
Total Body Irradiation Mitigates Inflammation and Extends the Therapeutic Time Window for Anti-Ricin Antibody Treatment against Pulmonary Ricinosis in Mice

Abstract
:1. Introduction
2. Results
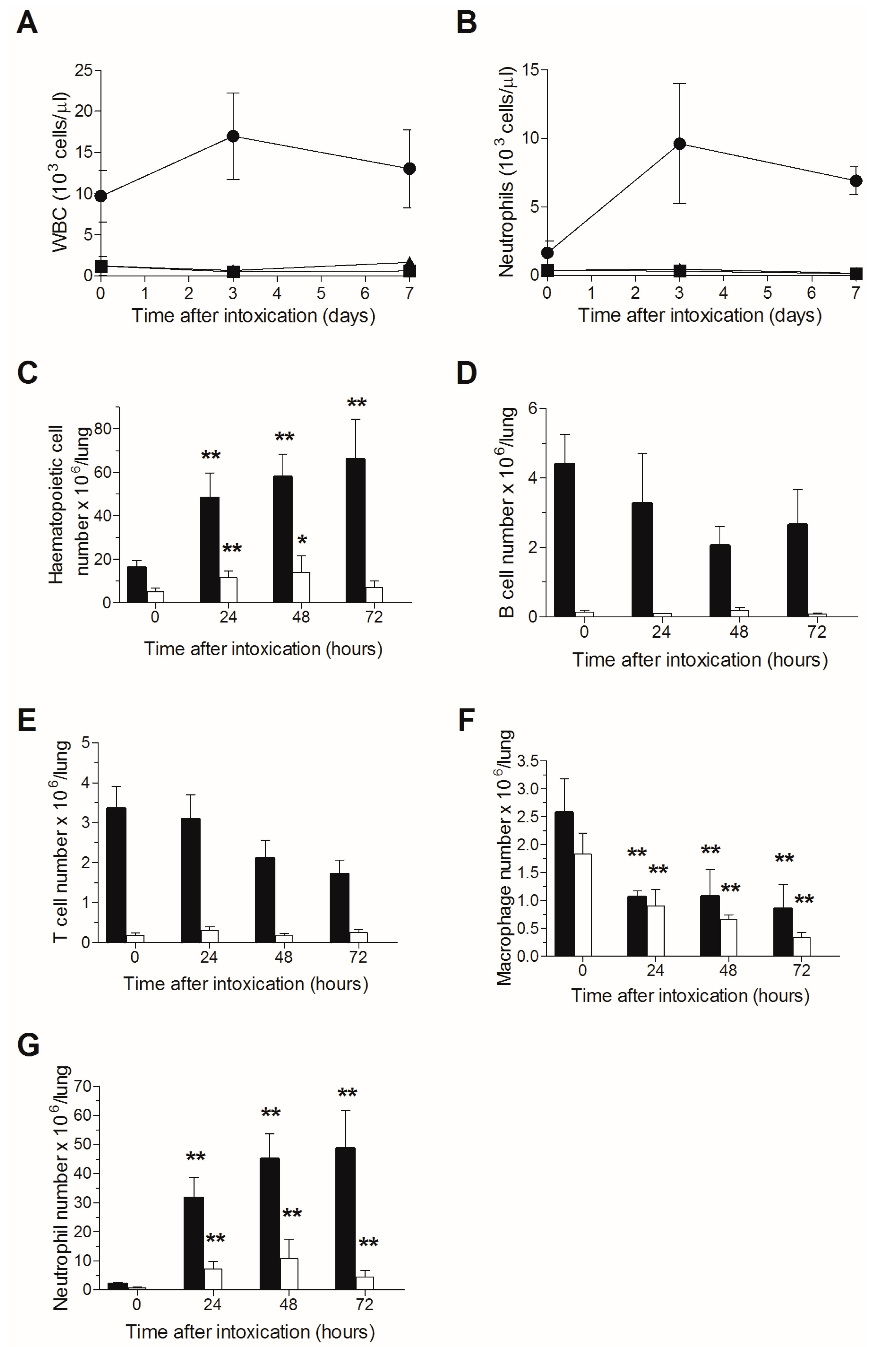
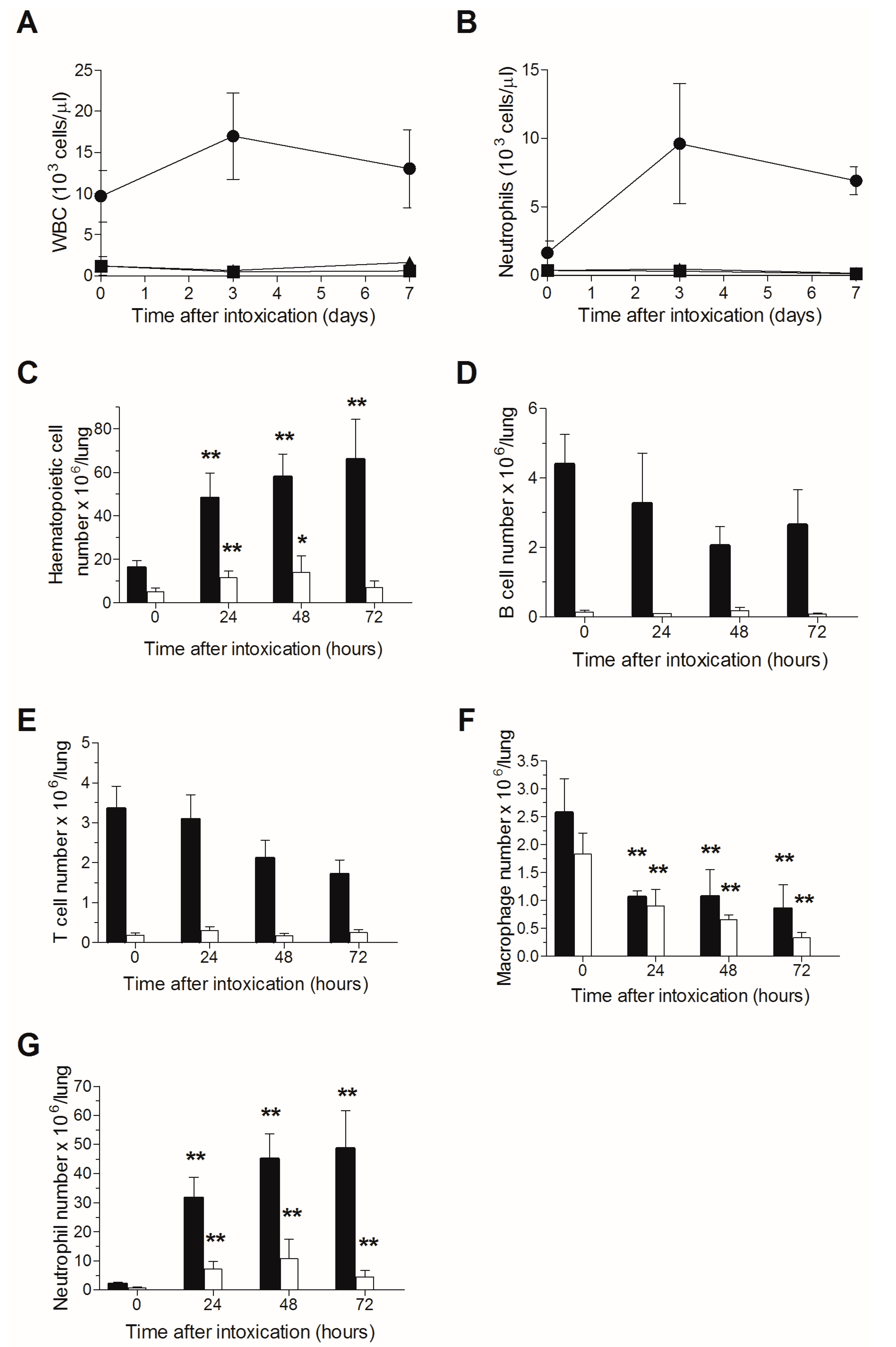
2.1. Effects of TBI on Peripheral Blood Count and Pulmonary Hematopoietic Cell Count Following Intranasal Ricin Intoxication
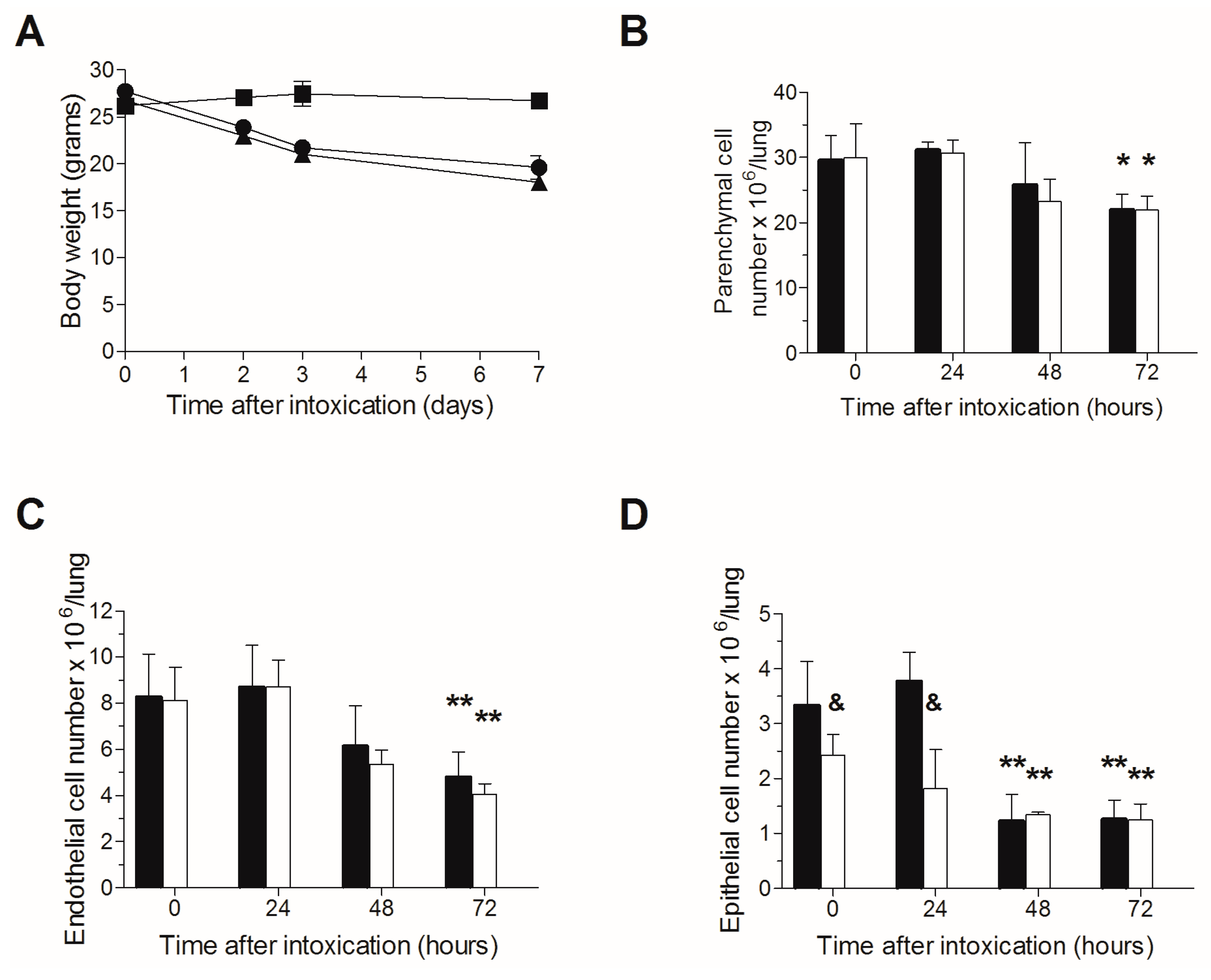
2.2. Lack of Additive Toxicity Following TBI in Ricin Intoxicated Mice
2.3. TBI-Induced Extension of the Mean Time to Death in Ricin Intoxicated Mice
2.4. Anti-Inflammatory Effects Induced by TBI Following Intranasal Ricin Intoxication
3. Discussion
4. Materials and Methods
4.1. Ricin Preparation
4.2. Anti-Ricin Antibodies
4.3. Animal Studies
4.4. Total Body Irradiation (TBI) Protocol
4.5. Peripheral Blood Counts
4.6. Flow Cytometry of the Lungs
4.7. Bronchoalveolar Lavage Fluid (BALF) Preparation and Analysis
4.8. Depurination Assay
4.9. Statistical Analysis
Author Contributions
Conflicts of Interest
References
- Olsnes, S.; Kozlov, J.V. Ricin. Toxicon 2001, 39, 1723–1728. [Google Scholar] [CrossRef]
- Greenfield, R.A.; Brown, B.R.; Hutchins, J.B.; Iandolo, J.J.; Jackson, R.; Slater, L.N.; Bronze, M.S. Microbiological, biological, and chemical weapons of warfare and terrorism. Am. J. Med. Sci. 2002, 323, 326–340. [Google Scholar] [CrossRef] [PubMed]
- Audi, J.; Belson, M.; Patel, M.; Schier, J.; Osterloh, J. Ricin poisoning: A comprehensive review. JAMA 2005, 294, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K. Rna n-glycosidase activity of ricin a-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar] [PubMed]
- Hartley, M.R.; Lord, J.M. Cytotoxic ribosome-inactivating lectins from plants. Biochim. Biophys. Acta 2004, 1701, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Olmo, N.; Turnay, J.; Gonzalez de Buitrago, G.; Lopez de Silanes, I.; Gavilanes, J.G.; Lizarbe, M.A. Cytotoxic mechanism of the ribotoxin alpha-sarcin. Induction of cell death via apoptosis. Eur. J. Biochem. 2001, 268, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Olsnes, S.; Fernandez-Puentes, C.; Carrasco, L.; Vazquez, D. Ribosome inactivation by the toxic lectins abrin and ricin. Kinetics of the enzymic activity of the toxin a-chains. Eur. J. Biochem. 1975, 60, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Prasad, V.; Valbonesi, P.; Srivastava, S.; Ghosal-Chowdhury, P.; Barbieri, L.; Bolognesi, A.; Stirpe, F. A systemic antiviral resistance-inducing protein isolated from clerodendrum inerme gaertn. Is a polynucleotide:Adenosine glycosidase (ribosome-inactivating protein). FEBS Lett. 1996, 396, 132–134. [Google Scholar] [CrossRef]
