
The Fungal bZIP Transcription Factor AtfB Controls Virulence-Associated Processes in Aspergillus parasiticus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
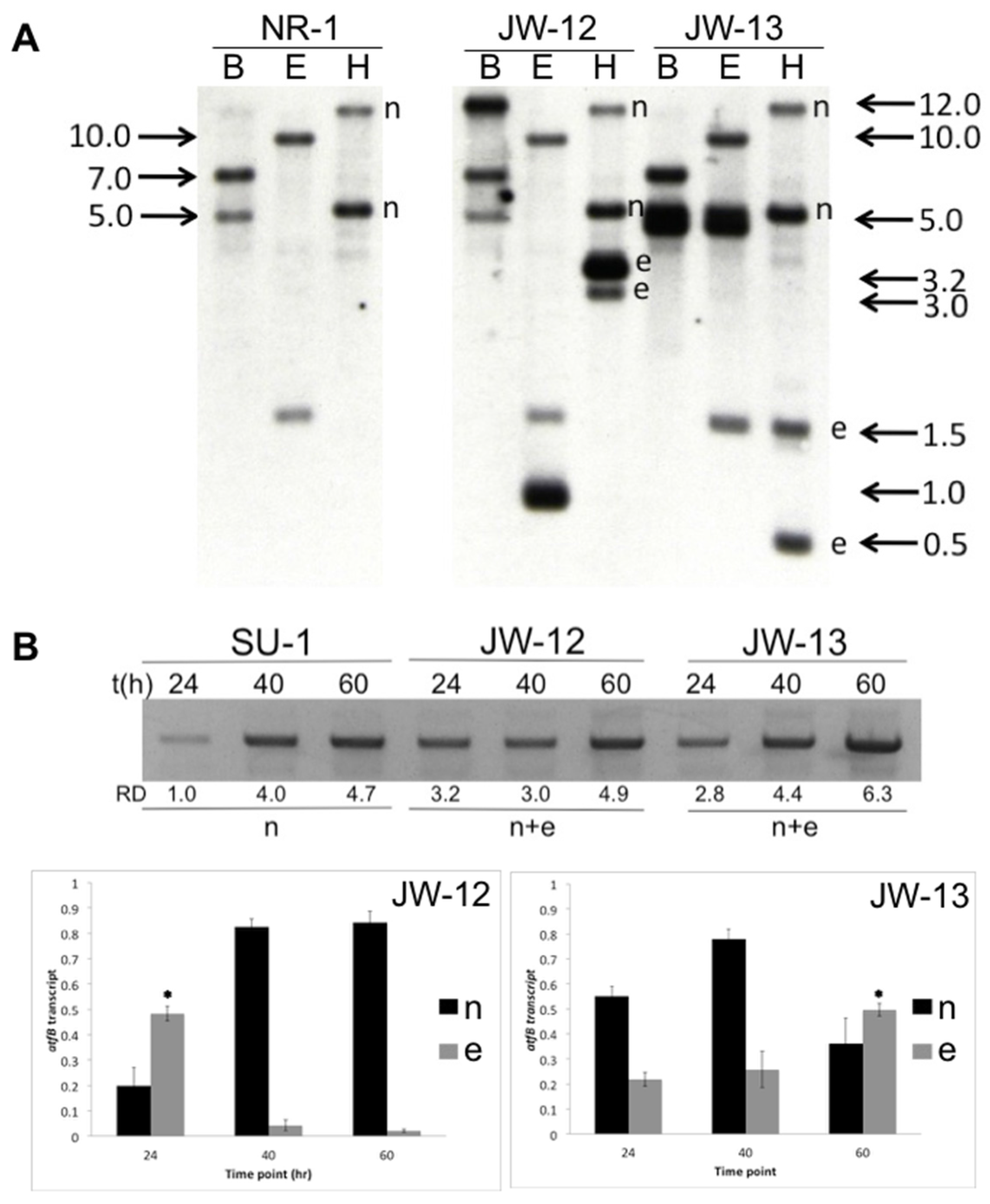
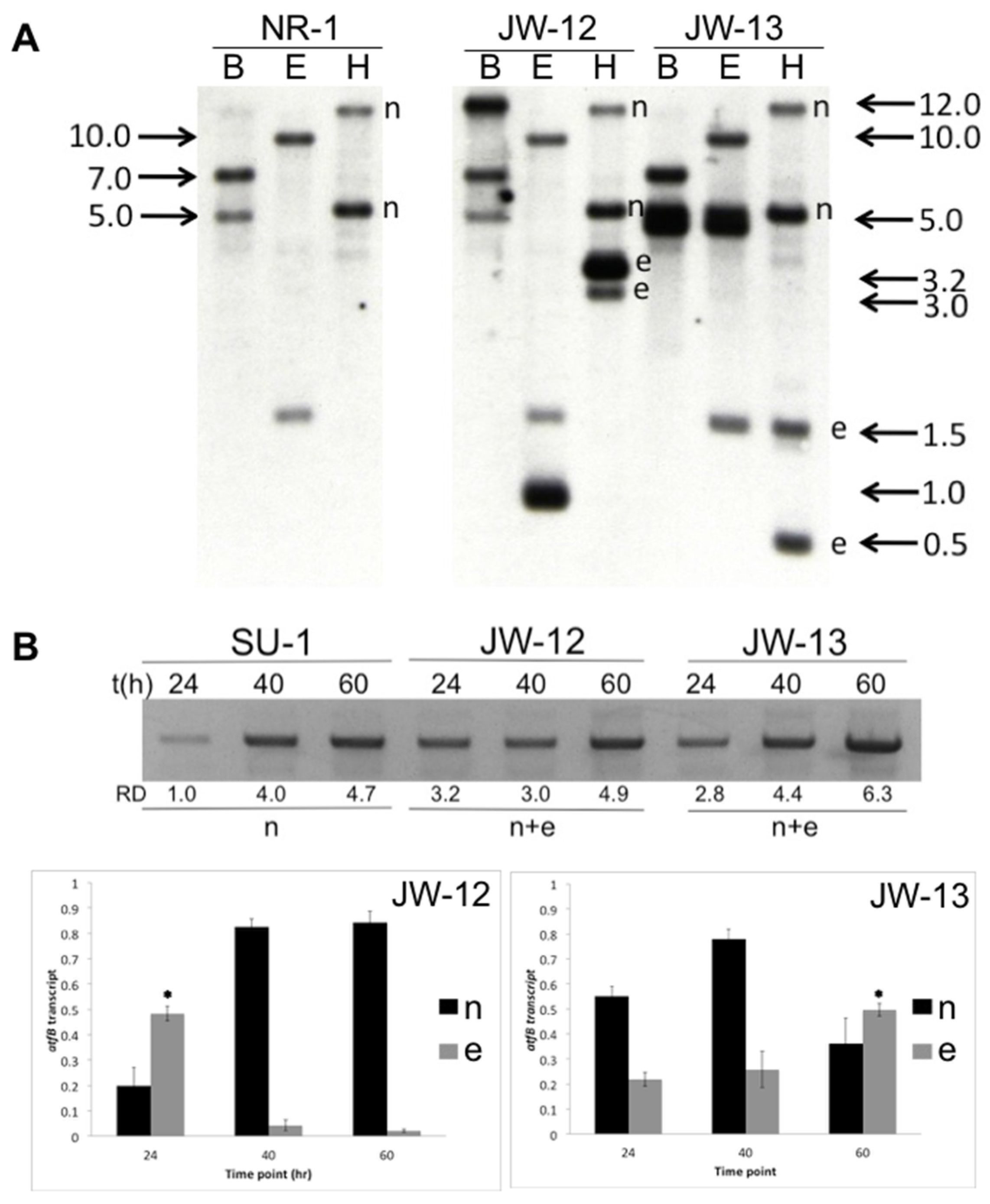
2.1. Molecular Analysis of AtfB-Silenced Strains
2.2. AtfB Directly Regulates Aflatoxin Biosynthesis and Fungal Development
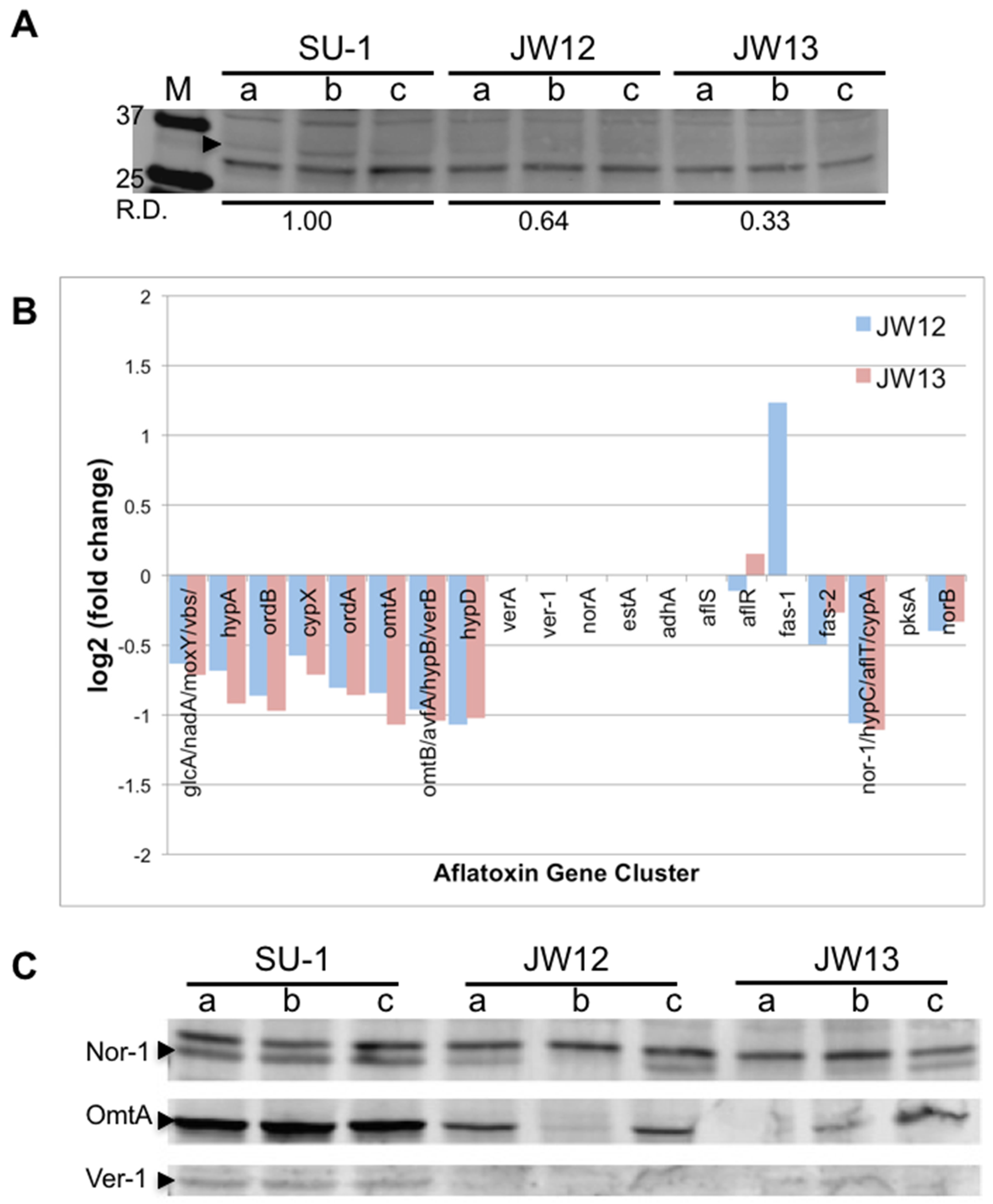
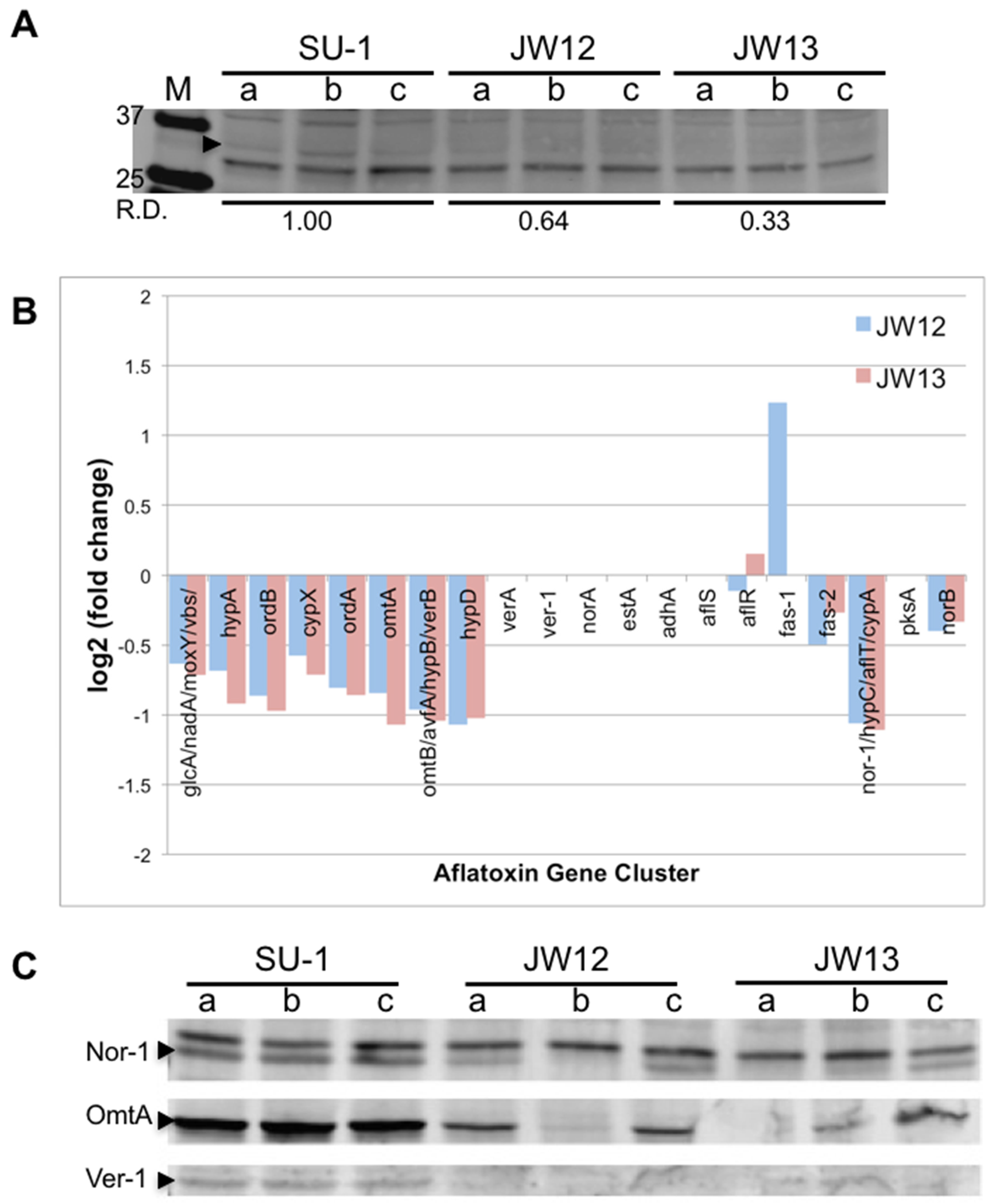
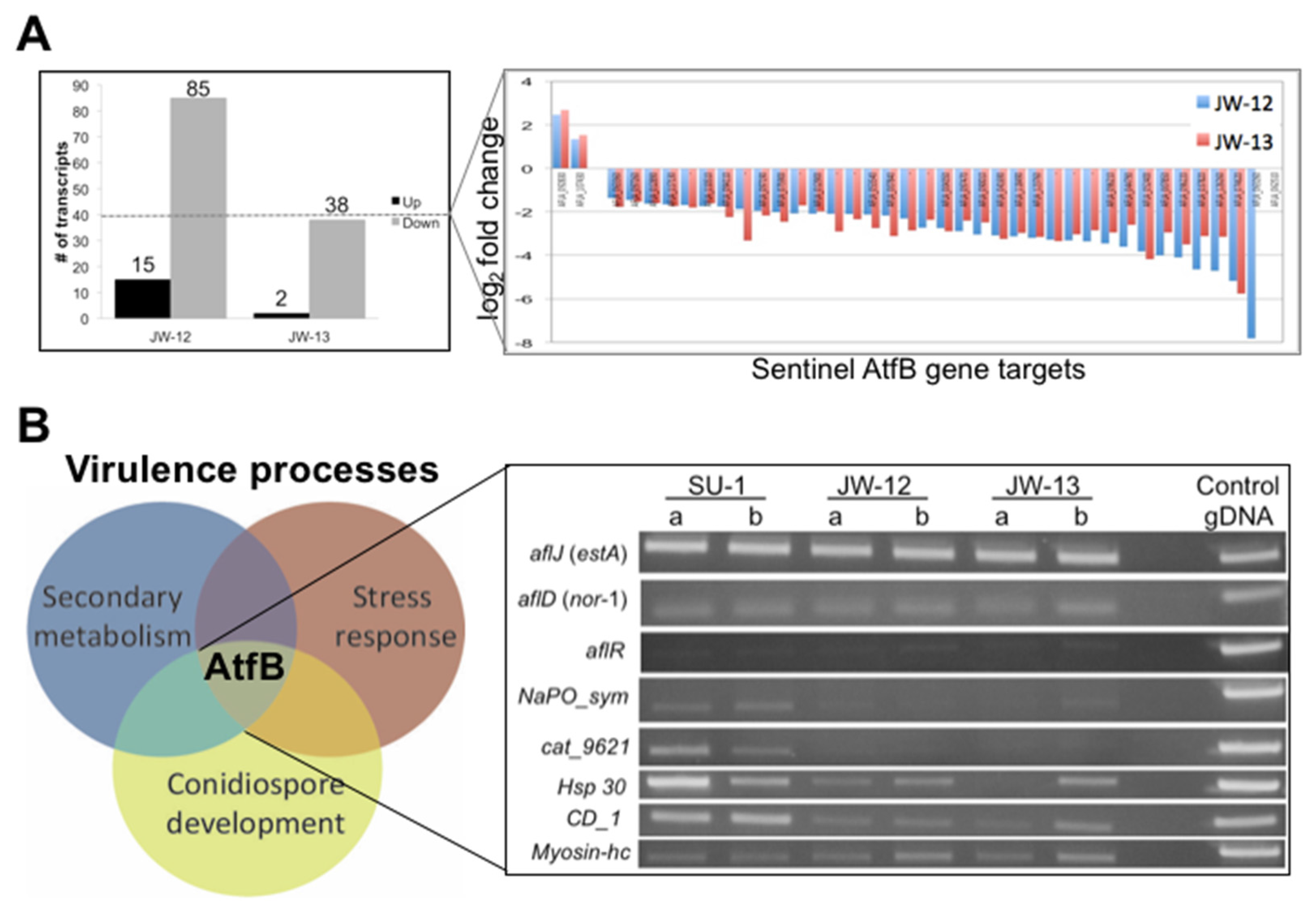
2.2.1. AtfB Directly Regulates Genes in the Aflatoxin Cluster
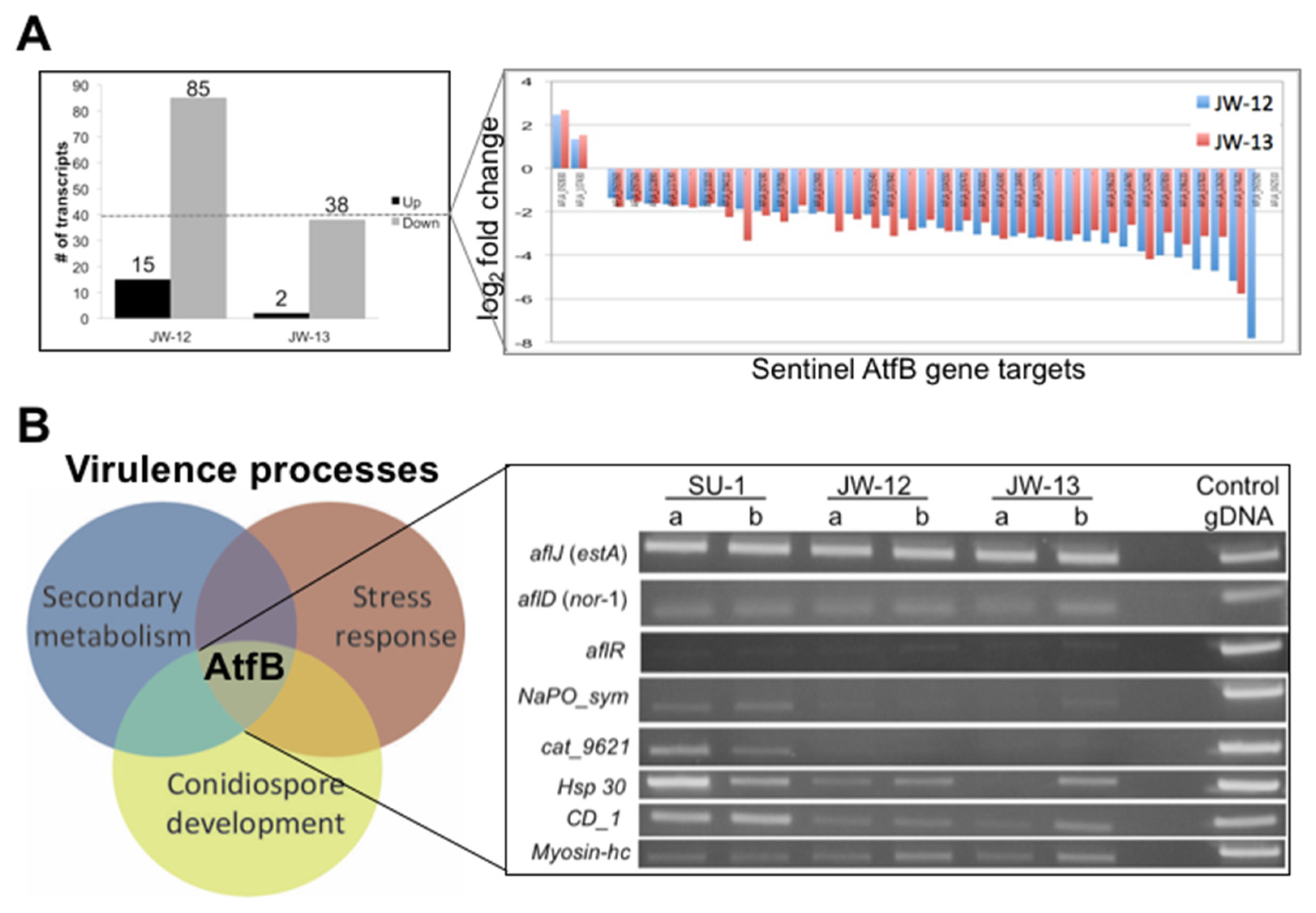
2.2.2. AtfB Regulates Expression of Sentinel Virulence-Associated Genes
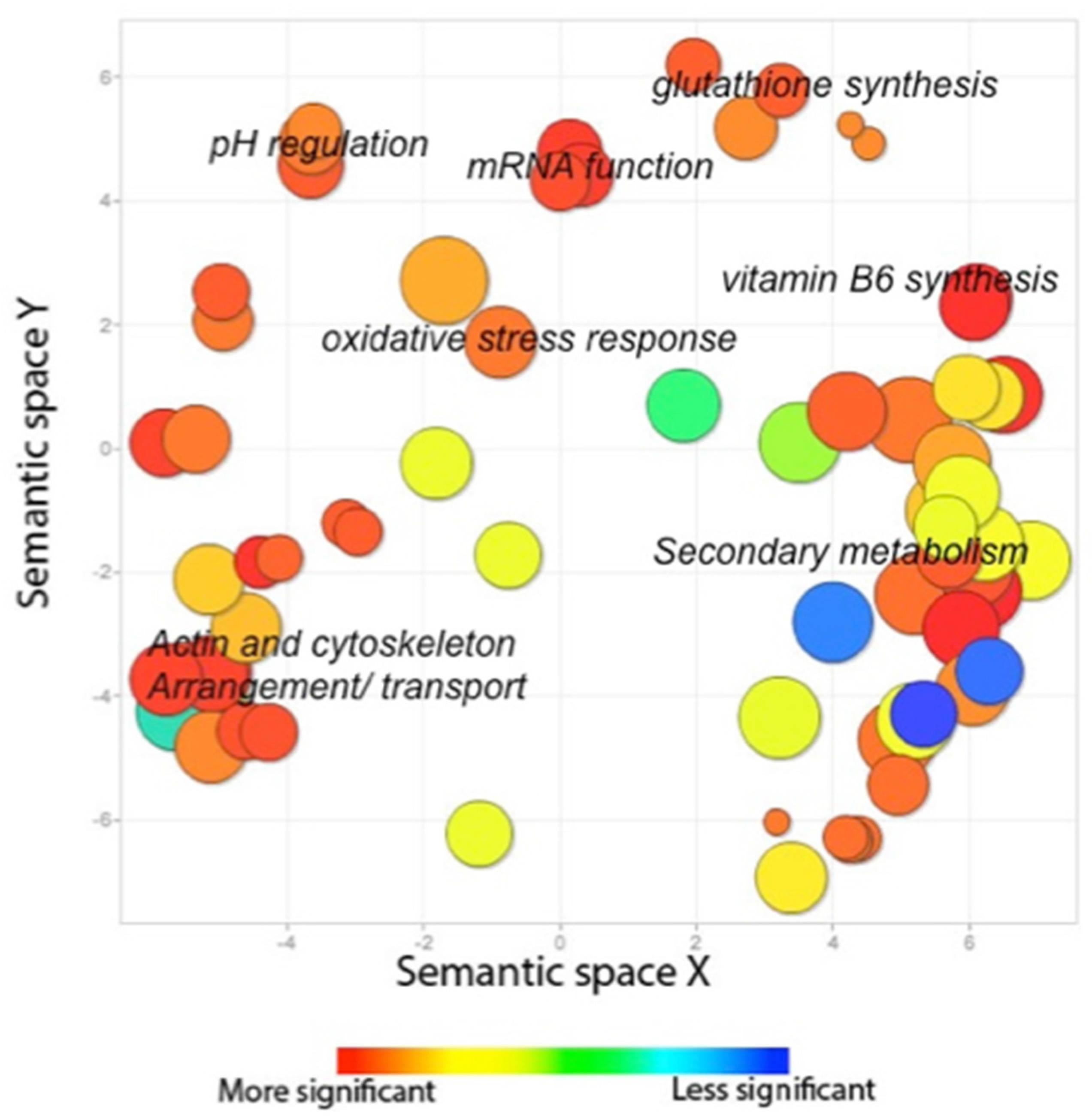
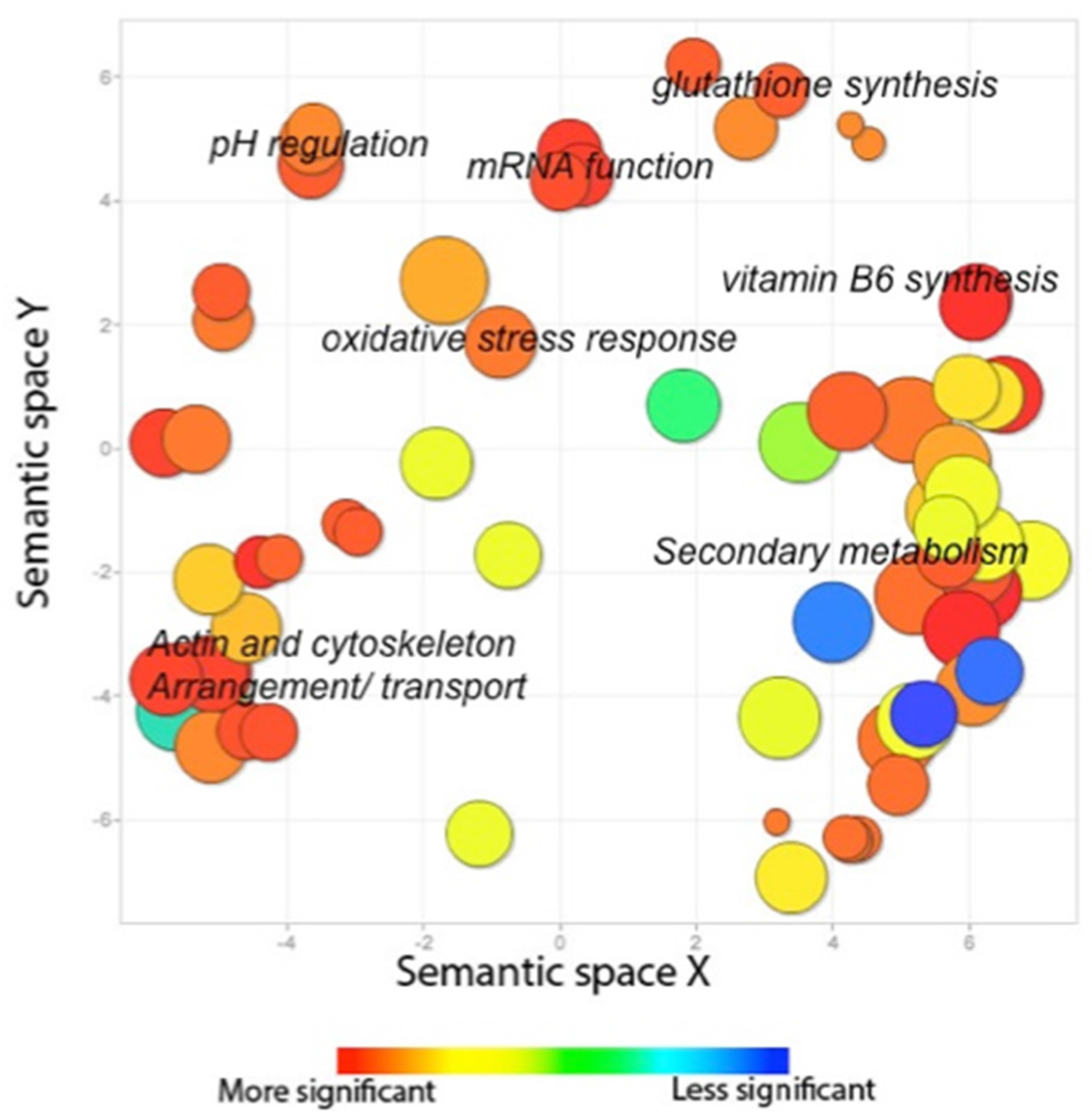
2.2.3. The AtfB Regulatory Network Extends beyond Canonical Oxidative Stress Response Pathways Associated with Fungal bZIPs
3. Discussion
3.1. Functional Significance of the AtfB Regulatory Network
3.2. Key Features of RNAi-Based Gene Silencing of AtfB
4. Conclusions
5. Materials and Methods
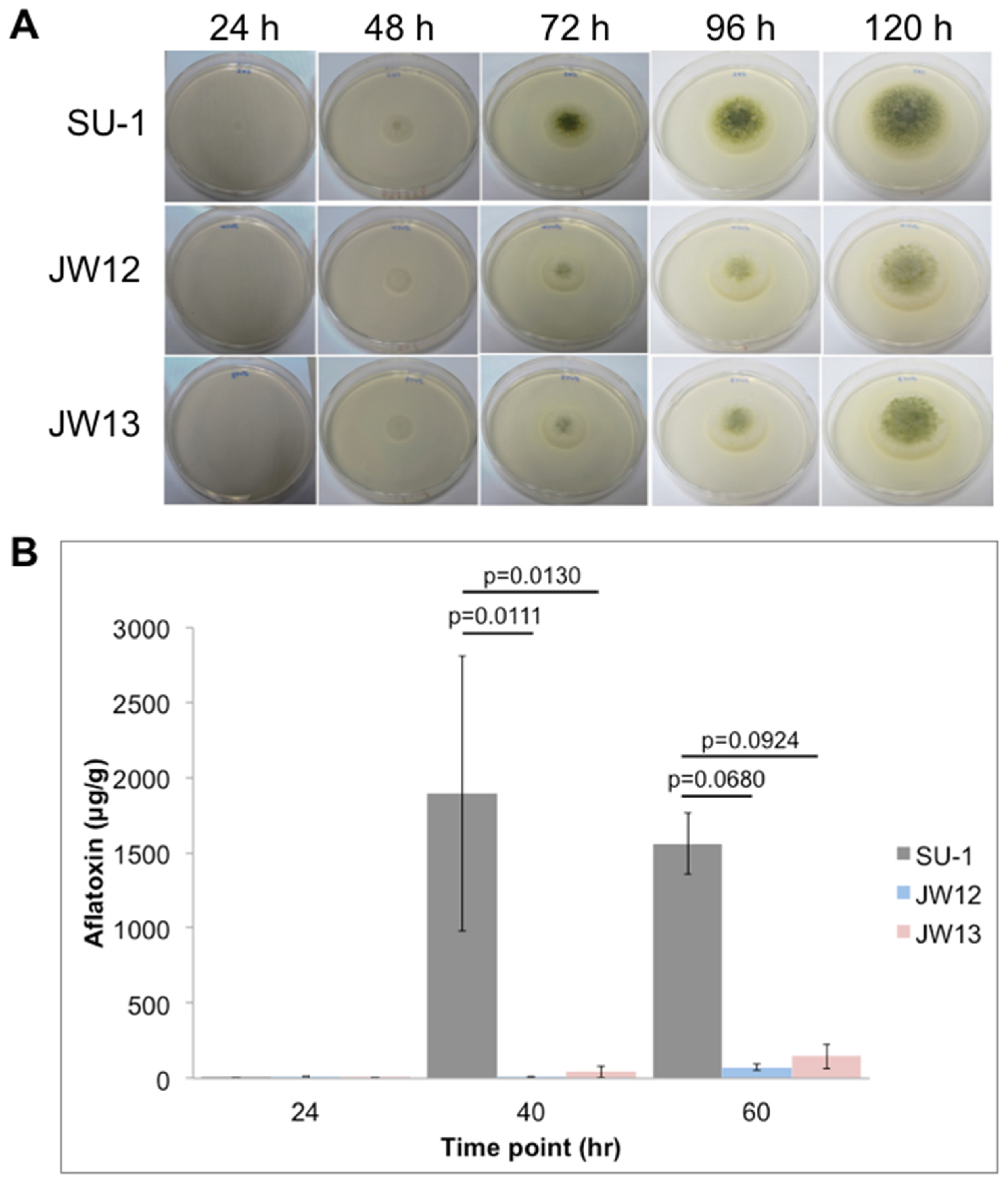
5.1. Fungal Strains and Growth Conditions
5.2. Creation of AtfB-Silenced Strains
5.2.1. Silencing Constructs
5.2.2. Fungal Transformation and Strain Confirmation
5.3. DNA Extraction and Southern Blot Analysis
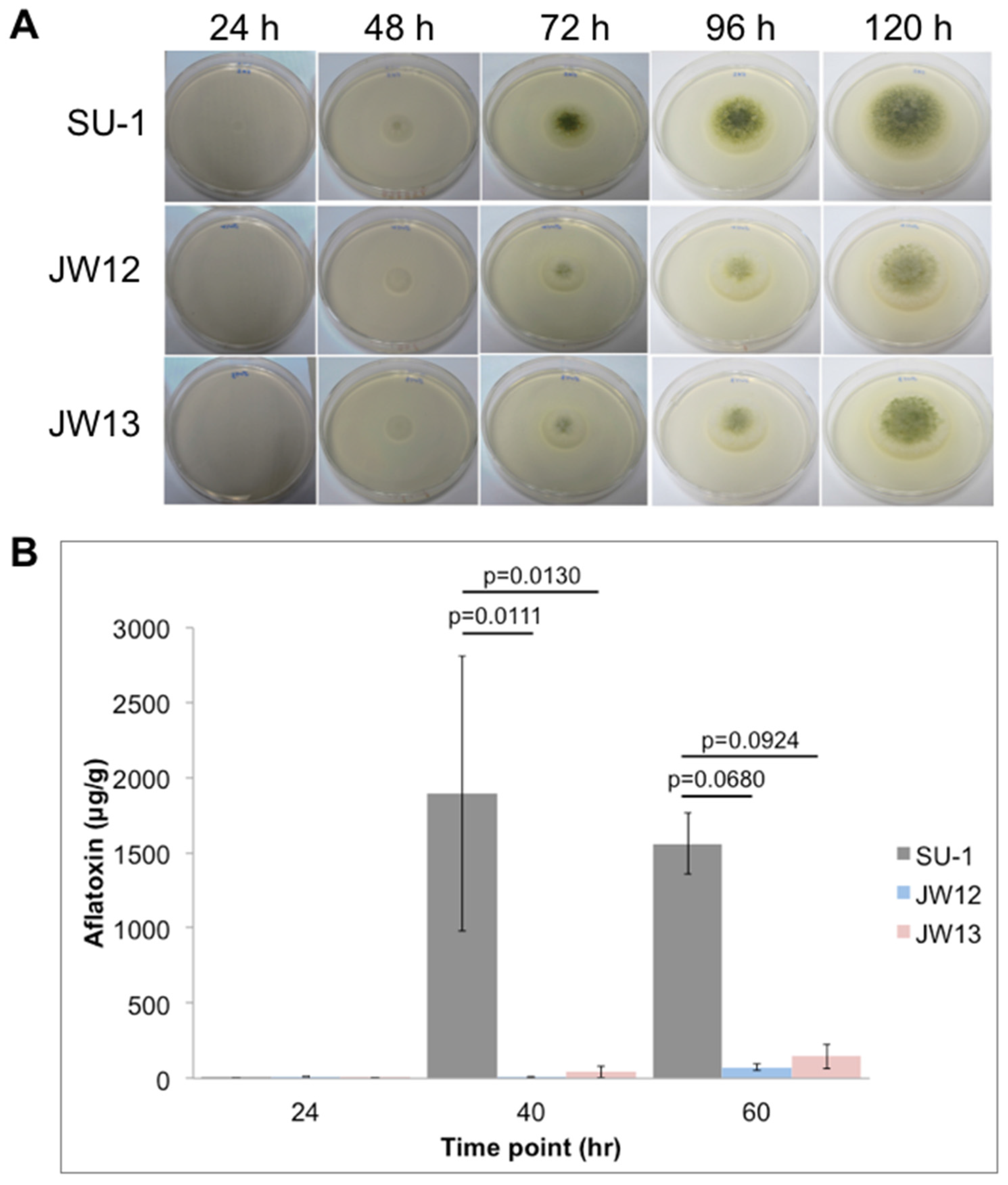
5.4. Aflatoxin Measurements in SU-1 and AtfB-Silenced Strains
5.5. RNA Isolation, Transcript Analysis, and HindIII Cutting Assay
5.6. Protein Extraction and Western Blot Analysis
5.7. RNA Isolation and cDNA Library Preparation for RNA Seq Analysis
5.8. RNA Seq Analysis
5.9. RT-PCR Analysis
5.10. Gene Ontology (GO) Pathway Analysis and REVIGO
