
Less Is More: Oligomer Extraction and Hydrothermal Annealing Increase PDMS Adhesion Forces for Materials Studies and for Biology-Focused Microfluidic Applications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Materials Characterization
3.2. Hydrothermal Annealing
3.3. PDMS Deformation and Oligomer Translocation
3.4. Fluidic Manipulation
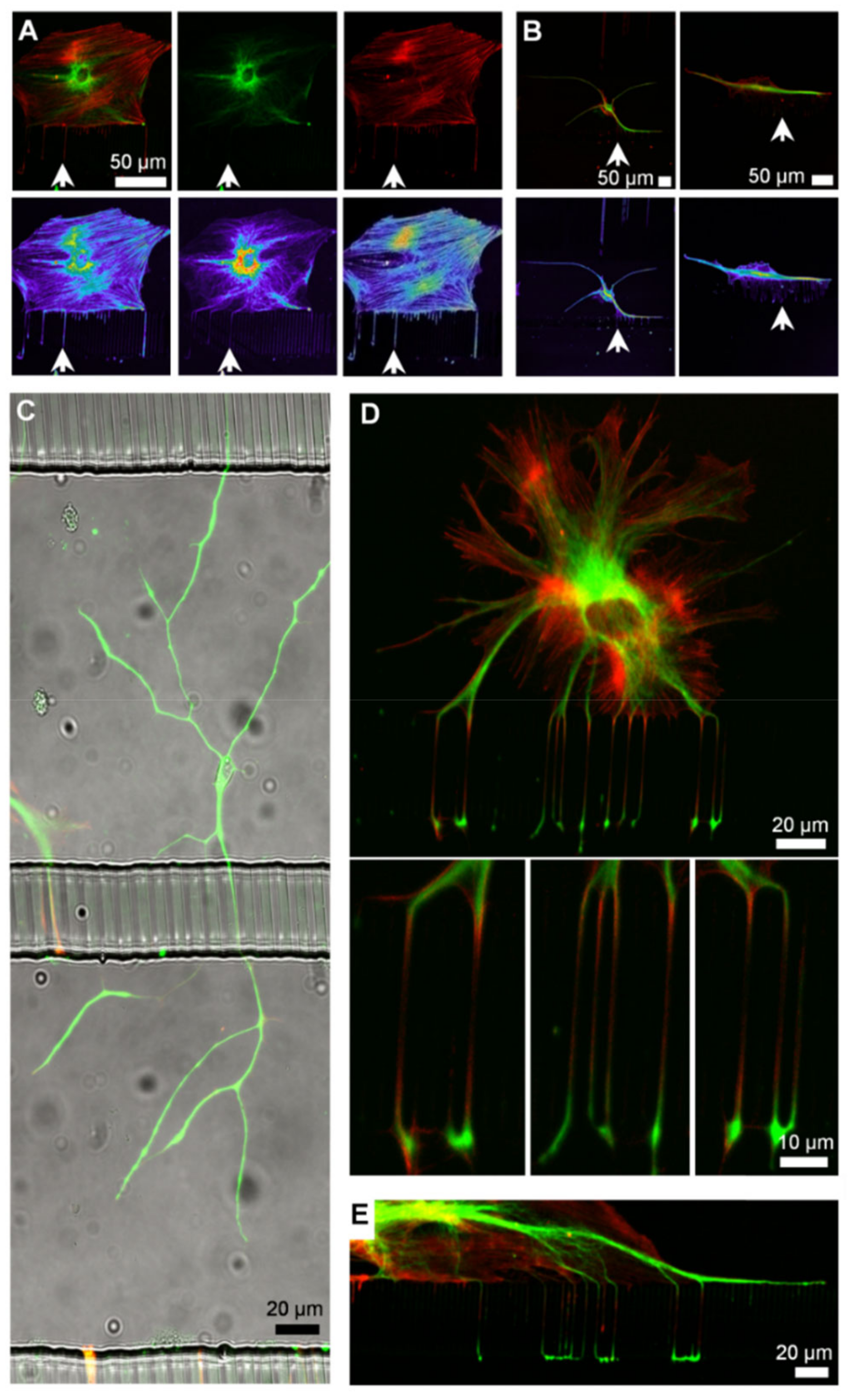
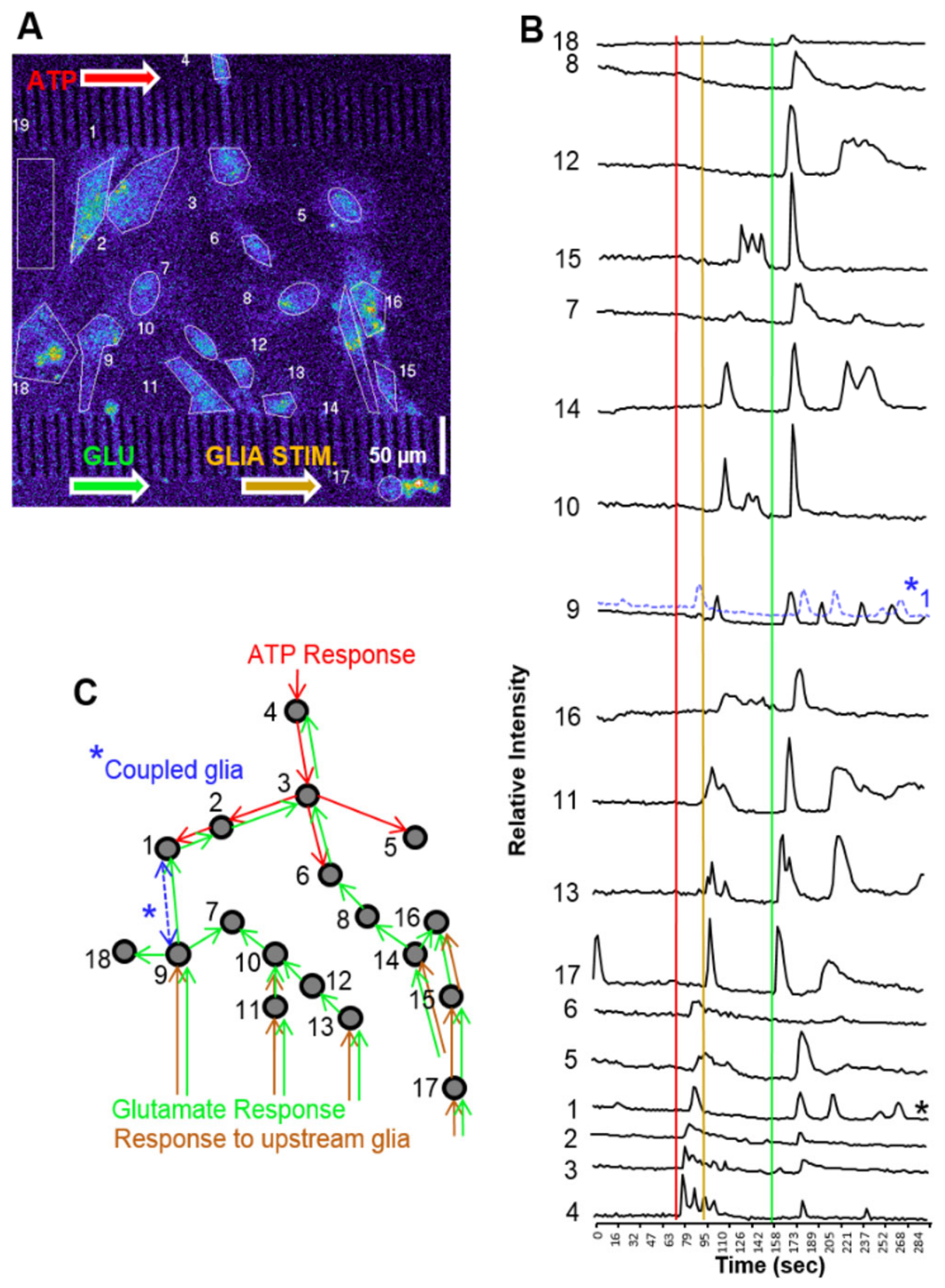
3.5. Cell Signaling in Microfluidics
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brimmo, A.T.; Menachery, A.; Sukumar, P.; Qasaimeh, M.A. Noncontact Multiphysics Probe for Spatiotemporal Resolved Single-Cell Manipulation and Analyses. Small 2021, 17, e2100801. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, S.; Guo, Y.; Zeng, X.; Liu, B.F. A review on microfluidics manipulation of the extracellular chemical microenvironment and its emerging application to cell analysis. Anal. Chim. Acta 2020, 1125, 94–113. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, M.D.; Gillette, M.U. Microfluidic Devices for Analysis of Neuronal Development. Eng. Biomater. Neural Appl. 2022, 169–185. [Google Scholar] [CrossRef]
- Low, L.A.; Mummery, C.; Berridge, B.R.; Austin, C.P.; Tagle, D.A. Organs-on-chips: Into the next decade. Nat. Rev. Drug Discov. 2021, 20, 345–361. [Google Scholar] [CrossRef]
- Millet, L.J.; Gillette, M.U. New perspectives on neuronal development via microfluidic environments. Trends Neurosci. 2012, 35, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Millet, L.J.; Gillette, M.U. Over a century of neuron culture: From the hanging drop to microfluidic devices. Yale J. Biol. Med. 2012, 85, 501–521. [Google Scholar]
- Selimović, Š.; Dokmeci, M.R.; Khademhosseini, A. Organs-on-a-chip for drug discovery. Curr. Opin. Pharmacol. 2013, 13, 829–833. [Google Scholar] [CrossRef]
- Taylor, A.M.; Dieterich, D.C.; Ito, H.T.; Kim, S.A.; Schuman, E.M. Microfluidic Local Perfusion Chambers for the Visualization and Manipulation of Synapses. Neuron 2010, 66, 57–68. [Google Scholar] [CrossRef]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension - how 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Nahavandi, S.; Tang, S.Y.; Baratchi, S.; Soffe, R.; Nahavandi, S.; Kalantar-Zadeh, K.; Mitchell, A.; Khoshmanesh, K. Microfluidic Platforms for the Investigation of Intercellular Signalling Mechanisms. Small 2014, 10, 4810–4826. [Google Scholar] [CrossRef]
