
Listeriolysin O Regulates the Expression of Optineurin, an Autophagy Adaptor That Inhibits the Growth of Listeria monocytogenes

 , and
, and
Abstract
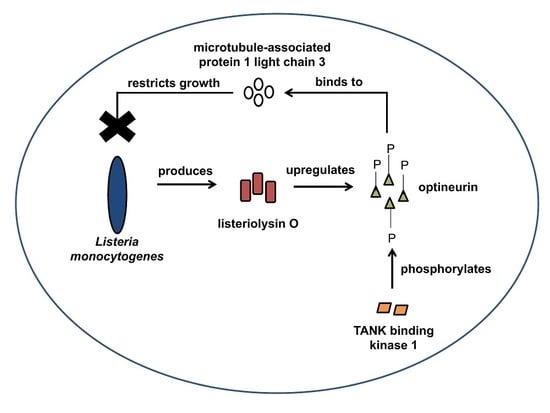
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
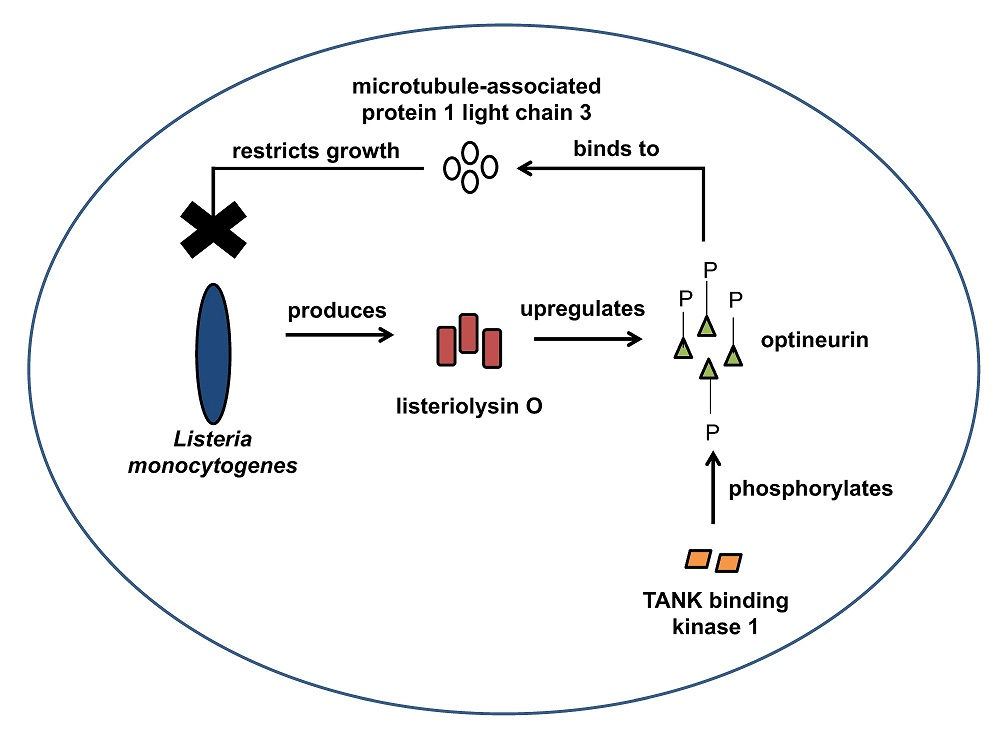
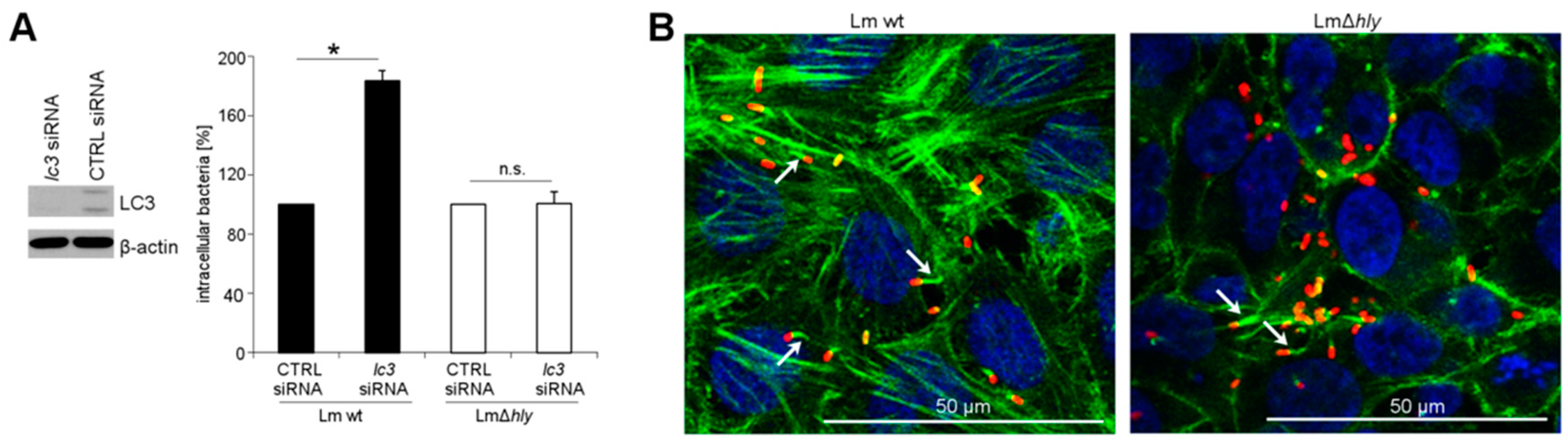
2.1. LC3 Is Essential for the Intracellular Growth Restriction of LLO-Producing L. monocytogenes
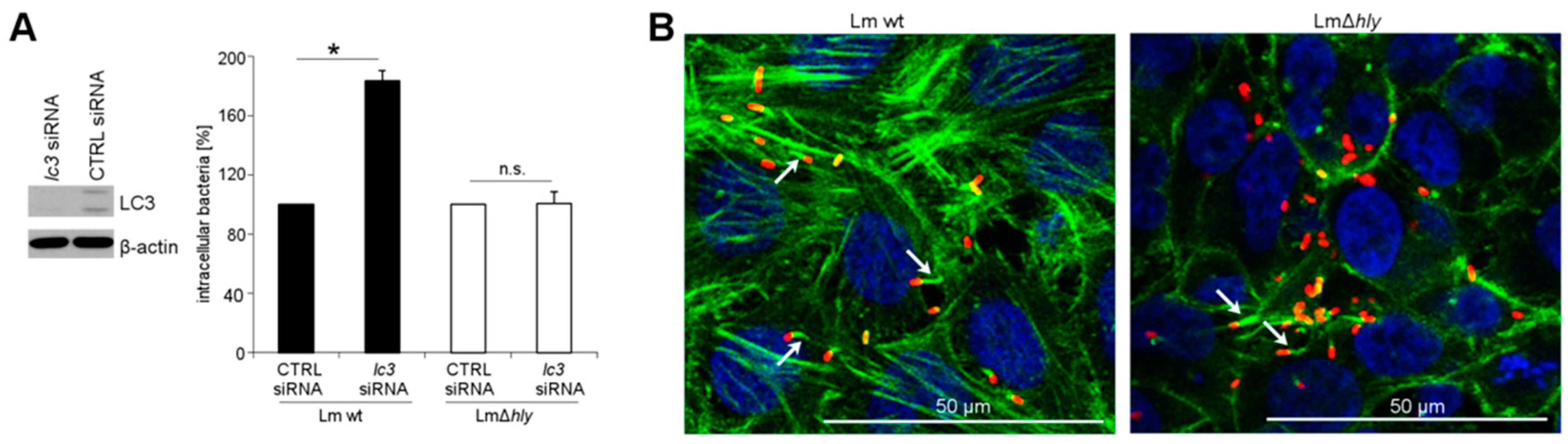
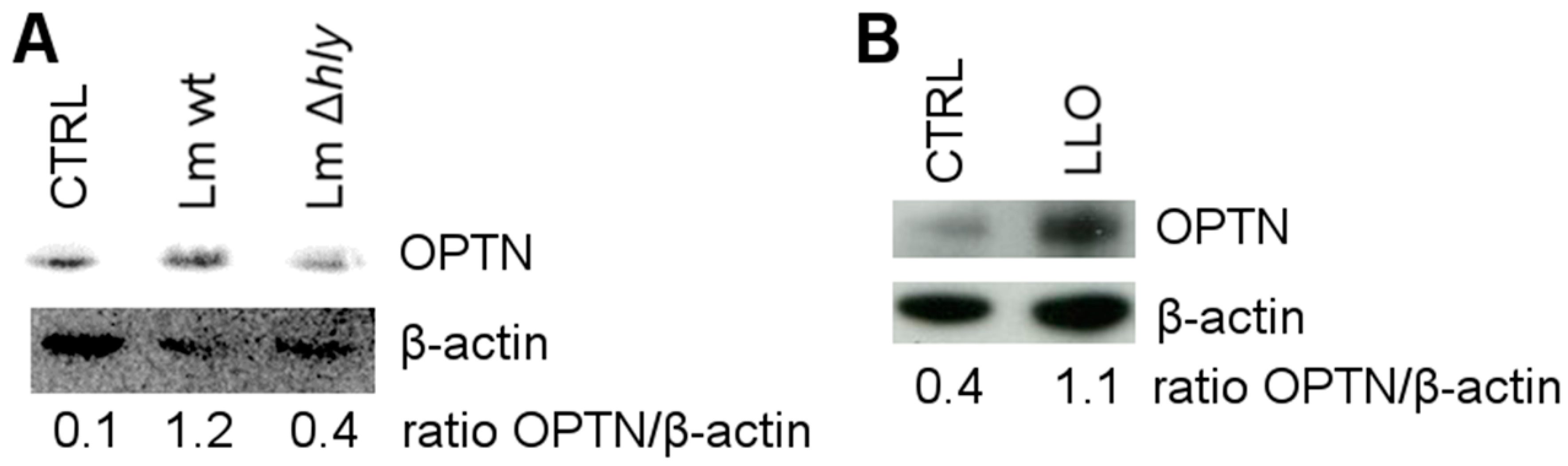
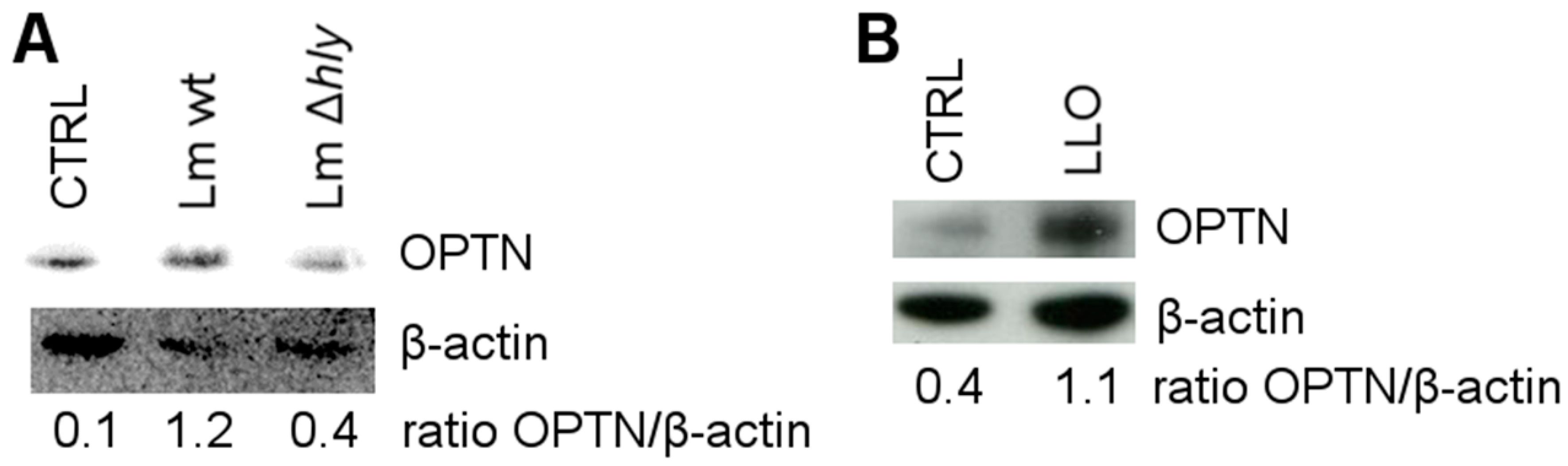
2.2. LLO Upregulates OPTN in HeLa Cells
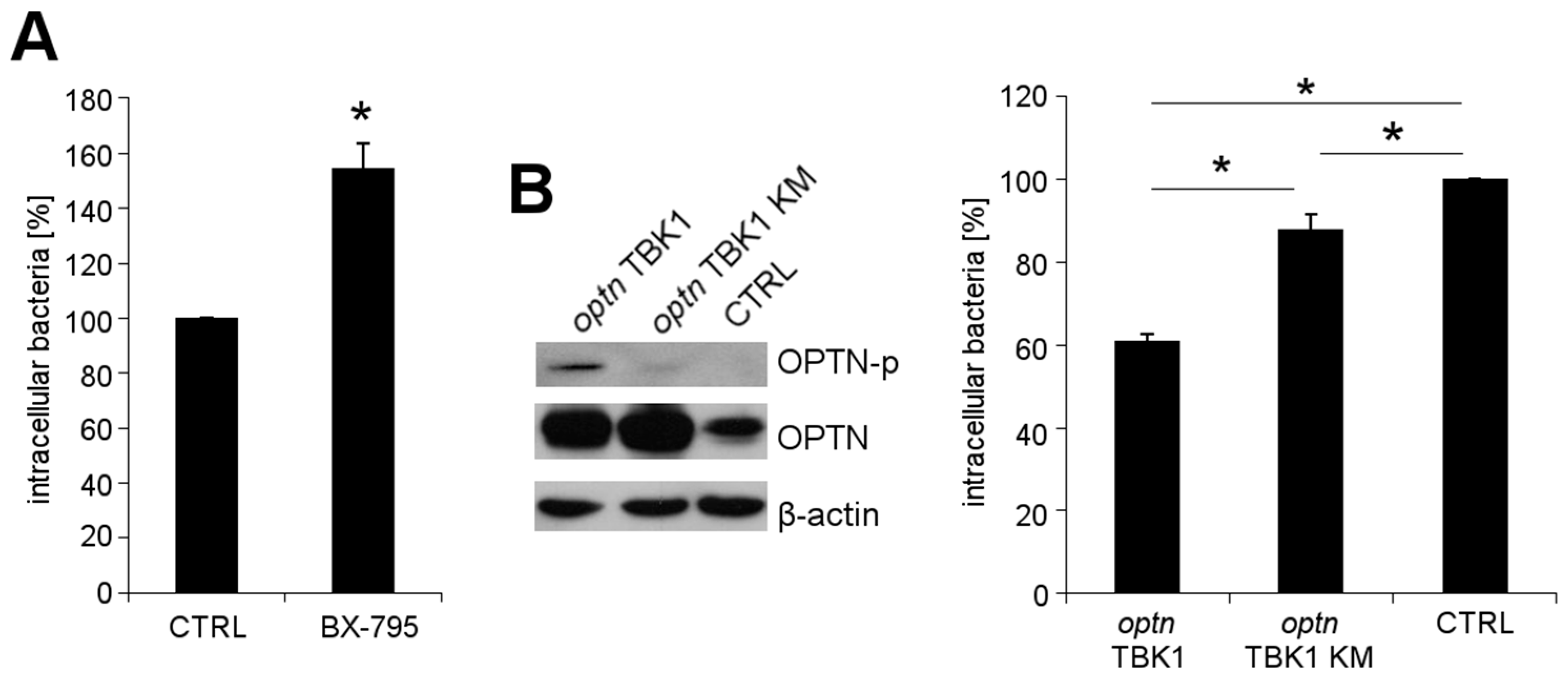
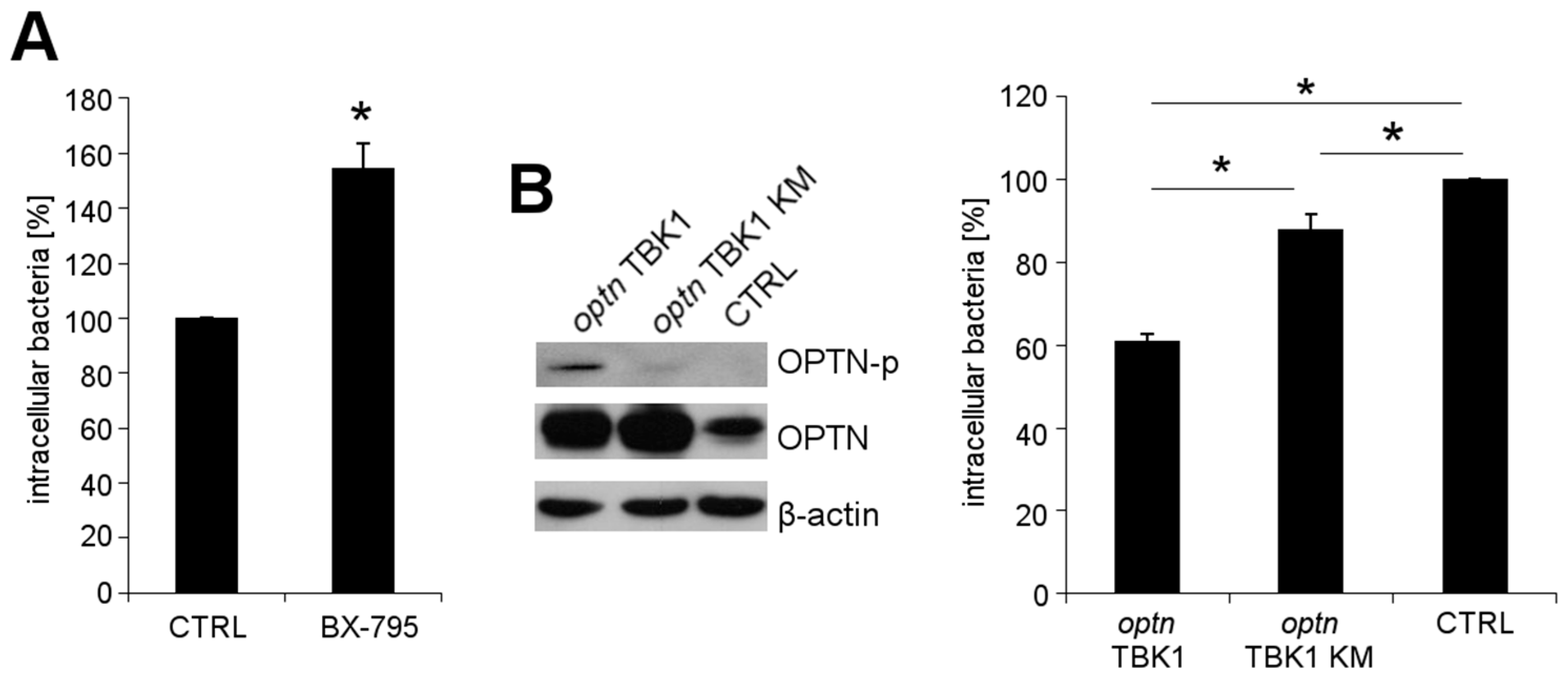
2.3. OPTN Phosphorylation by TBK1 Is Essential for the Growth Restriction of L. monocytogenes
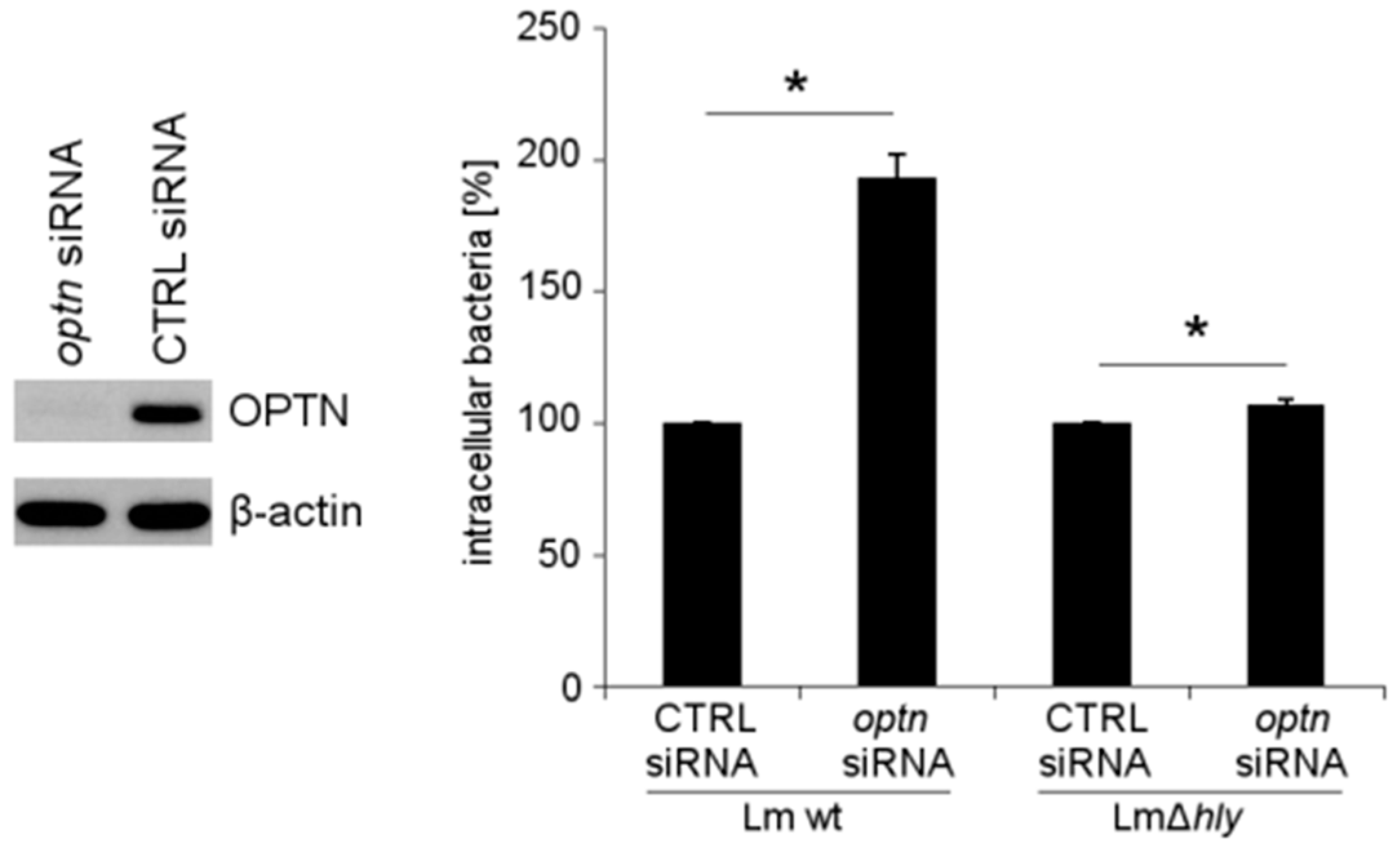
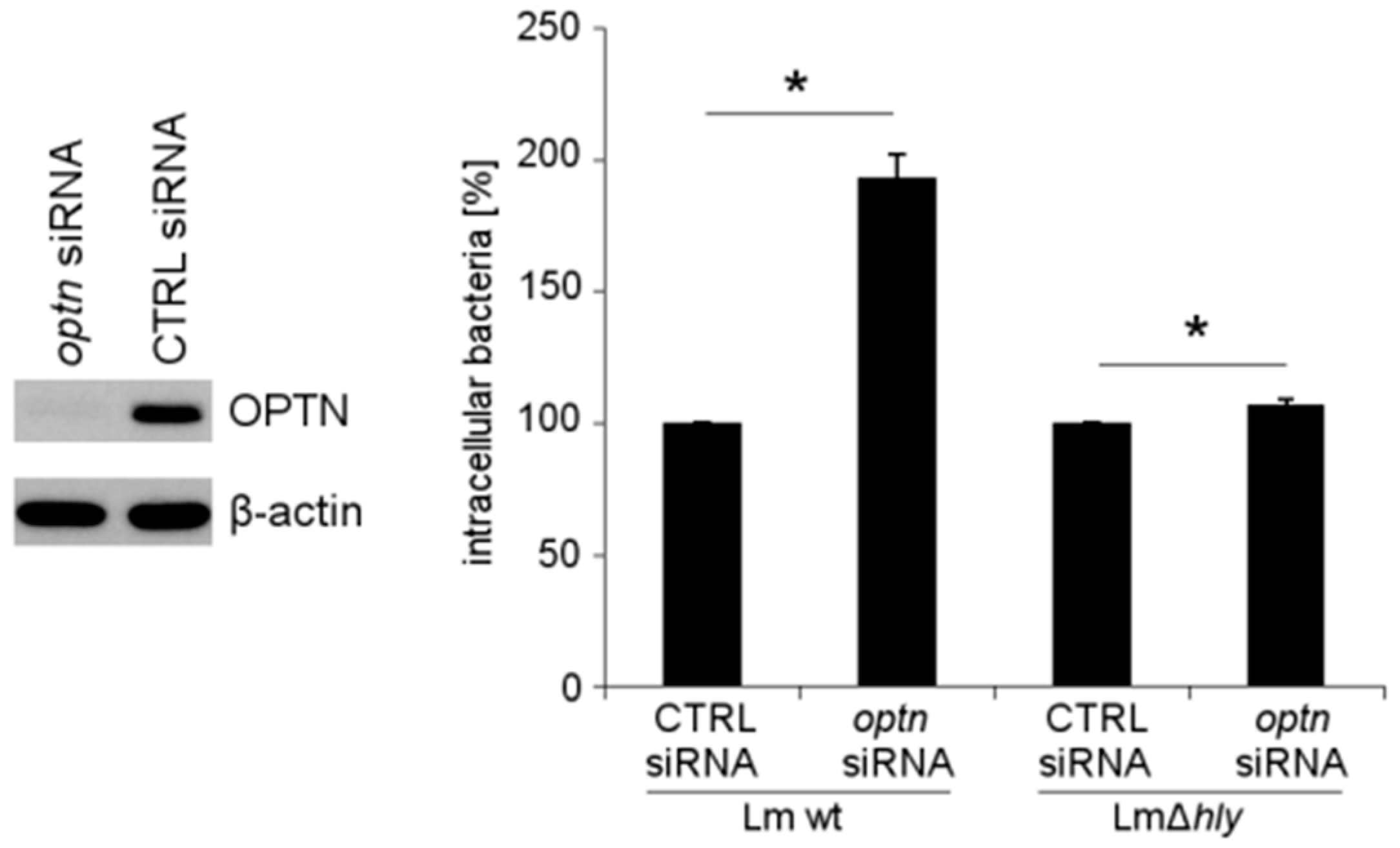
2.4. The Reduction of OPTN Promotes the Growth of Wt L. monocytogenes in an LLO-Dependent Manner
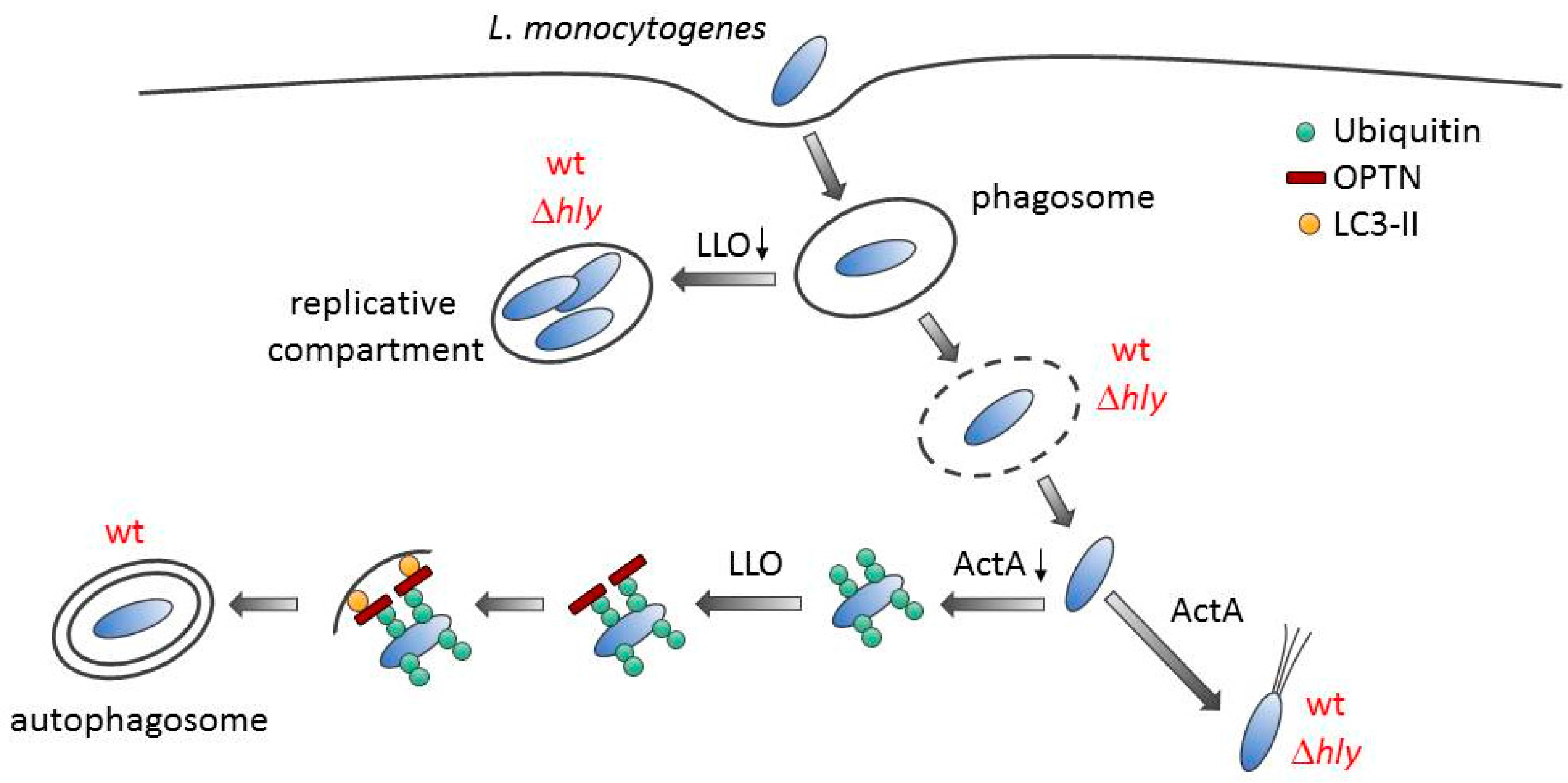
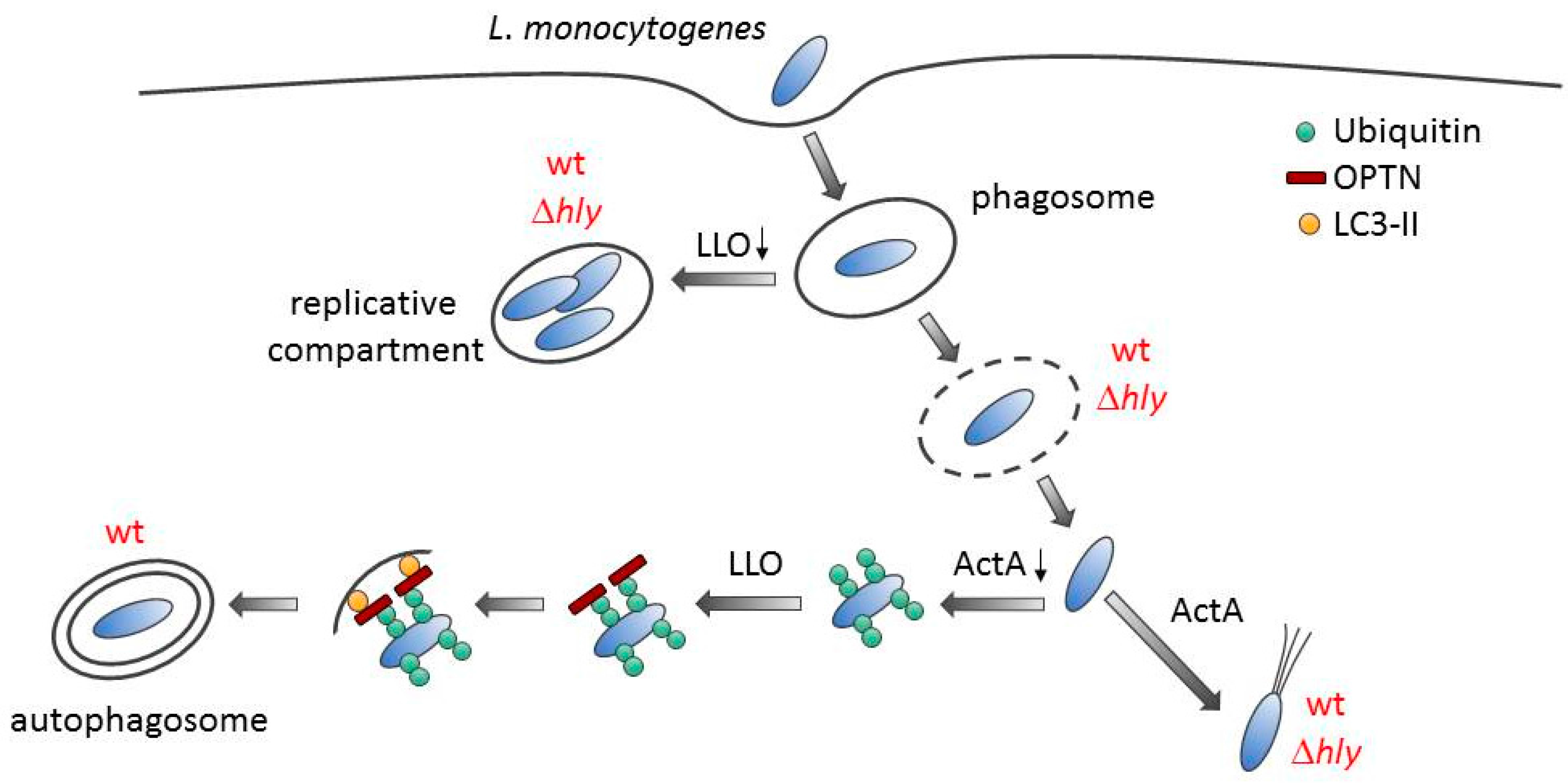
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Culture
5.2. RNAi Transfection
5.3. Plasmid Transfection
