; Haiser, H.J.; Turnbaugh, P.J. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 2013, 152, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Oudejans, J.J.; van den Brule, A.J.; Jiwa, N.M.; de Bruin, P.C.; Ossenkoppele, G.J.; van der Valk, P.; Walboomers, J.M.; Meijer, C.J. Bhrf1, the epstein-barr virus (ebv) homologue of the bcl-2 protooncogene, is transcribed in ebv-associated b-cell lymphomas and in reactive lymphocytes. Blood 1995, 86, 1893–1902. [Google Scholar] [PubMed]
- Hatton, O.L.; Harris-Arnold, A.; Schaffert, S.; Krams, S.M.; Martinez, O.M. The interplay between epstein-barr virus and b lymphocytes: Implications for infection, immunity, and disease. Immunol. Res. 2014, 58, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Wei, A.H.; Fletcher, J.I.; Willis, S.N.; Chen, L.; Roberts, A.W.; Huang, D.C.; Colman, P.M. Structural basis for apoptosis inhibition by epstein-barr virus bhrf1. PLoS Pathog. 2010, 6, e1001236. [Google Scholar] [CrossRef] [PubMed]
- Procko, E.; Berguig, G.Y.; Shen, B.W.; Song, Y.; Frayo, S.; Convertine, A.J.; Margineantu, D.; Booth, G.; Correia, B.E.; Cheng, Y.; et al. A computationally designed inhibitor of an epstein-barr viral bcl-2 protein induces apoptosis in infected cells. Cell 2014, 157, 1644–1656. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillere, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Zwielehner, J.; Lassl, C.; Hippe, B.; Pointner, A.; Switzeny, O.J.; Remely, M.; Kitzweger, E.; Ruckser, R.; Haslberger, A.G. Changes in human fecal microbiota due to chemotherapy analyzed by taqman-pcr, 454 sequencing and pcr-dgge fingerprinting. PLoS ONE 2011, 6, e28654. [Google Scholar] [CrossRef] [PubMed]
- Daillere, R.; Vetizou, M.; Waldschmitt, N.; Yamazaki, T.; Isnard, C.; Poirier-Colame, V.; Duong, C.P.M.; Flament, C.; Lepage, P.; Roberti, M.P.; et al. Enterococcus hirae and barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity 2016, 45, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti-pd-l1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by ctla-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of pd-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-pd-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct cellular mechanisms underlie anti-ctla-4 and anti-pd-1 checkpoint blockade. Cell 2017, 170, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Thiele Orberg, E.; Geis, A.L.; Chan, J.L.; Fu, K.; Shields, C.E.D.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe 2018, 23, 203–214 e205. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.R.; Abar, L.; Vingeliene, S.; Chan, D.S.; Aune, D.; Navarro-Rosenblatt, D.; Stevens, C.; Greenwood, D.; Norat, T. Fruits, vegetables and lung cancer risk: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Qin, S.; Zhang, T.; Song, X.; Zhang, S. The effect of fruit and vegetable intake on the development of lung cancer: A meta-analysis of 32 publications and 20,414 cases. Eur. J. Clin. Nutr. 2015, 69, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, J.; Leng, Y.; Lv, C. Intake of fruit and vegetables and risk of esophageal squamous cell carcinoma: A meta-analysis of observational studies. Int. J. Cancer 2013, 133, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Maasland, D.H.; van den Brandt, P.A.; Kremer, B.; Goldbohm, R.A.; Schouten, L.J. Consumption of vegetables and fruits and risk of subtypes of head-neck cancer in the netherlands cohort study. Int. J. Cancer 2015, 136, E396–E409. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Bergkvist, L.; Wolk, A. Fruit and vegetable consumption and incidence of gastric cancer: A prospective study. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1998–2001. [Google Scholar] [CrossRef] [PubMed]
