
Bacterial Toxins for Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
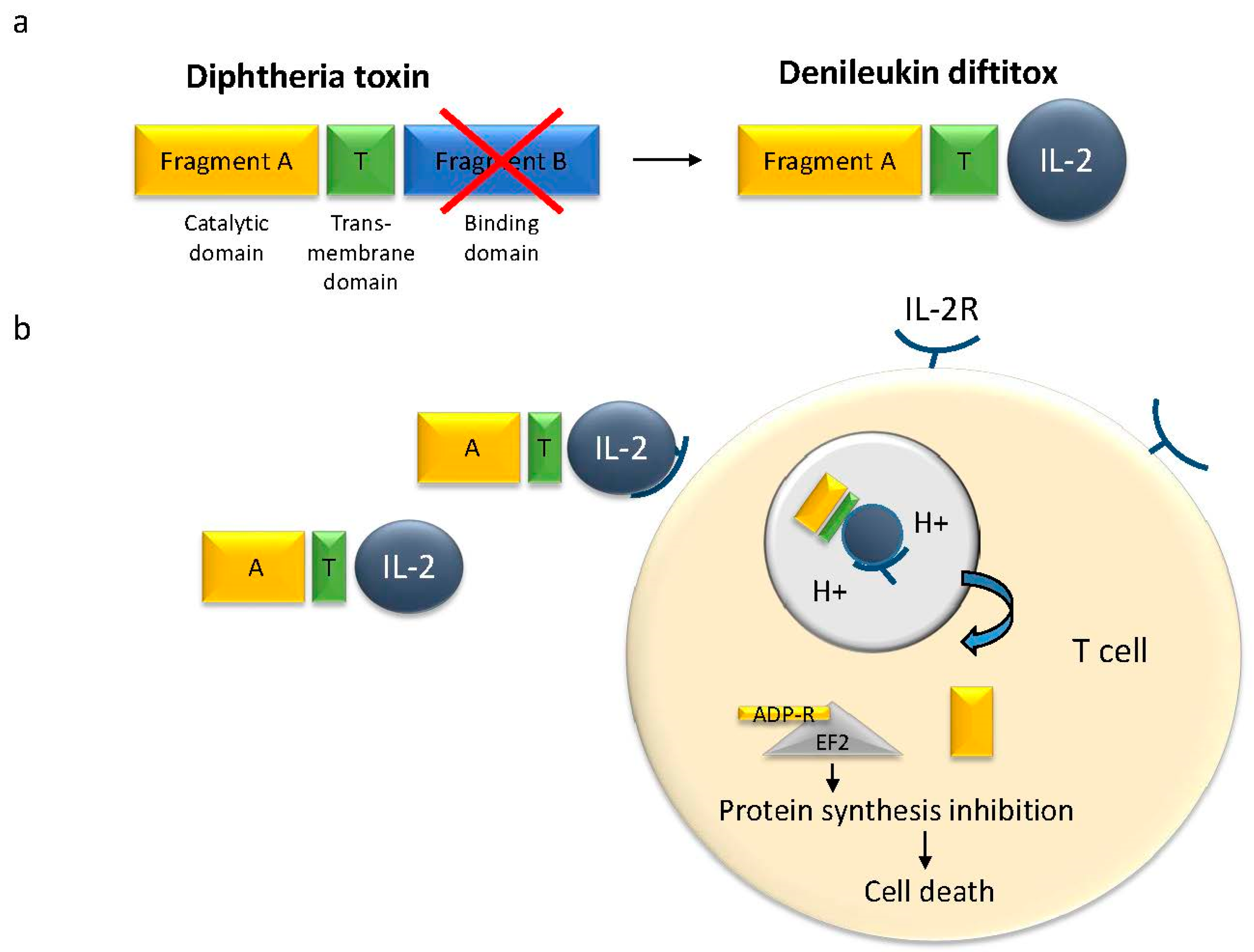
