Nano-Pulse Stimulation Ablates Orthotopic Rat Hepatocellular Carcinoma and Induces Innate and Adaptive Memory Immune Mechanisms that Prevent Recurrence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
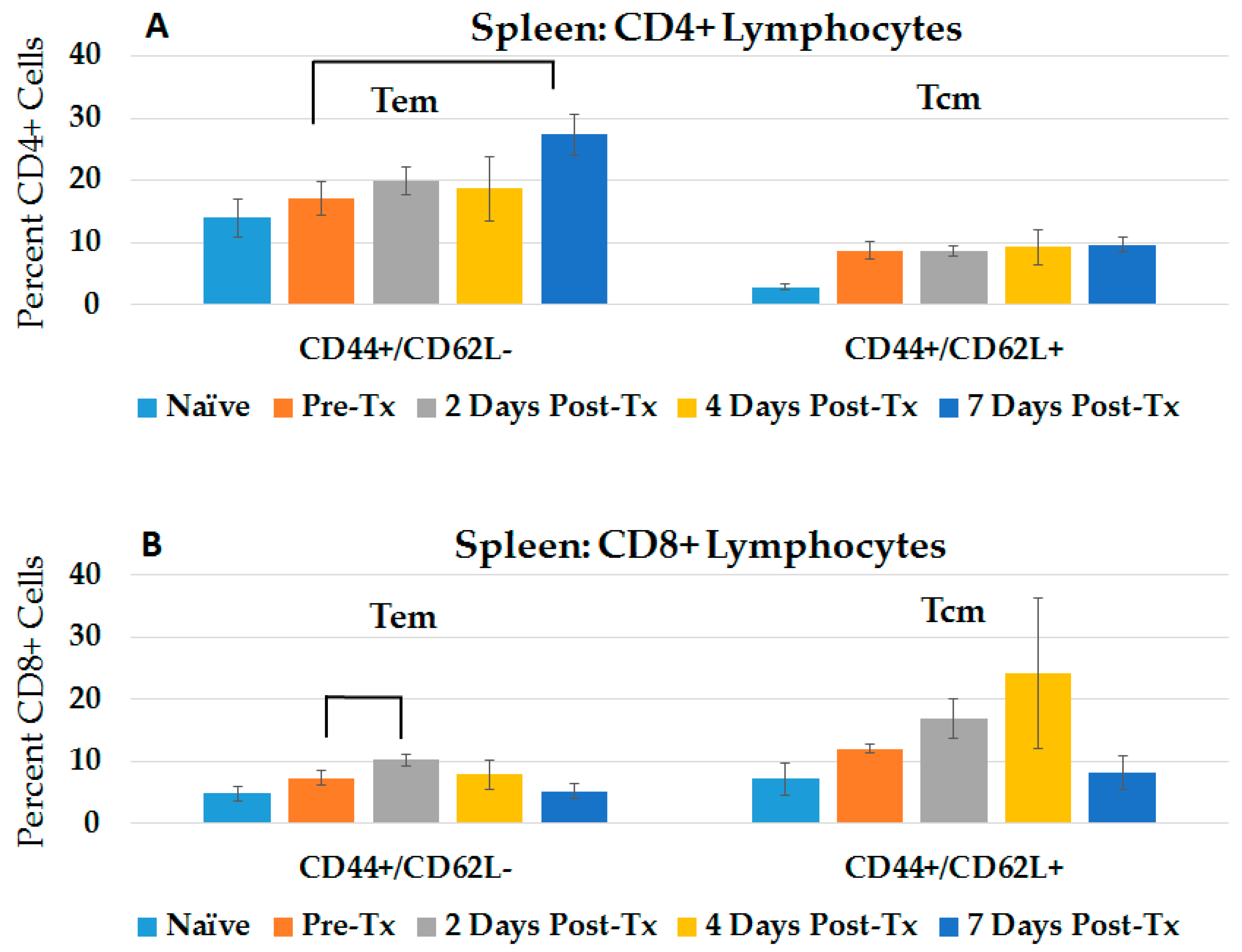
2.1. NPS Induces an Adaptive Immune Response in Blood and Spleen after HCC Ablation
2.2. NPS Induces Innate and Adaptive Immune Responses in Liver after HCC Ablation
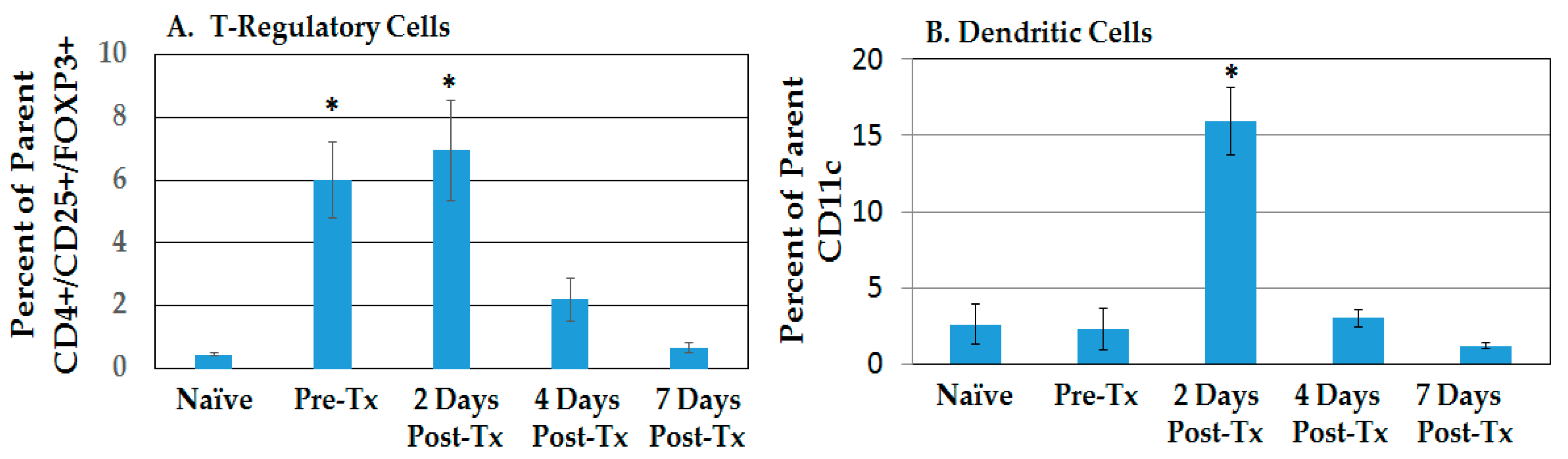
2.2.1. Resolution of Treg Immunosuppression and DC Infiltration in the TME after NPS
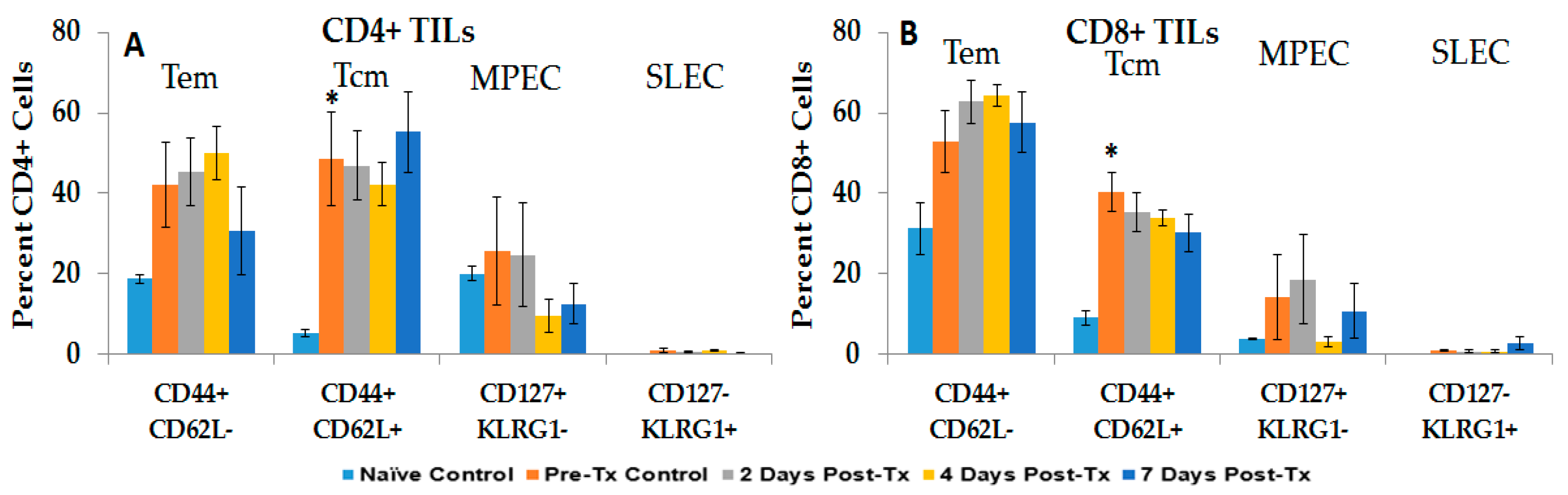
2.2.2. HCC and NPS Induce Multiple Phenotypic Changes in Liver Tumor-Infiltrating Lymphocytes (TILs)
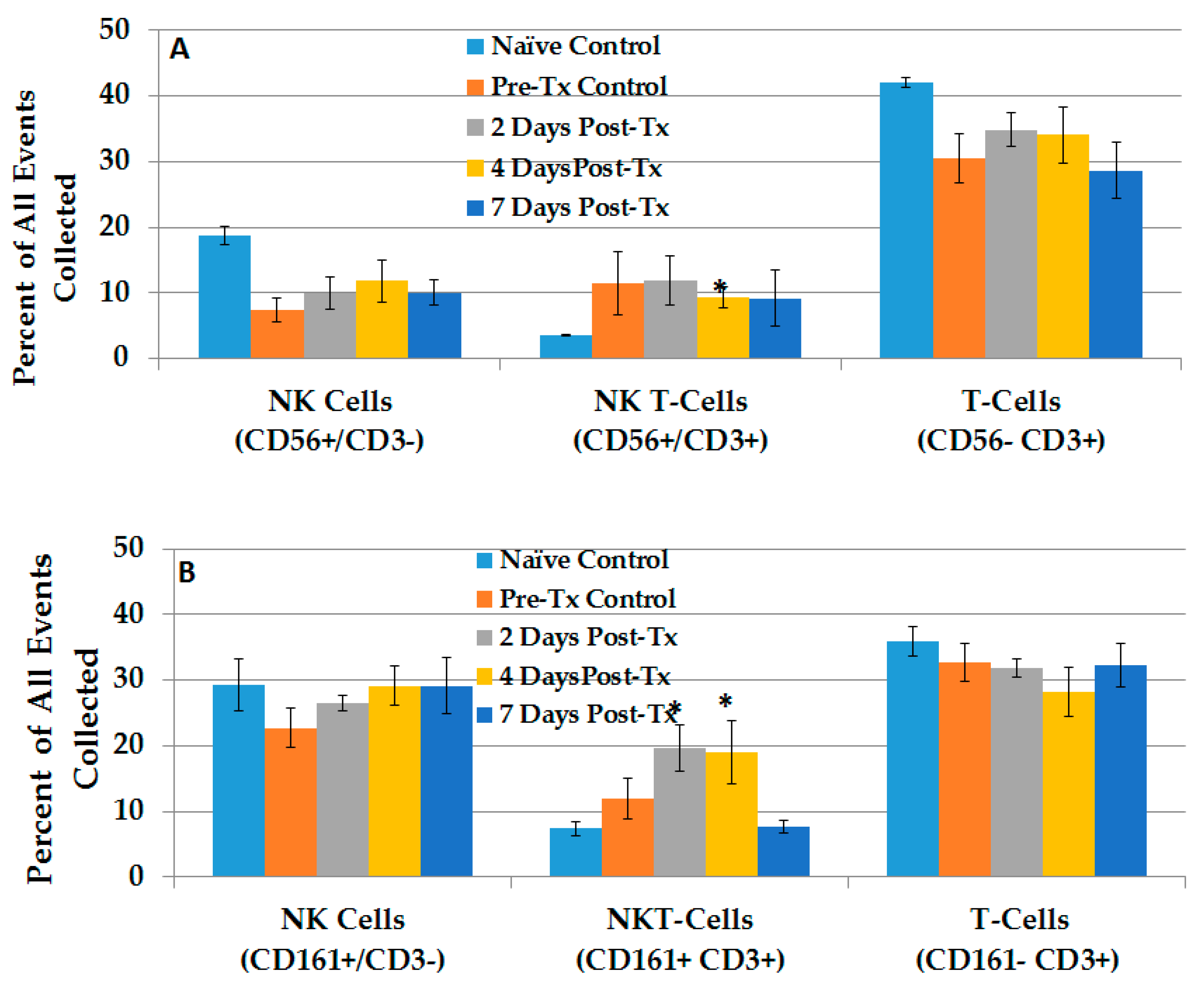
2.2.3. Dynamic Changes in Liver NK Cells, NKT-Cells and T-Lymphocytes in the TME after NPS
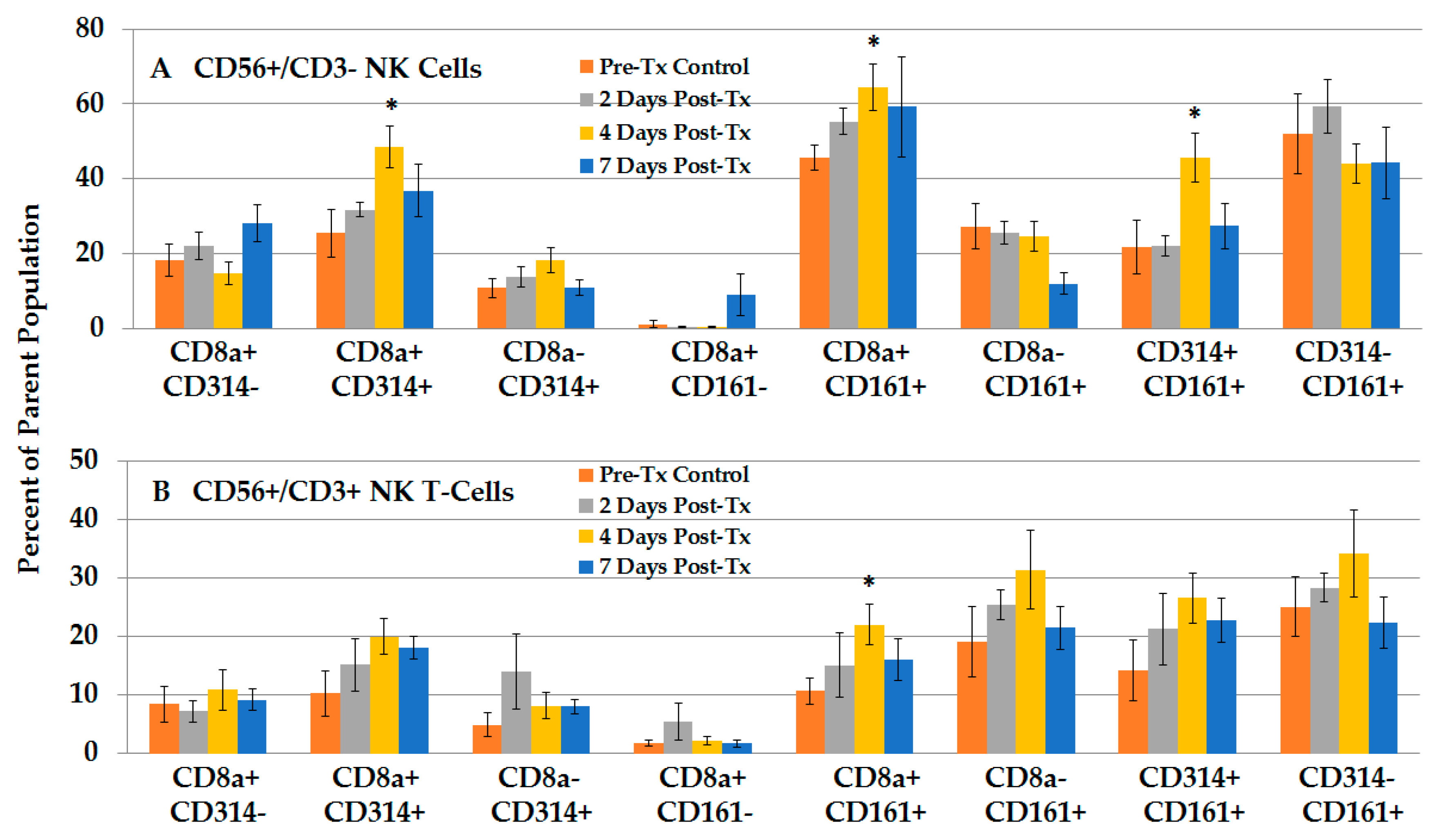
2.2.4. NPS Activates Distinct NK and NKT-Cell Phenotypes in the TME
2.3. Immune Responses Mediating the Protective, Vaccine Effect after Challenge Injections
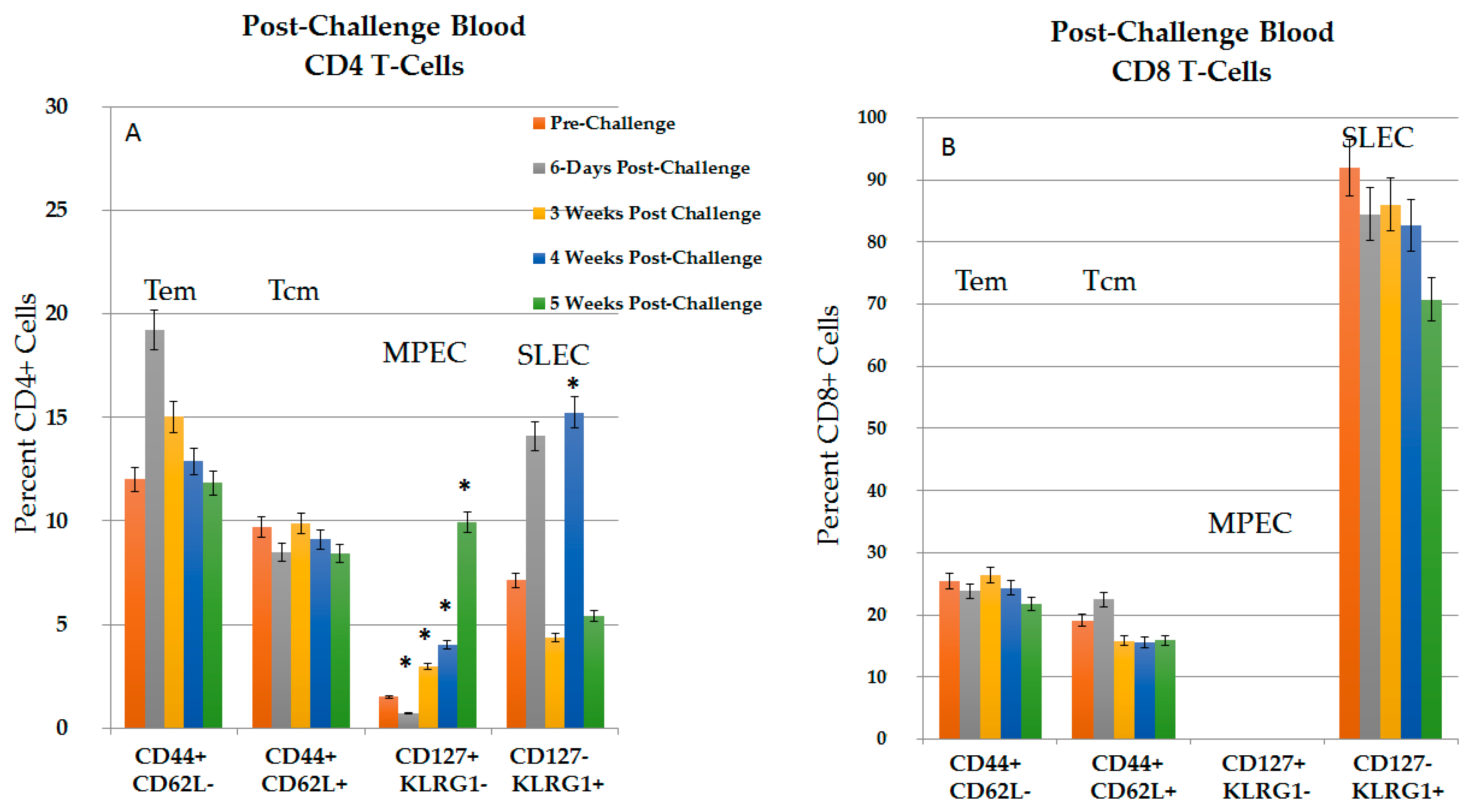
2.3.1. CD4+ and CD8+ Immune Response in Blood in the Post-Challenge, Vaccine Effect
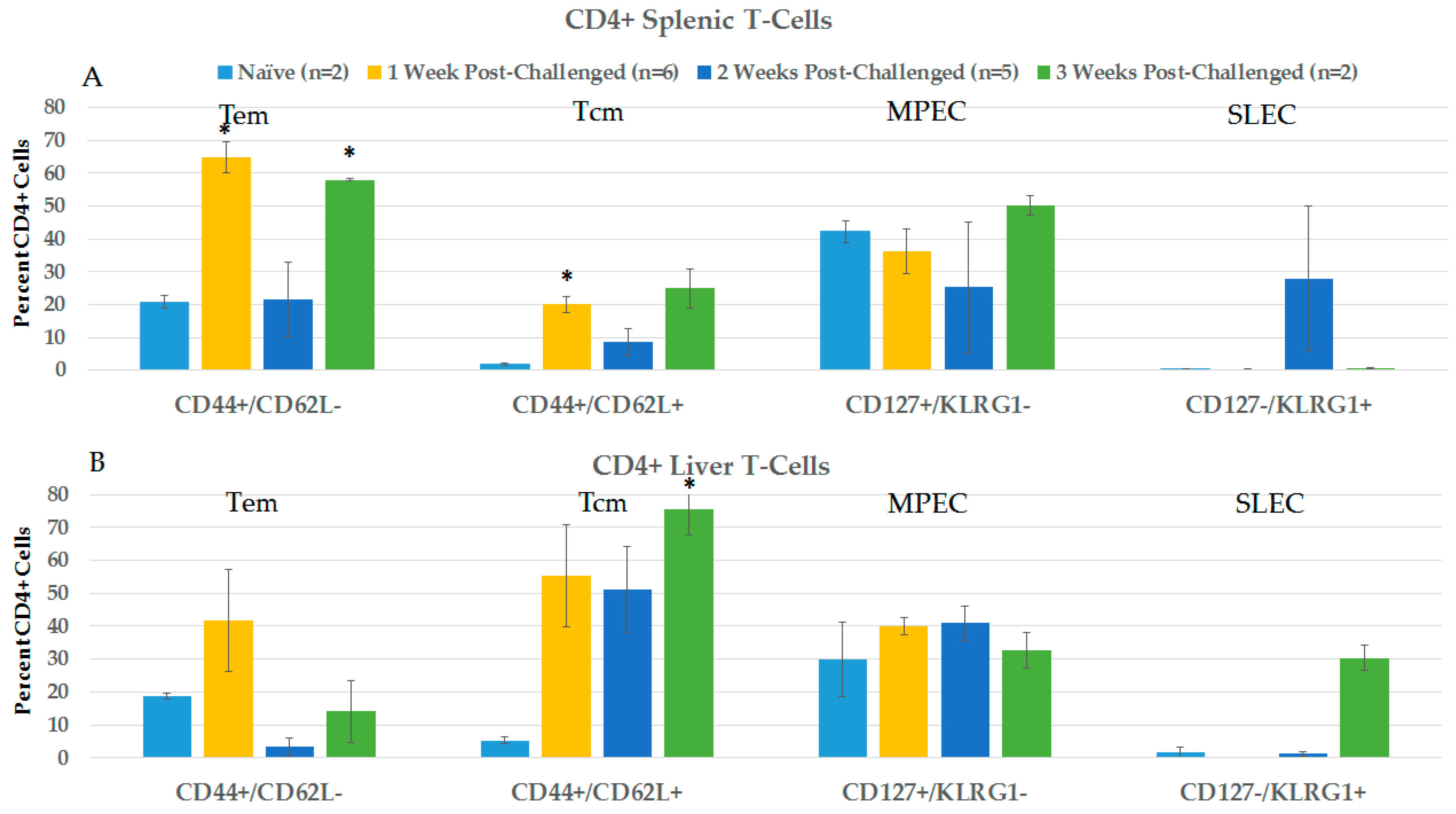
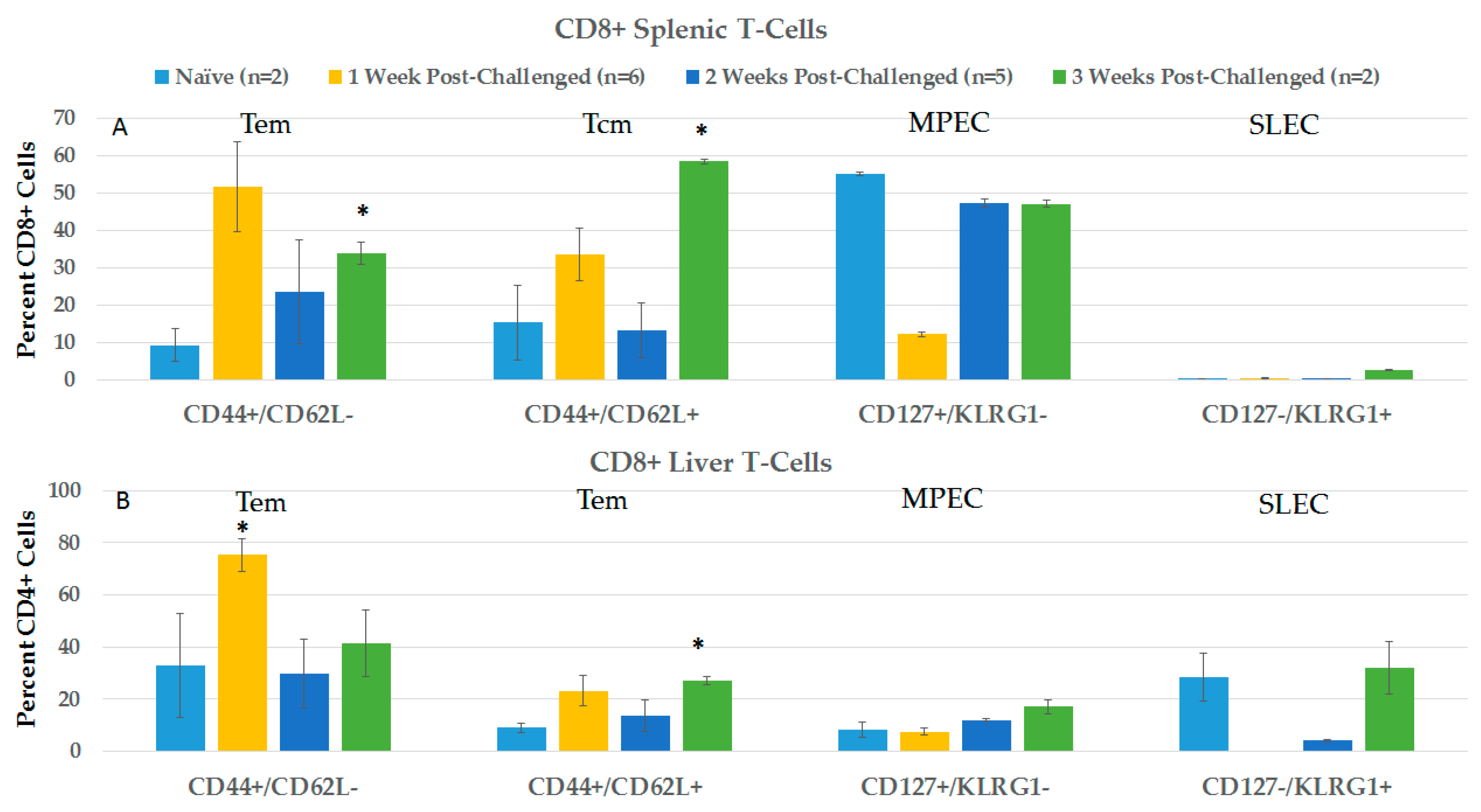
2.3.2. CD4+ and CD8+ Immune Responses in Spleen and Liver during the Post-Challenge Vaccine-Like Effect
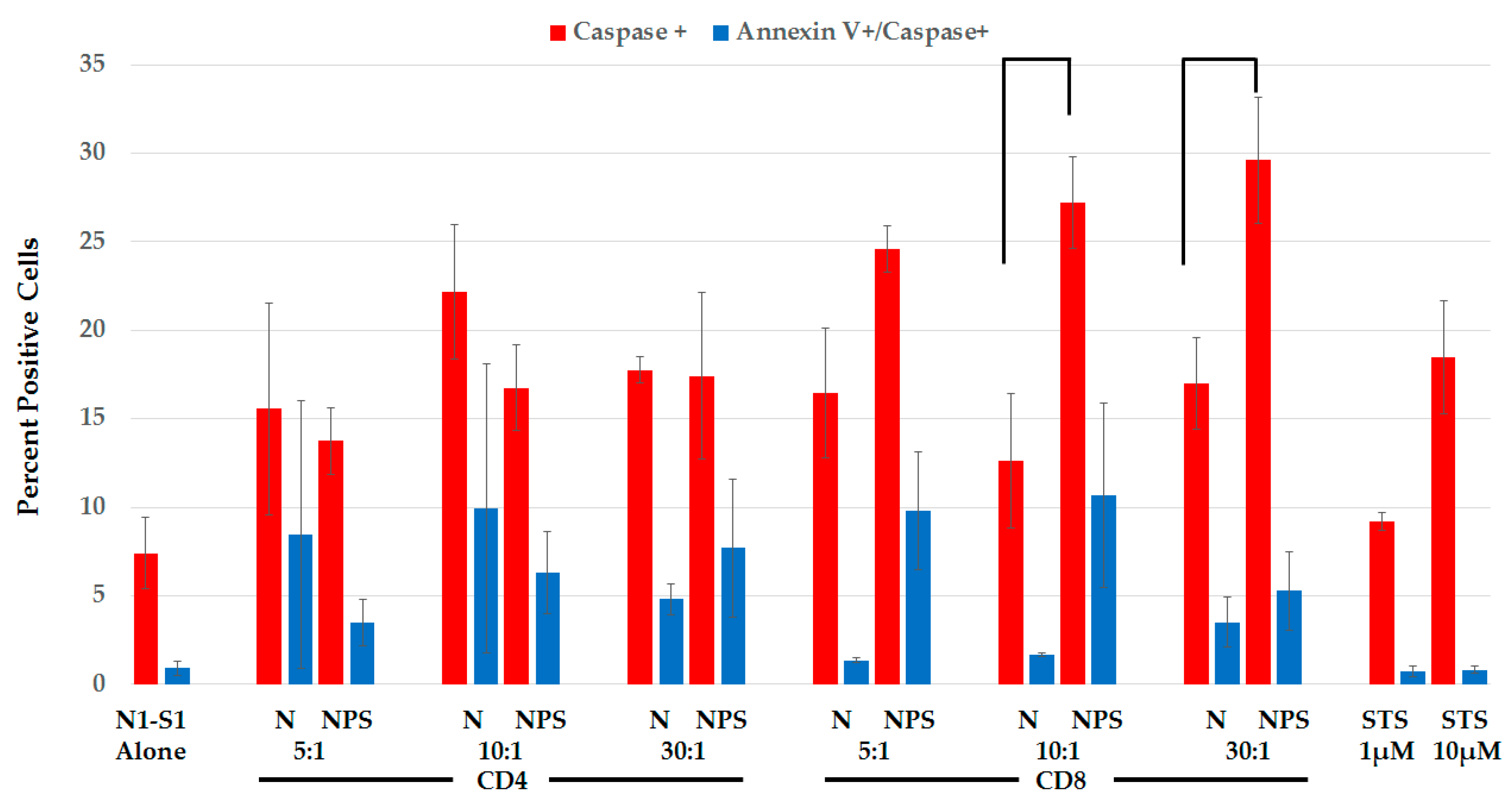
2.3.3. Splenic CD8+ Lymphocytes Are Cytotoxic after NPS-Induced Protection
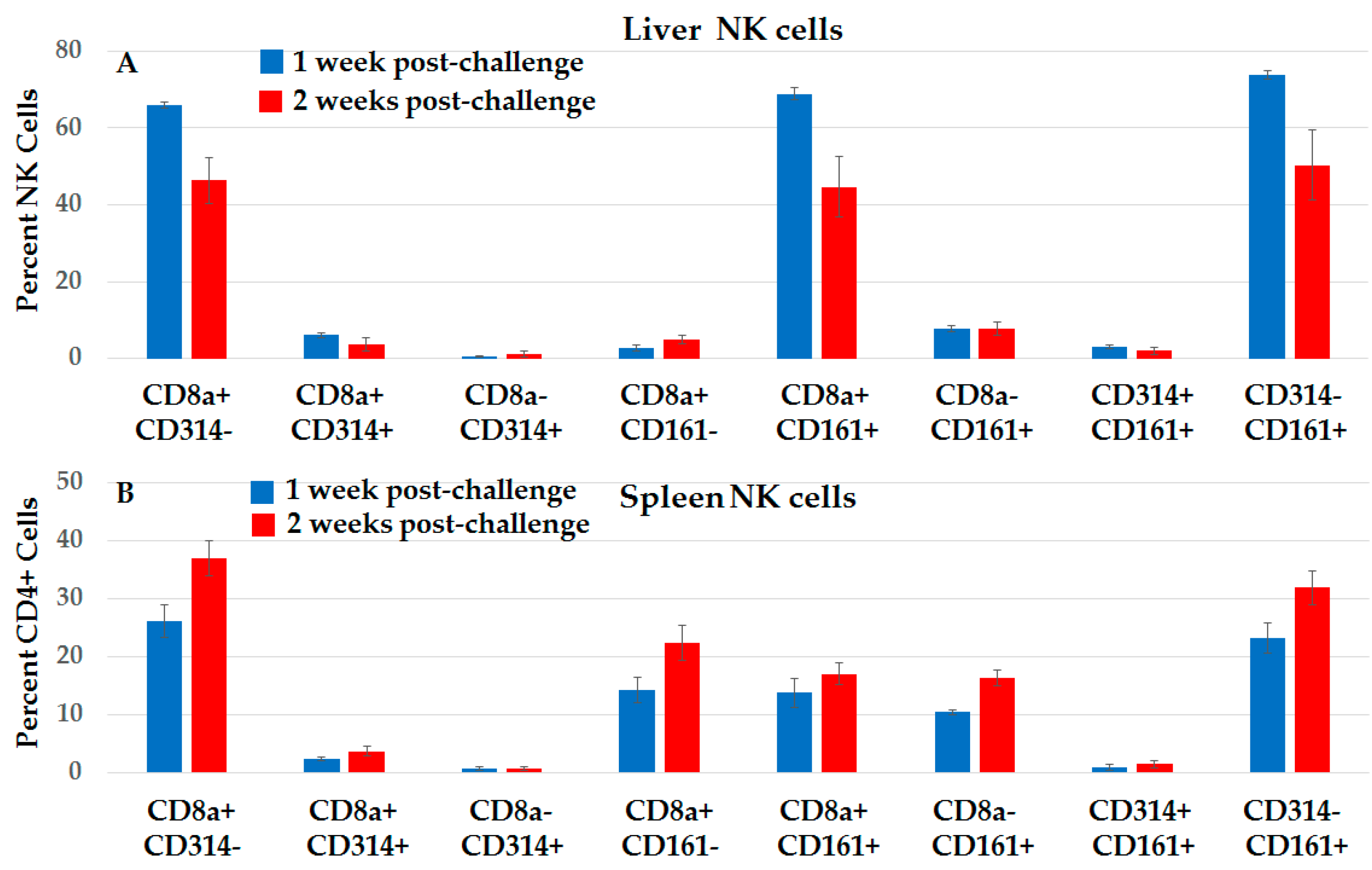
2.3.4. Distinct Subsets of NK Cells in Liver and Spleen in NPS-Induced Post-Challenge Vaccine-Like Effect
2.4. Analyses of NPS-Induced Response in Whole Animals