- Falach, R.; Sapoznikov, A.; Gal, Y.; Israeli, O.; Leitner, M.; Seliger, N.; Ehrlich, S.; Kronman, C.; Sabo, T. Quantitative profiling of the in vivo enzymatic activity of ricin reveals disparate depurination of different pulmonary cell types. Toxicol. Lett. 2016, 258, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.A.; Holgate, S.T. The airway epithelium: Structural and functional properties in health and disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Gal, Y.; Mazor, O.; Alcalay, R.; Seliger, N.; Aftalion, M.; Sapoznikov, A.; Falach, R.; Kronman, C.; Sabo, T. Antibody/doxycycline combined therapy for pulmonary ricinosis: Attenuation of inflammation improved survival of ricin-intoxicated mice. Toxicol. Rep. 2014, 1, 496–504. [Google Scholar] [CrossRef]
- Wilhelmsen, C.L.; Pitt, M.L. Lesions of acute inhaled lethal ricin intoxication in rhesus monkeys. Vet. Pathol. 1996, 33, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Katalan, S.; Falach, R.; Rosner, A.; Goldvaser, M.; Brosh-Nissimov, T.; Dvir, A.; Mizrachi, A.; Goren, O.; Cohen, B.; Gal, Y.; et al. A novel swine model of ricin-induced acute respiratory distress syndrome. Dis. Model. Mech. 2017, 10, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Gal, Y.; Sapoznikov, A.; Falach, R.; Ehrlich, S.; Aftalion, M.; Sabo, T.; Kronman, C. Potent antiedematous and protective effects of ciprofloxacin in pulmonary ricinosis. Antimicrob. Agents Chemother. 2016, 60, 7153–7158. [Google Scholar] [PubMed]
- DaSilva, L.; Cote, D.; Roy, C.; Martinez, M.; Duniho, S.; Pitt, M.L.; Downey, T.; Dertzbaugh, M. Pulmonary gene expression profiling of inhaled ricin. Toxicon 2003, 41, 813–822. [Google Scholar] [CrossRef]
- Lindauer, M.L.; Wong, J.; Iwakura, Y.; Magun, B.E. Pulmonary inflammation triggered by ricin toxin requires macrophages and il-1 signaling. J. Immunol. 2009, 183, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.A.; Rivera, V.R.; Pitt, M.L.; Vogel, P. Aerosolized specific antibody protects mice from lung injury associated with aerosolized ricin exposure. Toxicon 1996, 34, 1037–1044. [Google Scholar] [CrossRef]