- Guo, F.; French, J.B.; Li, P.; Zhao, H.; Chan, C.Y.; Fick, J.R.; Benkovic, S.J.; Huang, T.J. Probing cell-cell communication with microfluidic devices. Lab Chip 2013, 13, 3152–3162. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Du, C.; Acero, E.H.; Tang-Schomer, M.D.; Özkucur, N. A biofidelic 3D culture model to study the development of brain cellular systems. Sci. Rep. 2016, 6, 24953. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, T.B.; Zandén, C.; De Pablo, Y.; Kirchhoff, F.; Pekna, M.; Liu, J.; Pekny, M. Bioactive 3D cell culture system minimizes cellular stress and maintains the in vivo-like morphological complexity of astroglial cells. Glia 2013, 61, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Boyle, V.; Leibig, N.; Stark, G.B.; Penna, V. The Neuro-spheroid-A novel 3D in vitro model for peripheral nerve regeneration. J. Neurosci. Methods 2015, 246, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Gillette, M.U. Development of Microfluidic Devices for the Manipulation of Neuronal Synapses. In Microfluidic and Compartmentalized Platforms for Neurobiological Research; Humana Press: Totowa, NJ, USA, 2015; pp. 127–137. [Google Scholar]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting Organ-Level Lung Functions on a Chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Leslie, D.C.; Matthews, B.D.; Fraser, J.P.; Jurek, S.; Hamilton, G.A.; Thorneloe, K.S.; McAlexander, M.A.; Ingber, D.E. A Human Disease Model of Drug Toxicity-Induced Pulmonary Edema in a Lung-on-a-Chip Microdevice. Sci. Transl. Med. 2012, 4, 159ra147. [Google Scholar] [CrossRef] [PubMed]
- Sia, S.K.; Whitesides, G.M. Microfluidic devices fabricated in poly(dimethylsiloxane) for biological studies. Electrophoresis 2003, 24, 3563–3576. [Google Scholar]
- Mata, A.; Fleischman, A.J.; Roy, S. Characterization of polydimethylsiloxane (PDMS) properties for biomedical micro/nanosystems. Biomed. Microdevices 2005, 7, 281–293. [Google Scholar] [CrossRef]