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE Signaling Pathway: Key Mediator in Oxidative Stress and Potential Therapeutic Target in ALS. Neurol. Res. Int. 2012, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Roze, L.V.; Chanda, A.; Wee, J.; Awad, D.; Linz, J.E. Stress-related Transcription Factor AtfB Integrates Secondary Metabolism with Oxidative Stress Response in Aspergilli. J. Biol. Chem. 2011, 286, 35137–35148. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Arima, T.; Iwashita, K.; Yamada, O.; Gomi, K.; Akita, O. Aspergillus oryzae atfB encodes a transcription factor required for stress tolerance in conidia. Fungal Genet. Biol. 2008, 45, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Reverberi, M.; Zjalic, S.; Ricelli, A.; Punelli, F.; Camera, E.; Fabbri, C.; Picardo, M.; Fanelli, C.; Fabbri, A. Modulation of antioxidant defense in Aspergillus parasiticus is involved in aflatoxin biosynthesis: A role for the ApyapA gene. Eukaryot. Cell 2008, 7, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Iwashita, K.; Yamada, O.; Kobayashi, K.; Mizuno, A.; Akita, O.; Mikami, S.; Shimoi, H.; Gomi, K. Aspergillus oryzae atfA controls conidial germination and stress tolerance. Fungal Genet. Biol. 2009, 46, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.-B.; Amaike, S.; Wohlbach, D.J.; Gasch, A.P.; Chiang, Y.-M.; Wang, C.C.C.; Bok, J.W.; Rohlfs, M.; Keller, N.P. An Aspergillus nidulans bZIP response pathway hardwired for defensive secondary metabolism operates through aflR. Mol. Microbiol. 2012, 83, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-Y.; Roze, L.V.; Wee, J.; Linz, J.E. Evidence that a transcription factor regulatory network coordinates oxidative stress response and secondary metabolism in aspergilli. Microbiologyopen 2013, 2, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Etxebeste, O.; Herrero-García, E.; Araújo-Bazán, L.; Rodríguez-Urra, A.B.; Garzia, A.; Ugalde, U.; Espeso, E.A. The bZIP-type transcription factor FIbB regulates distinct morphogenetic stages of colony formation in Aspergillus nidulans. Mol. Microbiol. 