- Abraham, E.; Carmody, A.; Shenkar, R.; Arcaroli, J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1137–L1145. [Google Scholar] [PubMed]
- Till, G.O.; Johnson, K.J.; Kunkel, R.; Ward, P.A. Intravascular activation of complement and acute lung injury. Dependency on neutrophils and toxic oxygen metabolites. J. Clin. Investig. 1982, 69, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Chen, F.; Xu, L.; Xing, J.; Wang, X. HMGB1-TLR4-IL23-IL17A axis promotes paraquat-induced acute lung injury by mediating neutrophil infiltration in mice. Sci. Rep. 2017, 7, 597. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.P.; Liu, Q.; Broman, M.; Predescu, D.; Frey, R.S.; Malik, A.B. Inactivation of cd11b in a mouse transgenic model protects against sepsis-induced lung pmn infiltration and vascular injury. Physiol. Genom. 2005, 21, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Koma, T.; Yoshimatsu, K.; Nagata, N.; Sato, Y.; Shimizu, K.; Yasuda, S.P.; Amada, T.; Nishio, S.; Hasegawa, H.; Arikawa, J. Neutrophil depletion suppresses pulmonary vascular hyperpermeability and occurrence of pulmonary edema caused by hantavirus infection in c.B-17 scid mice. J. Virol. 2014, 88, 7178–7188. [Google Scholar] [CrossRef] [PubMed]
- Sercundes, M.K.; Ortolan, L.S.; Debone, D.; Soeiro-Pereira, P.V.; Gomes, E.; Aitken, E.H.; Neto, A.C.; Russo, M.; MR, D.I.L.; Alvarez, J.M.; et al. Targeting neutrophils to prevent malaria-associated acute lung injury/acute respiratory distress syndrome in mice. PLoS Pathog. 2016, 12, e1006054. [Google Scholar] [CrossRef] [PubMed]
- Segel, G.B.; Halterman, M.W.; Lichtman, M.A. The paradox of the neutrophil’s role in tissue injury. J. Leukoc. Biol. 2011, 89, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.L.; Moots, R.J.; Bucknall, R.C.; Edwards, S.W. Neutrophil function in inflammation and inflammatory diseases. Rheumatology 2010, 49, 1618–1631. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sharma, A.K.; LaPar, D.J.; Kron, I.L.; Ailawadi, G.; Liu, Y.; Jones, D.R.; Laubach, V.E.; Lau, C.L. Depletion of tissue plasminogen activator attenuates lung ischemia-reperfusion injury via inhibition of neutrophil extravasation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L718–L729. [Google Scholar] [CrossRef] [PubMed]
- Strickland, I.; Kisich, K.; Hauk, P.J.; Vottero, A.; Chrousos, G.P.; Klemm, D.J.; Leung, D.Y. High constitutive glucocorticoid receptor beta in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. J. Exp. Med. 2001, 193, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Cox, G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J. Immunol. 1995, 154, 4719–4725. [Google Scholar] [PubMed]