- Lunet, N.; Valbuena, C.; Vieira, A.L.; Lopes, C.; Lopes, C.; David, L.; Carneiro, F.; Barros, H. Fruit and vegetable consumption and gastric cancer by location and histological type: Case-control and meta-analysis. Eur. J. Cancer Prev. 2007, 16, 312–327. [Google Scholar] [CrossRef] [PubMed]
- Slavin, J.L.; Lloyd, B. Health benefits of fruits and vegetables. Adv. Nutr. 2012, 3, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.E.; Jacobs, E.T.; Baron, J.A.; Marshall, J.R.; Byers, T. Dietary supplements and cancer prevention: Balancing potential benefits against proven harms. J. Natl. Cancer Inst. 2012, 104, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012, CD007176. [Google Scholar] [CrossRef] [PubMed]
- Hehemann, J.H.; Correc, G.; Barbeyron, T.; Helbert, W.; Czjzek, M.; Michel, G. Transfer of carbohydrate-active enzymes from marine bacteria to japanese gut microbiota. Nature 2010, 464, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Vaikundamoorthy, R.; Krishnamoorthy, V.; Vilwanathan, R.; Rajendran, R. Structural characterization and anticancer activity (mcf7 and mda-mb-231) of polysaccharides fractionated from brown seaweed sargassum wightii. Int. J. Biol. Macromol. 2018, 111, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Abedin-Do, A.; Taherian-Esfahani, Z.; Ghafouri-Fard, S.; Ghafouri-Fard, S.; Motevaseli, E. Immunomodulatory effects of lactobacillus strains: Emphasis on their effects on cancer cells. Immunotherapy 2015, 7, 1307–1329. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chan, H.M.; Kubow, S. Kefir extracts suppress in vitro proliferation of estrogen-dependent human breast cancer cells but not normal mammary epithelial cells. J. Med. Food 2007, 10, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Fujiya, M.; Tanaka, H.; Ueno, N.; Moriichi, K.; Sasajima, J.; Ikuta, K.; Akutsu, H.; Tanabe, H.; Kohgo, Y. Probiotic-derived ferrichrome inhibits colon cancer progression via jnk-mediated apoptosis. Nat. Commun. 2016, 7, 12365. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, T.; Sequoia, J. Probiotics for gastrointestinal conditions: A summary of the evidence. Am. Fam. Phys. 2017, 96, 170–178. [Google Scholar]
- Chan, D.S.; Lau, R.; Aune, D.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Red and processed meat and colorectal cancer incidence: Meta-analysis of prospective studies. PLoS ONE 2011, 6, e20456. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Wolk, A. Meat consumption and risk of colorectal cancer: A meta-analysis of prospective studies. Int. J. Cancer 2006, 119, 2657–2664. [Google Scholar] [CrossRef] [PubMed]
- Norat, T.; Bingham, S.; Ferrari, P.; Slimani, N.; Jenab, M.; Mazuir, M.; Overvad, K.; Olsen, A.; Tjonneland, A.; Clavel, F.; et al. Meat, fish, and colorectal cancer risk: The european prospective investigation into cancer and nutrition. J. Natl. Cancer Inst. 2005, 97, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Loomis, D.; Guyton, K.Z.; Grosse, Y.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Mattock, H.; Straif, K.; International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 2015, 16, 1599–1600. [Google Scholar] [CrossRef]
- Larsson, S.C.; Wolk, A. Red and processed meat consumption and risk of pancreatic cancer: Meta-analysis of prospective studies. Br. J. Cancer 2012, 106, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Yang, X.; Zhang, C.; Zhu, C.; Tao, G.; Zhao, L.; Tang, S.; Shu, Z.; Cai, J.; Dai, S.; et al. Red and processed meat intake is associated with higher gastric cancer risk: A meta-analysis of epidemiological observational studies. PLoS ONE 2013, 8, e70955. [Google Scholar] [CrossRef] [PubMed]