2009, 73, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, A.W.; Irniger, S.; Braus, G.H. Induction of jlbA mRNA synthesis for a putative bZIP protein of Aspergillus nidulans by amino acid starvation. Curr. Genet. 2001, 39, 327–334. [Google Scholar] [PubMed]
- Amaike, S.; Affeldt, K.J.; Yin, W.B.; Franke, S.; Choithani, A.; Keller, N.P. The bZIP Protein MeaB Mediates Virulence Attributes in Aspergillus flavus. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Bok, J.W.; Wiemann, P.; Garvey, G.S.; Lim, F.Y.; Haas, B.; Wortman, J.; Keller, N.P. Illumina identification of RsrA, a conserved C2H2 transcription factor coordinating the NapA mediated oxidative stress signaling pathway in Aspergillus. BMC Genom. 2014, 15, 1011. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.-B.; Reinke, A.W.; Szilagyi, M.; Emri, T.; Chiang, Y.-M.; Keating, A.E.; Pocsi, I.; Wang, C.C.C.; Keller, N.P. bZIP transcription factors affecting secondary metabolism, sexual development and stress responses in Aspergillus nidulans. Microbiology 2013, 159, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Roze, L.V.; Miller, M.J.; Rarick, M.; Mahanti, N.; Linz, J.E. A Novel cAMP-response Element, CRE1, Modulates Expression of nor-1 in Aspergillus parasiticus. J. Biol. Chem. 2004, 279, 27428–27439. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Roze, L.V.; Trail, F.; Linz, J.E. Role of cis-acting sites NorL, a TATA box, and AflR1 in nor-1 transcriptional activation in Aspergillus parasiticus. Appl. Environ. Microbiol. 