2.4.1. NPS Induces a N1-S1-Specific Long-Lasting Post-HCC Ablation Vaccine-Like Effect in Rats
2.4.2. NPS Induces a Limited Abscopal Effect
3. Discussion
4. Materials and Methods
4.1. Animals and Cell Culture
4.2. The HCC Model and Nano-Pulse Stimulation Treatment
4.3. Splenocyte Preparation
4.4. Blood Preparation
4.5. Liver Preparation
4.6. Flow Cytometry and Antibodies
4.7. Cytotoxicity Assay
4.8. Statistics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Canbay, A.; Friedman, S.; Gores, G.J. Apoptosis: The nexus of liver injury and fibrosis. Hepatology 2004, 39, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Takehara, T.; Tatsumi, T.; Suzuki, T.; Rucker, E.B., 3rd; Hennighausen, L.; Jinushi, M.; Miyagi, T.; Kanazawa, Y.; Hayashi, N. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology 2004, 127, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Vick, B.; Weber, A.; Urbanik, T.; Maass, T.; Teufel, A.; Krammer, P.; Opferman, J.T.; Schuchmann, M.; Galle, P.R.; Schulze-Bergkamen, H. Knockout of myeloid cell leukemia-1 induces liver damage and increases apoptosis susceptibility of murine hepatocytes. Hepatology 2009, 49, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, C.E.; Bergmann, A. The Sound of Silence: Signaling by Apoptotic Cells. Curr. Top. Dev. Biol. 2015, 114, 241–265. [Google Scholar] [PubMed]
- Malhi, H.; Guicciardi, M.E.; Gores, G.J. Hepatocyte death: A clear and present danger. Physiol. Rev. 2010, 90, 1165–1194. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; Ribes, J.; Díaz, M.; Cléries, R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology 2004, 127, S5–S16. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Sun, B.; Satiroglu Tufan, N.L.; Liu, J.; Pan, J.; Lian, Z. Genetic mechanisms of hepatocarcinogenesis. Oncogene 2002, 21, 2593–2604. [Google Scholar] [CrossRef] [PubMed]