- Bylsma, L.C.; Alexander, D.D. A review and meta-analysis of prospective studies of red and processed meat, meat cooking methods, heme iron, heterocyclic amines and prostate cancer. Nutr. J. 2015, 14, 125. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.J.; Pollock, J.R.; Bingham, S.A. Haem, not protein or inorganic iron, is responsible for endogenous intestinal n-nitrosation arising from red meat. Cancer Res. 2003, 63, 2358–2360. [Google Scholar] [PubMed]
- Sinha, R.; Peters, U.; Cross, A.J.; Kulldorff, M.; Weissfeld, J.L.; Pinsky, P.F.; Rothman, N.; Hayes, R.B. Meat, meat cooking methods and preservation, and risk for colorectal adenoma. Cancer Res. 2005, 65, 8034–8041. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Bingham, S.A. Dietary fibre, fermentation and large bowel cancer. Cancer Surv. 1987, 6, 601–621. [Google Scholar] [PubMed]
- van der Beek, C.M.; Dejong, C.H.C.; Troost, F.J.; Masclee, A.A.M.; Lenaerts, K. Role of short-chain fatty acids in colonic inflammation, carcinogenesis, and mucosal protection and healing. Nutr. Rev. 2017, 75, 286–305. [Google Scholar] [CrossRef] [PubMed]
- Sivaprakasam, S.; Bhutia, Y.D.; Yang, S.; Ganapathy, V. Short-chain fatty acid transporters: Role in colonic homeostasis. Compr. Physiol. 2017, 8, 299–314. [Google Scholar] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor gpr43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Weitkunat, K.; Schumann, S.; Petzke, K.J.; Blaut, M.; Loh, G.; Klaus, S. Effects of dietary inulin on bacterial growth, short-chain fatty acid production and hepatic lipid metabolism in gnotobiotic mice. J. Nutr. Biochem. 2015, 26, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Bindels, L.B.; Porporato, P.; Dewulf, E.M.; Verrax, J.; Neyrinck, A.M.; Martin, J.C.; Scott, K.P.; Buc Calderon, P.; Feron, O.; Muccioli, G.G.; et al. Gut microbiota-derived propionate reduces cancer cell proliferation in the liver. Br. J. Cancer 2012, 107, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Dong, T.S.; Dalal, S.R.; Wu, F.; Bissonnette, M.; Kwon, J.H.; Chang, E.B. The microbe-derived short chain fatty acid butyrate targets mirna-dependent p21 gene expression in human colon cancer. PLoS ONE 2011, 6, e16221. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nosho, K.; Shima, K.; Baba, Y.; Irahara, N.; Kirkner, G.J.; Hazra, A.; De Vivo, I.; Giovannucci, E.L.; Meyerhardt, J.A.; et al. P21 expression in colon cancer and modifying effects of patient age and body mass index on prognosis. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2513–2521. [Google Scholar] [CrossRef] [PubMed]
- Aune, D.; Chan, D.S.; Lau, R.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Dietary fibre, whole grains, and risk of colorectal cancer: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2011, 343, d6617. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Holtzclaw, W.D.; Wehage, S.L.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Sulforaphane bioavailability from glucoraphanin-rich broccoli: Control by active endogenous myrosinase. PLoS ONE 2015, 10, e0140963. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Wehage, S.L.; Holtzclaw, W.D.; Kensler, T.W.; Egner, P.A.; Shapiro, T.A.; Talalay, P. Protection of humans by plant glucosinolates: Efficiency of conversion of glucosinolates to isothiocyanates by the gastrointestinal microflora. Cancer Prev. Res. 2012, 5, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, W.; Li, J.; Zhang, J.; Wang, X.; Wang, M. Sulforaphane reverses gefitinib tolerance in human lung cancer cells via modulation of sonic hedgehog signaling. Oncol. Lett. 2018, 15, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Egner, P.A.; Chen, J.G.; Wang, J.B.; Wu, Y.; Sun, Y.; Lu, J.H.; Zhu, J.; Zhang, Y.H.; Chen, Y.S.; Friesen, M.D.; et al. Bioavailability of sulforaphane from two broccoli sprout beverages: Results of a short-term, cross-over clinical trial in qidong, china. Cancer Prev. Res. 2011, 4, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.L.B.L.; He, C.X.; Zhao, Y.J.; Yang, X.L.; Pang, B.; Zhang, X.H.; Shan, Y.J. Sulforaphane inhibits human bladder cancer cell invasion by reversing epithelial-to-mesenchymal transition via directly targeting microrna-200c/zeb1 axis. J. Funct. Foods 2018, 41, 118–126. [Google Scholar] [CrossRef]
- Hullar, M.A.; Lancaster, S.M.; Li, F.; Tseng, E.; Beer, K.; Atkinson, C.; Wahala, K.; Copeland, W.K.; Randolph, T.W.; Newton, K.M.; et al. Enterolignan-producing phenotypes are associated with increased gut microbial diversity and altered composition in premenopausal women in the united states. Cancer Epidemiol. Biomark. Prev. 2015, 24, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Ward, H.A.; Kuhnle, G.G.; Mulligan, A.A.; Lentjes, M.A.; Luben, R.N.; Khaw, K.T. Breast, colorectal, and prostate cancer risk in the european prospective investigation into cancer and nutrition-norfolk in relation to phytoestrogen intake derived from an improved database. Am. J. Clin. Nutr. 2010, 91, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Praengam, K.; Sahasakul, Y.; Kupradinun, P.; Sakarin, S.; Sanitchua, W.; Rungsipipat, A.; Rattanapinyopituk, K.; Angkasekwinai, P.; Changsri, K.; Mhuantong, W.; et al. Brown rice and retrograded brown rice alleviate inflammatory response in dextran sulfate sodium (dss)-induced colitis mice. Food Funct. 2017, 8, 4630–4643. [Google Scholar] [CrossRef] [PubMed]
- Shivappa, N.; Godos, J.; Hebert, J.R.; Wirth, M.D.; Piuri, G.; Speciani, A.F.; Grosso, G. Dietary inflammatory index and colorectal cancer risk-a meta-analysis. Nutrients 2017, 9, 1043. [Google Scholar] [CrossRef] [PubMed]
- Shivappa, N.; Steck, S.E.; Hurley, T.G.; Hussey, J.R.; Hebert, J.R. Designing and developing a literature-derived, population-based dietary inflammatory index. Public Health Nutr. 2014, 17, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Vance, T.M.; Chun, O.K. Estimated intake and major food sources of flavonoids among us adults: Changes between 1999–2002 and 2007–2010 in nhanes. Eur. J. Nutr. 2016, 55, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, U.; Arias, N.; Boque, N.; Macarulla, M.T.; Portillo, M.P.; Martinez, J.A.; Milagro, F.I. Reshaping faecal gut microbiota composition by the intake of trans-resveratrol and quercetin in high-fat sucrose diet-fed rats. J. Nutr. Biochem. 2015, 26, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liu, L.; Ji, H.F. Regulative effects of curcumin spice administration on gut microbiota and its pharmacological implications. Food Nutr. Res. 2017, 61, 1361780. [Google Scholar] [CrossRef] [PubMed]