- Regehr, K.J.; Domenech, M.; Koepsel, J.T.; Carver, K.C.; Ellison-Zelski, S.J.; Murphy, W.L.; Schuler, L.A.; Alarid, E.T.; Beebe, D.J. Biological implications of polydimethylsiloxane-based microfluidic cell culture. Lab Chip 2009, 9, 2132–2139. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.M.; Zeringue, H.C.; Beebe, D.J. Microenvironment design considerations for cellular scale studies. Lab Chip 2004, 4, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Millet, L.J.; Stewart, M.E.; Sweedler, J.; Nuzzo, R.G.; Gillette, M.U. Microfluidic devices for culturing primary mammalian neurons at low densities. Lab Chip 2007, 7, 987–994. [Google Scholar] [CrossRef]
- Halldorsson, S.; Lucumi, E.; Gómez-Sjöberg, R.; Fleming, R.M.T. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens. Bioelectron. 2015, 63, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Łopacińska, J.M.; Emnéus, J.; Dufva, M. Poly(Dimethylsiloxane) (PDMS) Affects Gene Expression in PC12 Cells Differentiating into Neuronal-Like Cells. PLoS ONE 2013, 8, e53107. [Google Scholar] [CrossRef]
- Millet, L.J.; Stewart, M.E.; Nuzzo, R.G.; Gillette, M.U. Guiding neuron development with planar surface gradients of substrate cues deposited using microfluidic devices. Lab Chip 2010, 10, 1525–1535. [Google Scholar] [CrossRef]
- Toepke, M.W.; Beebe, D.J. PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip 2006, 6, 1484–1486. [Google Scholar] [CrossRef]
- Duffy, D.C.; McDonald, J.C.; Schueller, O.J.A.; Whitesides, G.M. Rapid prototyping of microfluidic systems in poly(dimethylsiloxane). Anal. Chem. 1998, 70, 4974–4984. [Google Scholar] [CrossRef] [PubMed]
- Langowski, B.A.; Uhrich, K.E. Oxygen plasma-treatment effects on Si transfer. Langmuir 2005, 21, 6366–6372. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Demirci, U.; Chen, Y.; Chen, P. Multiscale brain research on a microfluidic chip. Lab Chip 2020, 20, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzo, J.A.; Hart, R.P.; Zahn, J.D.; Pang, Z.P. Compartmentalized Devices as Tools for Investigation of Human Brain Network Dynamics. Dev. Dyn. 2019, 248, 65–77. [Google Scholar] [CrossRef]
- Frimat, J.-P.; Luttge, R. The Need for Physiological Micro-Nanofluidic Systems of the Brain. Front. Bioeng. Biotechnol. 2019, 7, 100. [Google Scholar] [CrossRef]