- Mishra, L.; Banker, T.; Murray, J.; Byers, S.; Thenappan, A.; He, A.R.; Shetty, K.; Johnson, L.; Reddy, E.P. Liver stem cells and hepatocellular carcinoma. Hepatology 2009, 49, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. Hepatic T cells and liver tolerance. Nat. Rev. Immunol. 2003, 3, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Jeong, W.I.; Tian, Z. Liver: An organ with predominant innate immunity. Hepatology 2008, 47, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. The liver as a lymphoid organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Doherty, D.G.; O’Farrelly, C. Innate and adaptive lymphoid cells in the human liver. Immunol. Rev. 2000, 174, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Beebe, S.J. Preclinical Studies with Nanosecond Pulses. In Handbook of Electroporation; Miklavčič, D., Ed.; Springer: Berlin, Germany, 2017; pp. 1543–, 1562; ISBN 978-3-319-26779-1. [Google Scholar]
- Djokic, M. Popovic, P., Kos, B., Dezman, R., Bosnjak, M., Zakelj, M.N., Miklavcic, D., Potrc, S., Stabuc, B., Eds.; et al. Electrochemotherapy as treatment option for hepatocellular carcinoma, a prospective pilot study. Eur. J. Surg. Oncol. 2018, doi: 10.1016/j.ejso, 2018; Cemazar. M.