2005, 71, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, K.C.; Montalbano, B.G.; Cary, J.W. Binding of the C6-zinc cluster protein, AFLR, to the promoters of aflatoxin pathway biosynthesis genes in Aspergillus parasiticus. Gene 1999, 230, 249–257. [Google Scholar] [CrossRef]
- Price, M.S.; Yu, J.; Nierman, W.C.; Kim, H.; Pritchard, B.; Jacobus, C.A.; Bhatnagar, D.; Cleveland, T.E.; Payne, G.A. The aflatoxin pathway regulator AflR induces gene transcription inside and outside of the aflatoxin biosynthetic cluster. FEMS Microbiol. Lett. 2006, 255, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Horng, J.S.; Chang, P.K.; Pestka, J.J.; Linz, J.E. Development of a homologous transformation system for Aspergillus parasiticus with the gene encoding nitrate reductase. Mol. Gen. Genet. 1990, 224, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Skory, C.D.; Chang, P.K.; Linz, J.E. Regulated expression of the nor-1 and ver-1 genes associated with aflatoxin biosynthesis. Appl. Environ. Microbiol. 1993, 59, 1642–1646. [Google Scholar] [PubMed]
- Linz, J.E.; Wee, J.; Roze, L.V. Aspergillus parasiticus SU-1 genome sequence, predicted chromosome structure, and comparative gene expression under aflatoxin-inducing conditions: evidence that differential expression contributes to species phenotype. Eukaryot. Cell 2014, 13, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Skory, C.D.; Horng, J.S.; Pestka, J.J.; Linz, J.E. Transformation of Aspergillus parasiticus with a homologous gene (pyrG) involved in pyrimidine biosynthesis. Appl. Environ. Microbiol. 1990, 56, 3315–3320. [Google Scholar] [PubMed]