- Daffern, P.J.; Jagels, M.A.; Hugli, T.E. Multiple epithelial cell-derived factors enhance neutrophil survival. Regulation by glucocorticoids and tumor necrosis factor-alpha. Am. J. Respir. Cell Mol. Biol. 1999, 21, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.Z.; Hisha, H.; Yang, G.X.; Fan, T.X.; Jin, T.; Li, Q.; Lian, Z.; Ikehara, S. Optimal protocol for total body irradiation for allogeneic bone marrow transplantation in mice. Bone Marrow Transplant. 2002, 30, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.; Lapidot, T.; Gozes, D.; Singer, T.S.; Reisner, Y. Abrogation of bone marrow allograft resistance in mice by increased total body irradiation correlates with eradication of host clonable t cells and alloreactive cytotoxic precursors. J. Immunol. 1987, 138, 460–465. [Google Scholar] [PubMed]
- Shen, H.; Yu, H.; Liang, P.H.; Cheng, H.; XuFeng, R.; Yuan, Y.; Zhang, P.; Smith, C.A.; Cheng, T. An acute negative bystander effect of gamma-irradiated recipients on transplanted hematopoietic stem cells. Blood 2012, 119, 3629–3637. [Google Scholar] [CrossRef] [PubMed]
- Neelis, K.J.; Visser, T.P.; Dimjati, W.; Thomas, G.R.; Fielder, P.J.; Bloedow, D.; Eaton, D.L.; Wagemaker, G. A single dose of thrombopoietin shortly after myelosuppressive total body irradiation prevents pancytopenia in mice by promoting short-term multilineage spleen-repopulating cells at the transient expense of bone marrow-repopulating cells. Blood 1998, 92, 1586–1597. [Google Scholar] [PubMed]
- Heissig, B.; Rafii, S.; Akiyama, H.; Ohki, Y.; Sato, Y.; Rafael, T.; Zhu, Z.; Hicklin, D.J.; Okumura, K.; Ogawa, H.; et al. Low-dose irradiation promotes tissue revascularization through vegf release from mast cells and mmp-9-mediated progenitor cell mobilization. J. Exp. Med. 2005, 202, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Sapoznikov, A.; Falach, R.; Mazor, O.; Alcalay, R.; Gal, Y.; Seliger, N.; Sabo, T.; Kronman, C. Diverse profiles of ricin-cell interactions in the lung following intranasal exposure to ricin. Toxins 2015, 7, 4817–4831. [Google Scholar] [CrossRef] [PubMed]
- Ghafoori, P.; Marks, L.B.; Vujaskovic, Z.; Kelsey, C.R. Radiation-induced lung injury. Assessment, management, and prevention. Oncology 2008, 22, 37–47; discussion 52–53. [Google Scholar] [PubMed]