- Li, X.; Xu, K.; Li, W.; Qiu, X.; Ma, B.; Fan, Q.; Li, Z. Immunologic response to tumor ablation with irreversible electroporation. PLoS ONE 2012, 7, e48749. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Sain, N.M.; Harlow, K.T.; Chen, Y.J.; Shires, P.K.; Heller, R.; Beebe, S.J. A protective effect after clearance of orthotopic rat hepatocellular carcinoma by nanosecond pulsed electric fields. Eur. J. Cancer 2014, 50, 2705–2713. [Google Scholar] [CrossRef] [PubMed]
- Nuccitelli, R. Berridge, J.C.; Mallon, Z.; Kreis, M.; Athos, B.; Nuccitelli, P. Nanoelectroablation of Murine Tumors Triggers a CD8-Dependent Inhibition of Secondary Tumor Growth. PLoS ONE 2015, 10, e0134364. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Jing, Y.; Burcus, N.I.; Lassiter, B.P.; Tanaz, R.; Heller, R.; Beebe, S.J. Nano-pulse stimulation induces potent immune responses, eradicating local breast cancer while reducing distant metastases. Int. J. Cancer 2017, 142, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Nuccitelli, R.; McDaniel, A.; Anand, S.; Cha, J.; Mallon, Z.; Berridge, J.C.; Uecker, D. Nano-Pulse Stimulation is a physical modality that can trigger immunogenic tumor cell death. J. Immunother. Cancer 2017, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Li, W.Q.; Hofmeister, R.R.; Young, H.A.; Hodge, D.R.; Keller, J.R.; Khaled, A.R.; Durum, S.K. Distinct regions of the interleukin-7 receptor regulate different Bcl2 family members. Mol. Cell. Biol. 2004, 2, 6501–6513. [Google Scholar] [CrossRef] [PubMed]
- Fergusson, J.R.; Fleming, V.M.; Klenerman, P. CD161-Expressing Human T Cells. Front. Immunol. 2011, 2, 36. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zeng, G. Cancer and innate immune system interactions: Translational potentials for cancer immunotherapy. J. Immunother. 