- Hong, S.Y.; Linz, J.E. Functional expression and sub-cellular localization of the early aflatoxin pathway enzyme Nor-1 in Aspergillus parasiticus. Mycol. Res. 2009, 113, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Roze, L.V.; Beaudry, R.M.; Keller, N.P.; Linz, J.E. Regulation of aflatoxin synthesis by FadA/cAMP/protein kinase A signaling in Aspergillus parasiticus. Mycopathologia 2004, 158, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.W.; Chiou, C.H.; Linz, J.E. Function of native OmtA in vivo and expression and distribution of this protein in colonies of Aspergillus parasiticus. Appl. Environ. Microbiol. 2002, 68, 5718–5727. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.-W.; Chiou, C.-H.; Klomparens, K.L.; Cary, J.W.; Linz, J.E. Subcellular localization of aflatoxin biosynthetic enzymes Nor-1, Ver-1, and OmtA in time-dependent fractionated colonies of Aspergillus parasiticus. Arch. Microbiol. 2004, 181, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-Y.; Linz, J.E. Functional expression and subcellular localization of the aflatoxin pathway enzyme Ver-1 fused to enhanced green fluorescent protein. Appl. Environ. Microbiol. 2008, 74, 6385–6396. [Google Scholar]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Roze, L.V.; Arthur, A.E.; Hong, S.-Y.; Chanda, A.; Linz, J.E. The initiation and pattern of spread of histone H4 acetylation parallel the order of transcriptional activation of genes in the aflatoxin cluster. Mol. Microbiol. 2007, 66, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Chanda, A.; Roze, L.V.; Kang, S.; Artymovich, K.A.; Hicks, G.R.; Raikhel, N.V.; Calvo, A.M.; Linz, J.E. A key role for vesicles in fungal secondary metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 19533–19538. [Google Scholar] [CrossRef] [PubMed]