- McFadden, R.M.; Larmonier, C.B.; Shehab, K.W.; Midura-Kiela, M.; Ramalingam, R.; Harrison, C.A.; Besselsen, D.G.; Chase, J.H.; Caporaso, J.G.; Jobin, C.; et al. The role of curcumin in modulating colonic microbiota during colitis and colon cancer prevention. Inflamm. Bowel Dis. 2015, 21, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Shim, Y.Y.; Cha, S.K.; Reaney, M.J.; Chee, K.M. Effect of lactobacillus acidophilus kfri342 on the development of chemically induced precancerous growths in the rat colon. J. Med. Microbiol. 2012, 61, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Guallar, E.; Stranges, S.; Mulrow, C.; Appel, L.J.; Miller, E.R., 3rd. Enough is enough: Stop wasting money on vitamin and mineral supplements. Ann. Intern. Med. 2013, 159, 850–851. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Gluud, L.L.; Nikolova, D.; Whitfield, K.; Krstic, G.; Wetterslev, J.; Gluud, C. Vitamin d supplementation for prevention of cancer in adults. Cochrane Database Syst. Rev. 2014, CD007469. [Google Scholar] [CrossRef] [PubMed]
- Vinceti, M.; Filippini, T.; Del Giovane, C.; Dennert, G.; Zwahlen, M.; Brinkman, M.; Zeegers, M.P.; Horneber, M.; D’Amico, R.; Crespi, C.M. Selenium for preventing cancer. Cochrane Database Syst. Rev. 2018, 1, CD005195. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Jofré, M.; Rueda, J.R.; Corsini-Muñoz, G.; Fonseca-Cortés, C.; Caraballoso, M.; Bonfill Cosp, X. Antioxidant drugs for preventing lung cancer in healthy people. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for preventing gastrointestinal cancers. Cochrane Database Syst. Rev. 2008, CD004183. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Kang, J.; Zhang, D. Microbial production of vitamin b12: A review and future perspectives. Microb. Cell Fact. 2017, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Reid, I.R.; Ibbertson, H.K. Calcium supplements in the prevention of steroid-induced osteoporosis. Am. J. Clin. Nutr. 1986, 44, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Valentin, M.; Coste Mazeau, P.; Zerah, M.; Ceccaldi, P.F.; Benachi, A.; Luton, D. Acid folic and pregnancy: A mandatory supplementation. Ann. D’endocrinol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Forrest, K.Y.; Stuhldreher, W.L. Prevalence and correlates of vitamin d deficiency in us adults. Nutr. Res. 2011, 31, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E. Acrylamide carcinogenicity. J. Agric. Food Chem. 2008, 56, 5984–5988. [Google Scholar] [CrossRef] [PubMed]
- Dearfield, K.L.; Abernathy, C.O.; Ottley, M.S.; Brantner, J.H.; Hayes, P.F. Acrylamide: Its metabolism, developmental and reproductive effects, genotoxicity, and carcinogenicity. Mutat. Res. 1988, 195, 45–77. [Google Scholar] [CrossRef]
- Bian, X.; Tu, P.; Chi, L.; Gao, B.; Ru, H.; Lu, K. Saccharin induced liver inflammation in mice by altering the gut microbiota and its metabolic functions. Food Chem. Toxicol. 2017, 107, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Soffritti, M.; Padovani, M.; Tibaldi, E.; Falcioni, L.; Manservisi, F.; Lauriola, M.; Belpoggi, F. Sucralose administered in feed, beginning prenatally through lifespan, induces hematopoietic neoplasias in male swiss mice. Int. J. Occup. Environ. Health 2016, 22, 7–17. [Google Scholar]
- Wang, J.L.; Lin, Y.W.; Chen, H.M.; Kong, X.; Xiong, H.; Shen, N.; Hong, J.; Fang, J.Y. Calcium prevents tumorigenesis in a mouse model of colorectal cancer. PLoS ONE 2011, 6, e22566. [Google Scholar] [CrossRef] [PubMed]
- Virk-Baker, M.K.; Nagy, T.R.; Barnes, S.; Groopman, J. Dietary acrylamide and human cancer: A systematic review of literature. Nutr. Cancer 2014, 66, 774–790. [Google Scholar] [CrossRef] [PubMed]
- Lipworth, L.; Sonderman, J.S.; Tarone, R.E.; McLaughlin, J.K. Review of epidemiologic studies of dietary acrylamide intake and the risk of cancer. Eur. J. Cancer Prev. 2012, 21, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Lohner, S.; Toews, I.; Meerpohl, J.J. Health outcomes of non-nutritive sweeteners: Analysis of the Research landscape. Nutr. J. 2017, 16, 55. [Google Scholar] [CrossRef] [PubMed]