2012, 35, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Arce-Sillas, A.; Álvarez-Luquín, D.D.; Tamaya-Domínguez, B.; Gomez-Fuentes, S.; Trejo-García, A.; Melo-Salas, M.; Cárdenas, G.; Rodríguez-Ramírez, J.; Adalid-Peralta, L. Regulatory T Cells: Molecular Actions on Effector Cells in Immune Regulation. J. Immunol. Res. 2016, 2016, 1720827. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.J.; Chang, W.A.; Huang, M.S.; Kuo, P.L. Tumor microenvironment: A new treatment target for cancer. ISRN Biochem. 2014, 2014, 351959. [Google Scholar] [CrossRef] [PubMed]
- Sauter, B.; Albert, M.L.; Francisco, L.; Larsson, M.; Somersan, S.; Bhardwaj, N. Consequences of cell death: Exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000, 191, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Blachère, N.E.; Darnell, R.B.; Albert, M.L. Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation. PLoS Biol. 2005, 3, e185. [Google Scholar] [CrossRef] [PubMed]
- Uhl, M.; Kepp, O.; Jusforgues-Saklani, H.; Vicencio, J.M.; Kroemer, G.; Albert, M.L. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ. 2009, 16, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8⁺ T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.B.; Lü, M.H.; Fan, Y.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Waheed, Y.; Manzoor, S.; Bilal, M.; Ashraf, W.; Ali, M.; Ashraf, M. Interaction of Hepatitis C virus proteins with pattern recognition receptors. Virol J. 2012, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Esteso, G.; Guerra, S.; Valés-Gómez, M.; Reyburn, H.T. Innate Immune Recognition of Double-stranded RNA Triggers Increased Expression of NKG2D Ligands after Virus Infection. J. Biol. Chem. 2017, 292, 20472–20480. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Rouce, R.H. The Application of Natural Killer Cell Immunotherapy for the Treatment of Cancer. Front. Immunol. 2015, 6, 578. [Google Scholar] [CrossRef] [PubMed]