- Sekonyela, R.; Palmer, J.M.; Bok, J.-W.; Jain, S.; Berthier, E.; Forseth, R.; Schroeder, F.; Keller, N.P. RsmA Regulates Aspergillus fumigatus Gliotoxin Cluster Metabolites Including Cyclo(L-Phe-L-Ser), a Potential New Diagnostic Marker for Invasive Aspergillosis. PLoS ONE 2013, 8, e62591. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P.; Turner, G.; Bennett, J.W. Fungal secondary metabolism—From biochemistry to genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Brakhage, A.A. Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 2013, 11, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Li, J.; Glass, N.L. Exploring the bZIP transcription factor regulatory network in Neurospora crassa. Microbiology 2011, 157, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Menke, J.; Weber, J.; Broz, K.; Kistler, H.C. Cellular development associated with induced mycotoxin synthesis in the filamentous fungus Fusarium graminearum. PLoS ONE 2013, 8, e63077. [Google Scholar] [CrossRef] [PubMed]
- Amare, M.G.; Keller, N.P. Molecular mechanisms of Aspergillus flavus secondary metabolism and development. Fungal Genet. Biol. 2014, 66, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.K.; Yu, J.H.; Keller, N.P.; Adams, T.H. Aspergillus sporulation and mycotoxin production both require inactivation of the FadA Gα protein-dependent signaling pathway. EMBO J. 1997, 16, 4916–4923. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.W.; Silverstein, R.B.; Kruger, S.J. Isolation and characterization of two nonaflatoxigenic classes of morphological variants of Aspergillus parasiticus. J. Am. Oil Chem. Soc. 1981, 58, A952–A955. [Google Scholar] [CrossRef]
- Kale, S.P.; Cary, J.W.; Bhatnagar, D.; Bennett, J.W. Characterization of experimentally induced, nonaflatoxigenic variant strains of Aspergillus parasiticus. Appl. Environ. Microbiol. 1996, 62, 3399–3404. [Google Scholar] [PubMed]