- Marks, L.B.; Yu, X.; Vujaskovic, Z.; Small, W., Jr.; Folz, R.; Anscher, M.S. Radiation-induced lung injury. Semin. Radiat. Oncol. 2003, 13, 333–345. [Google Scholar] [CrossRef]
- Medhora, M.; Gao, F.; Jacobs, E.R.; Moulder, J.E. Radiation damage to the lung: Mitigation by angiotensin-converting enzyme (ace) inhibitors. Respirology 2012, 17, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Mehta, V. Radiation pneumonitis and pulmonary fibrosis in non-small-cell lung cancer: Pulmonary function, prediction, and prevention. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Su, Y.; Hou, X.; He, H.; Liu, S.; Wu, J.; Rao, P. Protective effect of recombinant protein sod-tat on radiation-induced lung injury in mice. Life Sci. 2012, 91, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Tsoutsou, P.G.; Koukourakis, M.I. Radiation pneumonitis and fibrosis: Mechanisms underlying its pathogenesis and implications for future research. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Sabo, T.; Gal, Y.; Elhanany, E.; Sapoznikov, A.; Falach, R.; Mazor, O.; Kronman, C. Antibody treatment against pulmonary exposure to abrin confers significantly higher levels of protection than treatment against ricin intoxication. Toxicol. Lett. 2015, 237, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Hoidal, J.R.; Mukherjee, T.K. Role of tnfalpha in pulmonary pathophysiology. Respir. Res. 2006, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Finsterbusch, M.; Voisin, M.B.; Beyrau, M.; Williams, T.J.; Nourshargh, S. Neutrophils recruited by chemoattractants in vivo induce microvascular plasma protein leakage through secretion of tnf. J. Exp. Med. 2014, 211, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Komaki, Y.; Sugiura, H.; Koarai, A.; Tomaki, M.; Ogawa, H.; Akita, T.; Hattori, T.; Ichinose, M. Cytokine-mediated xanthine oxidase upregulation in chronic obstructive pulmonary disease’s airways. Pulm. Pharmacol. Ther. 2005, 18, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.M.; Ginger, L.A.; Kosila, N.; Elkins, N.D.; Essary, B.; McManaman, J.L.; Repine, J.E. Mononuclear phagocyte xanthine oxidoreductase contributes to cytokine-induced acute lung injury. Am. J. Respir. Cell Mol. Biol. 2004, 30, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Grosso, M.A.; Brown, J.M.; Viders, D.E.; Mulvin, D.W.; Banerjee, A.; Velasco, S.E.; Repine, J.E.; Harken, A.H. Xanthine oxidase-derived oxygen radicals induce pulmonary edema via direct endothelial cell injury. J. Surg. Res. 1989, 46, 355–360. [Google Scholar] [CrossRef]