- Kärre, K. Immunology. A perfect mismatch. Science 2002, 295, 2029–2031. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.S.; Hasegawa, J. Natural killer cell biology: An update and future directions. J. Allergy Clin. Immunol. 2013, 132, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition in the innate immune response. Biochem. J. 2009, 420, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Daussy, C.; Faurem, F.; Mayol, K.; Viel, S.; Gasteiger, G.; Charrier, E.; Bienvenu, J.; Henry, T.; Debien, E.; Hasan, U.A.; et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J. Exp. Med. 2014, 211, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Cretney, E.; Hayakawa, Y.; Ota, T.; Akiba, H.; Ogasawara, K.; Yagita, H.; Kinoshita, K.; Okumura, K.; Smyth, M.J. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood 2005, 105, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.Z.; Vermijlen, D.; Ahishali, B.; Triantis, V.; Plakoutsi, G.; Braet, F.; Vanderkerken, K.; Wisse, E. On the cell biology of pit cells, the liver-specific NK cells. World J. Gastroenterol. 2000, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vanderkerken, K.; Bouwens, L.; Wisse, E. Characterization of a phenotypically and functionally distinct subset of large granular lymphocytes (pit cells) in rat liver sinusoids. Hepatology 1990, 12, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Vanderkerken, K.; Bouwens, L.; Kuppen, P.J.; Baekeland, M.; Seynaeve, C.; Wisse, E. The