- Millar, B.J.; Deb, S. Effect of Autoclave Sterilisation on the Dimensional Stability and Tear Strength of Three Silicone Impression Materials. Open J. Stomatol. 2014, 4, 518. [Google Scholar] [CrossRef]
- Hillborg, H.; Sandelin, M.; Gedde, U.W. Hydrophobic recovery of polydimethylsiloxane after exposure to partial discharges as a function of crosslink density. Polymer 2001, 42, 7349–7362. [Google Scholar] [CrossRef]
- Hillborg, H.; Tomczak, N.; Olàh, A.; Schönherr, H.; Vancso, G.J. Nanoscale hydrophobic recovery: A chemical force microscopy study of UV/ozone-treated cross-linked poly(dimethylsiloxane). Langmuir 2004, 20, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.; Smith, P.J. Plasma treatment of polydimethylsiloxane. Adhes. Sci. Technol. 1994, 8, 1063. [Google Scholar] [CrossRef]
- Hillborg, H.; Ankner, J.F.; Gedde, U.W.; Smith, G.D.; Yasuda, H.K.; Wikstro, K. Crosslinked polydimethylsiloxane exposed to oxygen plasma studied by neutron reflectometry and other surface specific techniques. Polymer 2000, 41, 6851–6863. [Google Scholar] [CrossRef]
- Childs, W.R.; Motala, M.J.; Lee, K.J.; Nuzzo, R.G. Masterless soft lithography: Patterning UV/ozone-induced adhesion on poly(dimethylsiloxane) surfaces. Langmuir 2005, 21, 10096–10105. [Google Scholar] [CrossRef]
- DiBenedetto, A.T. Tailoring of interfaces in glass fiber reinforced polymer composites: A review. Mater. Sci. Eng. A 2001, 302, 74–82. [Google Scholar] [CrossRef]
- Stricher, A.M.; Rinaldi, R.G.; Barrès, C.; Ganachaud, F.; Chazeau, L.; Kelchtermans, M.; Wrobleski, D.A. How I met your elastomers: From network topology to mechanical behaviours of conventional silicone materials. RSC Adv. 2015, 5, 53713–53725. [Google Scholar] [CrossRef]
- Fritz, J.L.; Owen, M.J. Hydrophobic Recovery of Plasma-Treated Polydimethylsiloxane. J. Adhes. 1995, 54, 33–45. [Google Scholar] [CrossRef]
- Eddington, D.T.; Puccinelli, J.P.; Beebe, D.J. Thermal aging and reduced hydrophobic recovery of polydimethylsiloxane. Sens. Actuators B Chem. 2006, 114, 170–172. [Google Scholar] [CrossRef]
- Jo, K.; Heien, M.L.; Thompson, L.B.; Zhong, M.; Nuzzo, R.G.; Sweedler, J. Mass spectrometric imaging of peptide release from neuronal cells within microfluidic devices. Lab Chip 2007, 7, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Hassinger, T.D.; Guthrie, P.B.; Atkinson, P.B.; Bennett, M.; Kater, S.B. An extracellular signaling component in propagation of astrocytic calcium waves. Proc. Natl. Acad. Sci. USA 1996, 93, 13268–13273. [Google Scholar] [CrossRef] [PubMed]