5.4. Bacterial Culture and Infection
5.5. Determination of the Number of Intracellular Bacteria
5.6. Protein Preparation from Eukaryotic Cells and Immunoblotting
5.7. Immunofluorescence
5.8. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Disson, O.; Lecuit, M. In vitro and in vivo models to study human listeriosis: Mind the gap. Microbes Infect. 2013, 15, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Gründling, A.; Gonzalez, M.D.; Higgins, D.E. Requirement of the Listeria monocytogenes broad-range phospholipase PC-PLC during infection of human epithelial cells. J. Bacteriol. 2003, 185, 6295–6307. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, C.L.; Canadien, V.; Kaniuk, N.A.; Steinberg, B.E.; Higgins, D.E.; Brumell, J.H. Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature 2008, 451, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Lam, G.Y.; Cemma, M.; Muise, A.M.; Higgins, D.E.; Brumell, J.H. Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy 2013, 9, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Morse, N.; Robbins, J.R.; Rae, C.S.; Mochegova, S.N.; Swanson, M.S.; Zhao, Z.; Virgin, H.W.; Portnoy, D. Listeriolysin O is necessary and sufficient to induce autophagy during Listeria monocytogenes infection. PLoS ONE 2010, 5, e8610. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, C.L.; Canadien, V.; Gouin, E.; Troy, E.B.; Yoshimori, T.; Cossart, P.; Higgins, D.E.; Brumell, J.H. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 2007, 3, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Py, B.F.; Lipinski, M.M.; Yuan, J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy 2007, 3, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Baehrecke, E.H. Autophagy, cell death, and cancer. Mol. Cell Oncol. 2015, 2, e985913. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Moy, R.H.; Cherry, S. Antimicrobial autophagy: A conserved innate immune response in Drosophila. J. Innate Immun. 2013, 5, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian autophagy: How does it work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.T.; Shahnazari, S.; Brech, A.; Lamark, T.; Johansen, T.; Brumell, J.H. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 2009, 183, 5909–5916. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, I.; Amano, A.; Mizushima, N.; Yamamoto, A.; Yamaguchi, H.; Kamimoto, T.; Nara, A.; Funao, J.; Nakata, M.; Tsuda, K.; et al. Autophagy defends cells against invading group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Shi, J.; He, Q.; Hu, Q.; Wang, Y.Y.; Zhang, L.J.; Chan, W.T.; Chen, W.X. Streptococcus pneumoniae induces autophagy through the inhibition of the PI3K-I/Akt/mTOR pathway and ROS hypergeneration in A549 cells. PLoS ONE 2015, 10, e0122753. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, Y.; Ogawa, M.; Hain, T.; Yoshida, M.; Fukumatsu, M.; Kim, M.; Mimuro, H.; Nakagawa, I.; Yanagawa, T.; Ishii, T.; et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 2009, 11, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Mostowy, S.; Sancho-Shimizu, V.; Hamon, M.A.; Simeone, R.; Brosch, R.; Johansen, T.; Cossart, P. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J. Biol. Chem. 2011, 286, 26987–26995. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Mesquita, F.S.; Holden, D.W. The DUB-ious lack of ALIS in Salmonella infection: A Salmonella deubiquitinase regulates the autophagy of protein aggregates. Autophagy 2012, 8, 1824–1826. [Google Scholar] [CrossRef] [PubMed]