role of adhesion molecules in the recruitment of hepatic natural killer cells (pit cells) in rat liver. Hepatology 1996, 24, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Vermijlen, D.; Luo, D.; Froelich, C.J.; Medema, J.P.; Kummer, J.A.; Willems, E.; Braet, F.; Wisse, E. Hepatic natural killer cells exclusively kill splenic/blood natural killer-resistant tumor cells by the perforin/granzyme pathway. J. Leukoc. Biol. 2002, 72, 668–676. [Google Scholar] [PubMed]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs a gradient of Tbet expression that specifies memory precursor and short-lived effector CD8 T cell fates. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Fergusson, J.R.; Smith, K.E.; Fleming, V.M.; Rajoriya, N.; Newell, E.W.; Simmons, R.; Marchi, E.; Björkander, S.; Kang, Y.H.; Swadling, L.; et al. CD161 defines a transcriptional and Functional Phenotype across Distinct Human T Cell Lineages. Cell Rep. 2014, 9, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Santarlasci, V.; Capone, M.; Peired, A.; Frosali, F.; Crome, S.Q.; Querci, V.; Fambrini, M.; Liotta, F.; Levings, M.K.; et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur. J. Immunol. 2010, 40, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Adaptive immune features of natural killer cells. Nature 2009, 457, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Ugolini, S.; Vivier, E. Immunological memory within the innate immune system. EMBO J. 2014, 17, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed]