- Bristow, S.M.; Bolland, M.J.; MacLennan, G.S.; Avenell, A.; Grey, A.; Gamble, G.D.; Reid, I.R. Calcium supplements and cancer risk: A meta-analysis of randomised controlled trials. Br. J. Nutr. 2013, 110, 1384–1393. [Google Scholar] [CrossRef] [PubMed]
- Duda-Chodak, A.; Wajda, L.; Tarko, T.; Sroka, P.; Satora, P. A review of the interactions between acrylamide, microorganisms and food components. Food Funct. 2016, 7, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.J.; Abed, R.M.M. Biodegradation of polyacrylamide and its derivatives. Environ. Process. 2017, 4, 463–476. [Google Scholar]
- Suez, J.; Korem, T.; Zeevi, D.; Zilberman-Schapira, G.; Thaiss, C.A.; Maza, O.; Israeli, D.; Zmora, N.; Gilad, S.; Weinberger, A.; et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 2014, 514, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Uebanso, T.; Ohnishi, A.; Kitayama, R.; Yoshimoto, A.; Nakahashi, M.; Shimohata, T.; Mawatari, K.; Takahashi, A. Effects of low-dose non-caloric sweetener consumption on gut microbiota in mice. Nutrients 2017, 9, 560. [Google Scholar] [CrossRef] [PubMed]
- Palmnas, M.S.; Cowan, T.E.; Bomhof, M.R.; Su, J.; Reimer, R.A.; Vogel, H.J.; Hittel, D.S.; Shearer, J. Low-dose aspartame consumption differentially affects gut microbiota-host metabolic interactions in the diet-induced obese rat. PLoS ONE 2014, 9, e109841. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Chi, L.; Gao, B.; Tu, P.; Ru, H.; Lu, K. The artificial sweetener acesulfame potassium affects the gut microbiome and body weight gain in cd-1 mice. PLoS ONE 2017, 12, e0178426. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, D.C. Calcium signalling in bacteria. Mol. Microbiol. 2004, 54, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Fischer, J.; Krewer, G.; Akoh, C.C. Phenolic compounds from blueberries can inhibit colon cancer cell proliferation and induce apoptosis. J. Agric. Food Chem. 2005, 53, 7320–7329. [Google Scholar] [CrossRef] [PubMed]
- Hassimotto, N.M.; Genovese, M.I.; Lajolo, F.M. Antioxidant activity of dietary fruits, vegetables, and commercial frozen fruit pulps. J. Agric. Food Chem. 2005, 53, 2928–2935. [Google Scholar] [CrossRef] [PubMed]
- Henning, S.M.; Niu, Y.; Lee, N.H.; Thames, G.D.; Minutti, R.R.; Wang, H.; Go, V.L.; Heber, D. Bioavailability and antioxidant activity of tea flavanols after consumption of green tea, black tea, or a green tea extract supplement. Am. J. Clin. Nutr. 2004, 80, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Mukhtar, H. Cancer and metastasis: Prevention and treatment by green tea. Cancer Metastasis Rev. 2010, 29, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Sartippour, M.R.; Heber, D.; Ma, J.; Lu, Q.; Go, V.L.; Nguyen, M. Green tea and its catechins inhibit breast cancer xenografts. Nutr. Cancer 2001, 40, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Ankolekar, C.; Johnson, D.; Pinto Mda, S.; Johnson, K.; Labbe, R.; Shetty, K. Inhibitory potential of tea polyphenolics and influence of extraction time against helicobacter pylori and lack of inhibition of beneficial lactic acid bacteria. J. Med. Food 2011, 14, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Boehm, K.; Borrelli, F.; Ernst, E.; Habacher, G.; Hung, S.K.; Milazzo, S.; Horneber, M. Green tea (camellia sinensis) for the prevention of cancer. Cochrane Database Syst. Rev. 2009, CD005004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; Xu, Q.; Lu, J.; Wang, P.; Zhang, H.W.; Zhou, L.; Ma, X.Q.; Zhou, Y.H. Tea consumption and the incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Eur. J. Cancer Prev. 2015, 24, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Rady, I.M.H.; Rady, M.; Siddiqui, I.A.; Mukhtar, H. Cancer preventive and therapeutic effects of egcg, the major polyphenol in green tea. Egypt. J. Basic Appl. Sci. 2017, 5, 1–23. [Google Scholar] [CrossRef]