- Calvo, A.M.; Bok, J.; Brooks, W.; Keller, N.P. veA is required for toxin and sclerotial production in Aspergillus parasiticus. Appl. Environ. Microbiol. 2004, 70, 4733–4739. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, N.A. Epistasis and Genetic Regulation of Variation in the Arabidopsis Metabolome. Plant Cell 2008, 20, 1185–1186. [Google Scholar] [CrossRef]
- Monnahan, P.J.; Kelly, J.K. Epistasis Is a Major Determinant of the Additive Genetic Variance in Mimulus guttatus. PLoS Genet. 2015, 11, e1005201. [Google Scholar] [CrossRef] [PubMed]
- Chanda, A.; Roze, L.V.; Linz, J.E. A Possible Role for Exocytosis in Aflatoxin Export in Aspergillus parasiticus. Eukaryot. Cell 2010, 9, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Linz, J.E.; Chanda, A.; Hong, S.-Y.; Whitten, D.A.; Wilkerson, C.; Roze, L.V. Proteomic and Biochemical Evidence Support a Role for Transport Vesicles and Endosomes in Stress Response and Secondary Metabolism in Aspergillus parasiticus. J. Proteome Res. 2012, 11, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Kistler, H.C.; Broz, K. Cellular compartmentalization of secondary metabolism. Front Microbiol. 2015, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Linz, J.E.; Wee, J.; Roze, L.V. Aflatoxin biosynthesis: regulation and subcellular localization. In Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites; Martín, J.-F., García-Estrada, C., Zeilinger, S., Eds.; Springer: New York, NY, USA, 2014; pp. 89–110. [Google Scholar]
- Suelmann, R.; Fischer, R. Mitochondrial movement and morphology depend on an intact actin cytoskeleton in Aspergillus nidulans. Cell Motil. Cytoskelet. 2000, 45, 42–50. [Google Scholar] [CrossRef]