- Faggioni, R.; Gatti, S.; Demitri, M.T.; Delgado, R.; Echtenacher, B.; Gnocchi, P.; Heremans, H.; Ghezzi, P. Role of xanthine oxidase and reactive oxygen intermediates in lps- and tnf-induced pulmonary edema. J. Lab. Clin. Med. 1994, 123, 394–399. [Google Scholar] [PubMed]
- Arbibe, L.; Koumanov, K.; Vial, D.; Rougeot, C.; Faure, G.; Havet, N.; Longacre, S.; Vargaftig, B.B.; Bereziat, G.; Voelker, D.R.; et al. Generation of lyso-phospholipids from surfactant in acute lung injury is mediated by type-ii phospholipase a2 and inhibited by a direct surfactant protein a-phospholipase a2 protein interaction. J. Clin. Investig. 1998, 102, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Touqui, L.; Arbibe, L. A role for phospholipase a2 in ards pathogenesis. Mol. Med. Today 1999, 5, 244–249. [Google Scholar] [CrossRef]
- Costa-Junior, H.M.; Hamaty, F.C.; da Silva Farias, R.; Einicker-Lamas, M.; da Silva, M.H.; Persechini, P.M. Apoptosis-inducing factor of a cytotoxic t cell line: Involvement of a secretory phospholipase a2. Cell Tissue Res. 2006, 324, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.A.; Bolognese, B.; Roshak, A. Respective roles of the 14 kda and 85 kda phospholipase a2 enzymes in human monocyte eicosanoid formation. Adv. Exp. Med. Biol. 1999, 469, 215–219. [Google Scholar] [PubMed]
- Rosenthal, M.D.; Gordon, M.N.; Buescher, E.S.; Slusser, J.H.; Harris, L.K.; Franson, R.C. Human neutrophils store type ii 14-kda phospholipase a2 in granules and secrete active enzyme in response to soluble stimuli. Biochem. Biophys. Res. Commun. 1995, 208, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Von Allmen, C.E.; Schmitz, N.; Bauer, M.; Hinton, H.J.; Kurrer, M.O.; Buser, R.B.; Gwerder, M.; Muntwiler, S.; Sparwasser, T.; Beerli, R.R.; et al. Secretory phospholipase a2-iid is an effector molecule of cd4+cd25+ regulatory t cells. Proc. Natl. Acad. Sci. USA 2009, 106, 11673–11678. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Quinton, L.J.; Simms, B.T.; Lupa, M.M.; Kogan, M.S.; Mizgerd, J.P. Roles of interleukin-6 in activation of stat proteins and recruitment of neutrophils during escherichia coli pneumonia. J. Infect. Dis. 2006, 193, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Leemans, J.C.; Vervoordeldonk, M.J.; Florquin, S.; van Kessel, K.P.; van der Poll, T. Differential role of interleukin-6 in lung inflammation induced by lipoteichoic acid and peptidoglycan from staphylococcus aureus. Am. J. Respir. Crit. Care Med. 2002, 165, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Rijneveld, A.W.; van den Dobbelsteen, G.P.; Florquin, S.; Standiford, T.J.; Speelman, P.; van Alphen, L.; van der Poll, T. Roles of interleukin-6 and macrophage inflammatory protein-2 in pneumolysin-induced lung inflammation in mice. J. Infect. Dis. 2002, 185, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Granell, S.; Gironella, M.; Bulbena, O.; Panes, J.; Mauri, M.; Sabater, L.; Aparisi, L.; Gelpi, E.; Closa, D. Heparin mobilizes xanthine oxidase and induces lung inflammation in acute pancreatitis. Crit. Care Med. 2003, 31, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, V.G.; Tan, S.; Weinbroum, A.; McCammon, A.T.; Samuelson, P.N.; Gelman, S.; Parks, D.A. Lung injury after hepatoenteric ischemia-reperfusion: Role of xanthine oxidase. Am. J. Respir. Crit. Care Med. 1996, 154, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Terada, L.S.; Dormish, J.J.; Shanley, P.F.; Leff, J.A.; Anderson, B.O.; Repine, J.E. Circulating xanthine oxidase mediates lung neutrophil sequestration after intestinal ischemia-reperfusion. Am. J. Physiol. 1992, 263, L394–L401. [Google Scholar] [PubMed]
- Lee, Y.M.; Hybertson, B.M.; Terada, L.S.; Repine, A.J.; Cho, H.G.; Repine, J.E. Mepacrine decreases lung leak in rats given interleukin-1 intratracheally. Am. J. Respir. Crit. Care Med. 1997, 155, 1624–1628. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.M.; Meliton, A.Y.; Meliton, L.N.; Dudek, S.M.; Leff, A.R. Secretory group v phospholipase a2 regulates acute lung injury and neutrophilic inflammation caused by lps in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L879–L887. [Google Scholar] [CrossRef] [PubMed]
- Burgos, R.A.; Hidalgo, M.A.; Figueroa, C.D.; Conejeros, I.; Hancke, J.L. New potential targets to modulate neutrophil function in inflammation. Mini Rev. Med. Chem. 2009, 9, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Liu, S.Y. Studies on the antitumor lectins isolated from the seeds of ricinus communis (castor bean). Toxicon 1986, 24, 757–765. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intoxication Time | Blood Neutrophil Count (103 Cells/µL) | MTTD (Days) | |
|---|---|---|---|