- Bowser, D.N.; Khakh, B.S. Two forms of single-vesicle astrocyte exocytosis imaged with total internal reflection fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2007, 104, 4212–4217. [Google Scholar] [CrossRef]
- Bowser, D.N.; Khakh, B.S. Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J. Gen. Physiol. 2007, 129, 485–491. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Orkand, R.K.; Kettenmann, H. Glial calcium: Homeostasis and signaling function. Physiol. Rev. 1998, 78, 99–141. [Google Scholar] [CrossRef]
- Zonta, M.; Angulo, M.C.; Gobbo, S.; Rosengarten, B.; Hossmann, K.-A.; Pozzan, T.; Carmignoto, G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 2003, 6, 43–50. [Google Scholar] [CrossRef]
- Parri, H.R.; Gould, T.M.; Crunelli, V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat. Neurosci. 2001, 4, 803–812. [Google Scholar] [CrossRef]
- James, L.R.; Andrews, S.; Walker, S.; de Sousa, P.R.S.; Ray, A.; Russell, N.A.; Bellamy, T.C. High-throughput analysis of calcium signalling kinetics in astrocytes stimulated with different neurotransmitters. PLoS ONE 2011, 6, e26889. [Google Scholar] [CrossRef]
- Nett, W.J.; Oloff, S.H.; Mccarthy, K.D. Hippocampal astrocytes in situ exhibit calcium oscillations that occur independent of neuronal activity. J. Neurophysiol. 2002, 87, 528–537. [Google Scholar] [CrossRef]
- Fiacco, T.A.; McCarthy, K.D. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J. Neurosci. 2004, 24, 722–732. [Google Scholar] [CrossRef]
- Finkbeiner, S.M. Glial calcium. Glia 1993, 9, 83–104. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, W.T.; Cornell-Bell, A.H.; Sontheimer, H. Coupling in Glial Cells: Who Is Coupled, and Why? Glia 1994, 11, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science 1994, 263, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Hosmane, S.; Yang, I.H.; Ruffin, A.; Thakor, N.; Venkatesan, A. Circular compartmentalized microfluidic platform: Study of axon–glia interactions. Lab Chip 2010, 10, 741–747. [Google Scholar] [CrossRef]