- Wee, J. Regulation and Subcellular Localization of Aflatoxin Biosynthesis in Aspergillus Parasiticus; Springer: New York, NY, USA, 2015. [Google Scholar]
- Cogoni, C.; Romano, N.; Macino, G. Suppression of gene expression by homologous transgenes. Antonie Van Leeuwenhoek 1994, 65, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Goldoni, M.; Azzalin, G.; Macino, G.; Cogoni, C. Efficient gene silencing by expression of double stranded RNA in Neurospora crassa. Fungal Genet Biol. 2004, 41, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Fulci, V.; Macino, G. Quelling: post-transcriptional gene silencing guided by small RNAs in Neurospora crassa. Curr. Opin. Microbiol. 2007, 10, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Masanga, J.O.; Matheka, J.M.; Omer, R.A.; Ommeh, S.C.; Monda, E.O.; Alakonya, A.E. Downregulation of transcription factor aflR in Aspergillus flavus confers reduction to aflatoxin accumulation in transgenic maize with alteration of host plant architecture. Plant Cell Rep. 2015, 34, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wee, J.; Hong, S.-Y.; Roze, L.V.; Day, D.M.; Chanda, A.; Linz, J.E. The Fungal bZIP Transcription Factor AtfB Controls Virulence-Associated Processes in Aspergillus parasiticus. Toxins 2017, 9, 287. https://doi.org/10.3390/toxins9090287
Wee J, Hong S-Y, Roze LV, Day DM, Chanda A, Linz JE. The Fungal bZIP Transcription Factor AtfB Controls Virulence-Associated Processes in Aspergillus parasiticus. Toxins. 2017; 9(9):287. https://doi.org/10.3390/toxins9090287
Chicago/Turabian StyleWee, Josephine, Sung-Yong Hong, Ludmila V. Roze, Devin M. Day, Anindya Chanda, and John E. Linz. 2017. "The Fungal bZIP Transcription Factor AtfB Controls Virulence-Associated Processes in Aspergillus parasiticus" Toxins 9, no. 9: 287. https://doi.org/10.3390/toxins9090287