- Falk, H.; Forde, P.F.; Bay, M.L.; Mangalanathan, U.M.; Hojman, P.; Soden, D.M.; Gehl, J. Calcium electroporation induces tumor eradication, long-lasting immunity and cytokine responses in the CT26 colon cancer mouse model. Oncoimmunology 2017, 6, e1301332. [Google Scholar] [CrossRef] [PubMed]
- Falk, H.; Lambaa, S.; Johannesen, H.H.; Wooler, G.; Venzo, A.; Gehl, J. Electrochemotherapy and calcium electroporation inducing a systemic immune response with local and distant remission of tumors in a patient with malignant melanoma-A case report. Acta Oncol. 2017, 56, 1126–1131. [Google Scholar] [CrossRef] [PubMed]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lassiter, B.P.; Guo, S.; Beebe, S.J. Nano-Pulse Stimulation Ablates Orthotopic Rat Hepatocellular Carcinoma and Induces Innate and Adaptive Memory Immune Mechanisms that Prevent Recurrence. Cancers 2018, 10, 69. https://doi.org/10.3390/cancers10030069
Lassiter BP, Guo S, Beebe SJ. Nano-Pulse Stimulation Ablates Orthotopic Rat Hepatocellular Carcinoma and Induces Innate and Adaptive Memory Immune Mechanisms that Prevent Recurrence. Cancers. 2018; 10(3):69. https://doi.org/10.3390/cancers10030069
Chicago/Turabian StyleLassiter, Brittany P., Siqi Guo, and Stephen J. Beebe. 2018. "Nano-Pulse Stimulation Ablates Orthotopic Rat Hepatocellular Carcinoma and Induces Innate and Adaptive Memory Immune Mechanisms that Prevent Recurrence" Cancers 10, no. 3: 69. https://doi.org/10.3390/cancers10030069
APA StyleLassiter, B. P., Guo, S., & Beebe, S. J. (2018). Nano-Pulse Stimulation Ablates Orthotopic Rat Hepatocellular Carcinoma and Induces Innate and Adaptive Memory Immune Mechanisms that Prevent Recurrence. Cancers, 10(3), 69. https://doi.org/10.3390/cancers10030069