- Ghai, R. Transcriptional Response of Murine Bone Marrow Macrophages to Listeriolysin, the Pore-Forming Toxin of Listeria monocytogenes. Ph.D. Thesis, Justus Liebig University, Giessen, Germany, 2006. [Google Scholar]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; Dötsch, V.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Rich, K.A.; Burkett, C.; Webster, P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol. 2003, 5, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Dortet, L.; Mostowy, S.; Samba-Louaka, A.; Gouin, E.; Nahori, M.A.; Wiemer, E.A.; Dussurget, O.; Cossart, P. Recruitment of the major vault protein by InlK: A Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog. 2011, 7, e1002168. [Google Scholar] [CrossRef]
- Mitchell, G.; Ge, L.; Huang, Q.; Chen, C.; Kianian, S.; Roberts, M.F.; Schekman, R.; Portnoy, D.A. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect. Immun. 2015, 83, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Pillich, H.; Puri, M.; Chakraborty, T. ActA of Listeria monocytogenes and Its Manifold Activities as an Important Listerial Virulence Factor. Curr. Top. Microbiol. Immunol. 2017, 399, 113–132. [Google Scholar] [PubMed]
- Bakshi, S.; Taylor, J.; Strickson, S.; McCartney, T.; Cohen, P. Identification of TBK1 complexes required for the phosphorylation of IRF3 and the production of interferon β. Biochem. J. 2017, 474, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Alsharifi, M.; Müllbacher, A.; Regner, M. Interferon type I responses in primary and secondary infections. Immunol. Cell Biol. 2008, 86, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Lundkvist, G.; Dons, L.; Kristensson, K.; Rottenberg, M.E. Interferon-gamma mediates neuronal killing of intracellular bacteria. Scand. J. Immunol. 2004, 60, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Bakowski, M.A.; Braun, V.; Brumell, J.H. Salmonella-containing vacuoles: Directing traffic and nesting to grow. Traffic 2008, 9, 2022–2031. [Google Scholar] [CrossRef] [PubMed]
- Kreibich, S.; Emmenlauer, M.; Fredlund, J.; Rämö, P.; Münz, C.; Dehio, C.; Enninga, J.; Hardt, W.D. Autophagy proteins promote repair of endosomal membranes damaged by the Salmonella type three secretion system 1. Cell Host Microbe 2015, 18, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Hotze, E.M.; Tweten, R.K. Membrane assembly of the cholesterol-dependent cytolysin pore complex. Biochim. Biophys. Acta 2012, 1818, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Pillich, H.; Loose, M.; Zimmer, K.P.; Chakraborty, T. Activation of the unfolded protein response by Listeria monocytogenes. Cell Microbiol. 2012, 14, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Glaser, P.; Frangeul, L.; Buchrieser, C.; Rusniok, C.; Amend, A.; Baquero, F.; Berche, P.; Bloecker, H.; Brandt, P.; Chakraborty, T.; et al. Comparative genomics of Listeria species. Science 2001, 294, 849–852. [Google Scholar] [PubMed]
- Guzman, C.A.; Rohde, M.; Chakraborty, T.; Domann, E.; Hudel, M.; Wehland, J.; Timmis, K.N. Interaction of Listeria monocytogenes with mouse dendritic cells. Infect. Immun. 1995, 63, 3665–3673. [Google Scholar] [PubMed]
- Loose, M.; Hudel, M.; Zimmer, K.P.; Garcia, E.; Hammerschmidt, S.; Lucas, R.; Chakraborty, T.; Pillich, H. Pneumococcal hydrogen peroxide-induced stress signaling regulates inflammatory genes. J. Infect. Dis. 2015, 211, 306–316. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puri, M.; La Pietra, L.; Mraheil, M.A.; Lucas, R.; Chakraborty, T.; Pillich, H. Listeriolysin O Regulates the Expression of Optineurin, an Autophagy Adaptor That Inhibits the Growth of Listeria monocytogenes. Toxins 2017, 9, 273. https://doi.org/10.3390/toxins9090273
Puri M, La Pietra L, Mraheil MA, Lucas R, Chakraborty T, Pillich H. Listeriolysin O Regulates the Expression of Optineurin, an Autophagy Adaptor That Inhibits the Growth of Listeria monocytogenes. Toxins. 2017; 9(9):273. https://doi.org/10.3390/toxins9090273
Chicago/Turabian StylePuri, Madhu, Luigi La Pietra, Mobarak Abu Mraheil, Rudolf Lucas, Trinad Chakraborty, and Helena Pillich. 2017. "Listeriolysin O Regulates the Expression of Optineurin, an Autophagy Adaptor That Inhibits the Growth of Listeria monocytogenes" Toxins 9, no. 9: 273. https://doi.org/10.3390/toxins9090273
APA StylePuri, M., La Pietra, L., Mraheil, M. A., Lucas, R., Chakraborty, T., & Pillich, H. (2017). Listeriolysin O Regulates the Expression of Optineurin, an Autophagy Adaptor That Inhibits the Growth of Listeria monocytogenes. Toxins, 9(9), 273. https://doi.org/10.3390/toxins9090273