| At Exposure Day | 3 Days Post Exposure | ||
| Non irradiated mice | 1.53 ± 0.60 (n = 12) | 13.43 ± 1.56 ** (n = 3) | 6.3 (n = 18) |
| 3 days post irradiation | 0.18 ± 0.09 (n = 9) | 0.40 ± 0.45 (n = 4) | 8.1 (n = 11) |
| 9 days post irradiation | 0.29 ± 0.26 (n = 3) | 0.50 ± 0.23 (n = 2) | 8.4 (n = 14) |
| 18 days post irradiation | 0.35 ± 0.12 (n = 3) | 11.36 ± 7.25 * (n = 4) | 5.6 (n = 9) |
| Marker | Pre-Intoxication Treatment | Time after Exposure (h) | |
|---|---|---|---|
| 0 | 72 | ||
| TNFα (pg/mL) | none | 37 ± 16 | 80 ± 2 7 * |
| TBI | 35 ± 5 | 106 ± 32 * | |
| IL-1β (pg/mL) | none | 0 ± 0 | 108 ± 76 |
| TBI | 3 ± 4 | 4 ± 6 # | |
| IL-6 (pg/mL) | none | 3 ± 2 | 1695 ± 711 ** |
| TBI | 0 ± 0 | 363 ± 99 *## | |
| ChE (mU/mL) | none | 3 ± 1 | 273 ± 38 ** |
| TBI | 5 ± 1 | 130 ± 38 **## | |
| Protein (mg/mL) | none | 0.4 ± 0.1 | 5.2 ± 1.1 ** |
| TBI | 0.7 ± 0.1 | 2.8 ± 0.5 **## | |
| XO (mU/mL) | none | 0.3 ± 0.3 | 3.7 ± 0.6 ** |
| TBI | 0.2 ± 0.1 | 2.4 ± 0.6 **## | |
| sPLA2 (U/mL) | none | 7.8 ± 5.8 | 24.5 ± 8.7 * |
| TBI | 1.8 ± 1.8 | 1.6 ± 3.2 ## | |
| Anti-Ricin Ab Treatment Following i.n. Exposure (2LD50) | ||||||
|---|---|---|---|---|---|---|
| Time of Ab Treatment (h) | --- | 48 | 72 | |||
| Pretreatment of Mice | none | TBI | none | TBI | none | TBI |
| % survival (survivors/total) | 1 (1/70) | 1 (1/76) | 4 (1/23) | 42 (10/24) | 0 (0/7) | 36 (4/11) |
| MTTD (days) | 6.4 | 9.4 | 6.0 | 8.7 | 3.6 | 8.4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gal, Y.; Sapoznikov, A.; Falach, R.; Ehrlich, S.; Aftalion, M.; Kronman, C.; Sabo, T. Total Body Irradiation Mitigates Inflammation and Extends the Therapeutic Time Window for Anti-Ricin Antibody Treatment against Pulmonary Ricinosis in Mice. Toxins 2017, 9, 278. https://doi.org/10.3390/toxins9090278
Gal Y, Sapoznikov A, Falach R, Ehrlich S, Aftalion M, Kronman C, Sabo T. Total Body Irradiation Mitigates Inflammation and Extends the Therapeutic Time Window for Anti-Ricin Antibody Treatment against Pulmonary Ricinosis in Mice. Toxins. 2017; 9(9):278. https://doi.org/10.3390/toxins9090278
Chicago/Turabian StyleGal, Yoav, Anita Sapoznikov, Reut Falach, Sharon Ehrlich, Moshe Aftalion, Chanoch Kronman, and Tamar Sabo. 2017. "Total Body Irradiation Mitigates Inflammation and Extends the Therapeutic Time Window for Anti-Ricin Antibody Treatment against Pulmonary Ricinosis in Mice" Toxins 9, no. 9: 278. https://doi.org/10.3390/toxins9090278