- Shi, M.; Majumdar, D.; Gao, Y.; Brewer, B.M.; Goodwin, C.R.; McLean, J.; Li, D.; Webb, D.J. Glia co-culture with neurons in microfluidic platforms promotes the formation and stabilization of synaptic contacts. Lab Chip 2013, 13, 3008–3021. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Bushell, T.J.; Zagnoni, M. Chemically induced synaptic activity between mixed primary hippocampal co-cultures in a microfluidic system. Integr. Biol. 2014, 6, 636–644. [Google Scholar] [CrossRef]
- Park, J.; Koito, H.; Li, J.; Han, A. Multi-compartment neuron–glia co-culture platform for localized CNS axon–glia interaction study. Lab Chip 2012, 12, 3296. [Google Scholar] [CrossRef]
- Hosmane, S.; Tegenge, M.A.; Rajbhandari, L.; Uapinyoying, P.; Kumar, N.G.; Thakor, N.; Venkatesan, A. Toll/interleukin-1 receptor domain-containing adapter inducing interferon-β mediates microglial phagocytosis of degenerating axons. J. Neurosci. 2012, 32, 7745–7757. [Google Scholar] [CrossRef]
- Majumdar, D.; Gao, Y.; Li, D.; Webb, D.J. Co-culture of neurons and glia in a novel microfluidic platform. J. Neurosci. Methods 2011, 196, 38–44. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millet, L.J.; Jain, A.; Gillette, M.U. Less Is More: Oligomer Extraction and Hydrothermal Annealing Increase PDMS Adhesion Forces for Materials Studies and for Biology-Focused Microfluidic Applications. Micromachines 2023, 14, 214. https://doi.org/10.3390/mi14010214
Millet LJ, Jain A, Gillette MU. Less Is More: Oligomer Extraction and Hydrothermal Annealing Increase PDMS Adhesion Forces for Materials Studies and for Biology-Focused Microfluidic Applications. Micromachines. 2023; 14(1):214. https://doi.org/10.3390/mi14010214
Chicago/Turabian StyleMillet, Larry J., Anika Jain, and Martha U. Gillette. 2023. "Less Is More: Oligomer Extraction and Hydrothermal Annealing Increase PDMS Adhesion Forces for Materials Studies and for Biology-Focused Microfluidic Applications" Micromachines 14, no. 1: 214. https://doi.org/10.3390/mi14010214
APA StyleMillet, L. J., Jain, A., & Gillette, M. U. (2023). Less Is More: Oligomer Extraction and Hydrothermal Annealing Increase PDMS Adhesion Forces for Materials Studies and for Biology-Focused Microfluidic Applications. Micromachines, 14(1), 214. https://doi.org/10.3390/mi14010214