- Cardona, F.; Andres-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuno, M.I. Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.M.; Li, T.C.; Jhan, Y.L.; Weng, J.H.; Chou, C.H. The impact of microbial biotransformation of catechin in enhancing the allelopathic effects of rhododendron formosanum. PLoS ONE 2013, 8, e85162. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Chen, J.H.; Chou, F.P.; Wang, C.J. Protocatechuic acid inhibits cancer cell metastasis involving the down-regulation of ras/akt/nf-kappab pathway and mmp-2 production by targeting rhob activation. Br. J. Pharmacol. 2011, 162, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Batra, P.; Sharma, A.K. Anti-cancer potential of flavonoids: Recent trends and future perspectives. 3 Biotech 2013, 3, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Chen, X.; Jassbi, A.R.; Xiao, J. Microbial biotransformation of bioactive flavonoids. Biotechnol. Adv. 2015, 33, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Anthony, M.S.; Clarkson, T.B.; Hughes, C.L., Jr.; Morgan, T.M.; Burke, G.L. Soybean isoflavones improve cardiovascular risk factors without affecting the reproductive system of peripubertal rhesus monkeys. J. Nutr. 1996, 126, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Whitten, P.L.; Patisaul, H.B. Cross-species and interassay comparisons of phytoestrogen action. Environ. Health Perspect. 2001, 109 (Suppl. 1), 5–20. [Google Scholar] [CrossRef] [PubMed]
- Lephart, E.D.; Setchell, K.D.; Handa, R.J.; Lund, T.D. Behavioral effects of endocrine-disrupting substances: Phytoestrogens. ILAR J. 2004, 45, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.W. Health effects of soy protein and isoflavones in humans. J. Nutr. 2008, 138, 1244S–1249S. [Google Scholar] [CrossRef] [PubMed]
- Tsuchihashi, R.; Sakamoto, S.; Kodera, M.; Nohara, T.; Kinjo, J. Microbial metabolism of soy isoflavones by human intestinal bacterial strains. J. Nat. Med. 2008, 62, 456–460. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armstrong, H.; Bording-Jorgensen, M.; Dijk, S.; Wine, E. The Complex Interplay between Chronic Inflammation, the Microbiome, and Cancer: Understanding Disease Progression and What We Can Do to Prevent It. Cancers 2018, 10, 83. https://doi.org/10.3390/cancers10030083
Armstrong H, Bording-Jorgensen M, Dijk S, Wine E. The Complex Interplay between Chronic Inflammation, the Microbiome, and Cancer: Understanding Disease Progression and What We Can Do to Prevent It. Cancers. 2018; 10(3):83. https://doi.org/10.3390/cancers10030083
Chicago/Turabian StyleArmstrong, Heather, Michael Bording-Jorgensen, Stephanie Dijk, and Eytan Wine. 2018. "The Complex Interplay between Chronic Inflammation, the Microbiome, and Cancer: Understanding Disease Progression and What We Can Do to Prevent It" Cancers 10, no. 3: 83. https://doi.org/10.3390/cancers10030083
APA StyleArmstrong, H., Bording-Jorgensen, M., Dijk, S., & Wine, E. (2018). The Complex Interplay between Chronic Inflammation, the Microbiome, and Cancer: Understanding Disease Progression and What We Can Do to Prevent It. Cancers, 10(3), 83. https://doi.org/10.3390/cancers10030083