2. Single Chain Bacterial Toxins
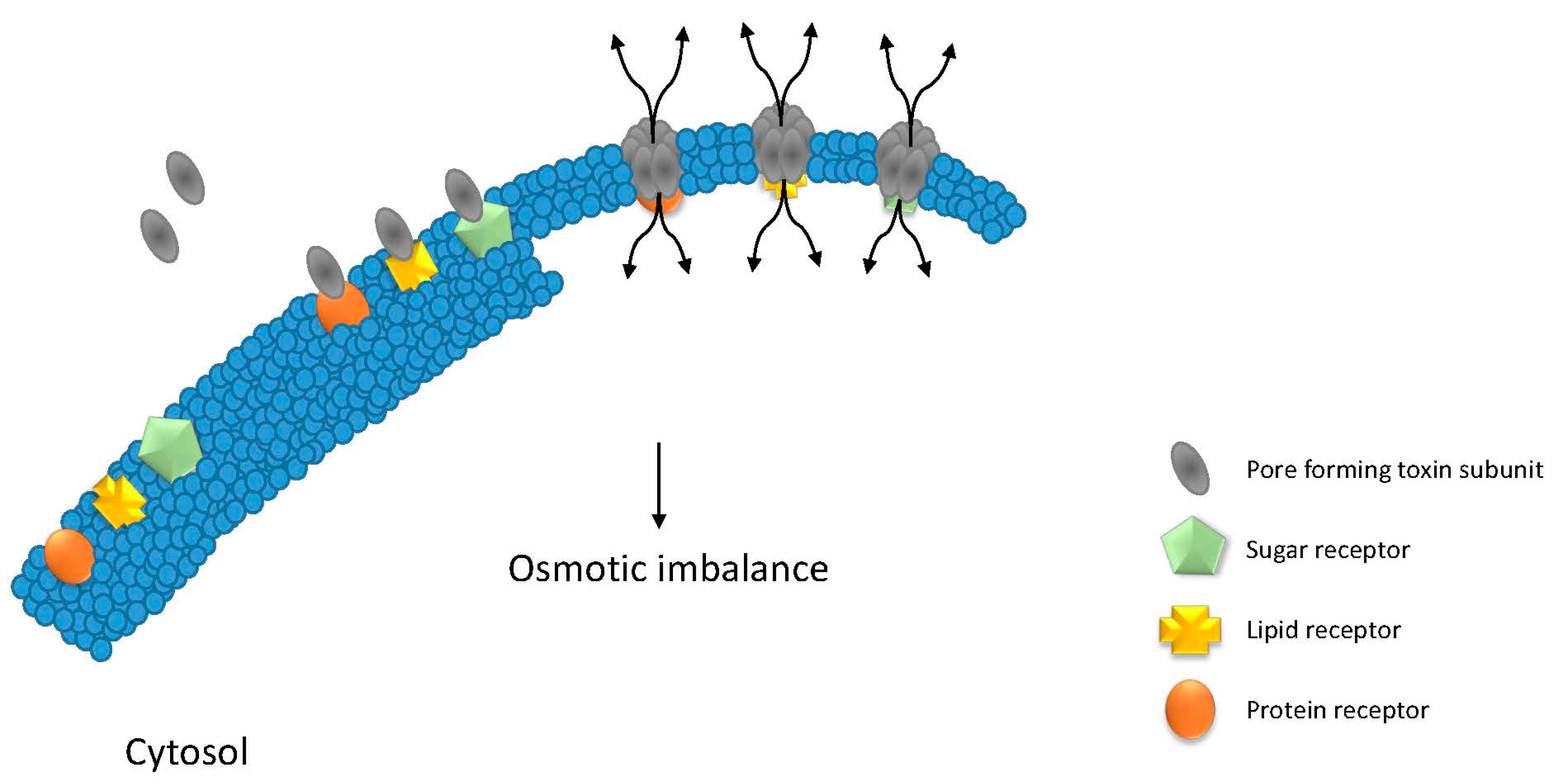
3. Pore Forming Toxins
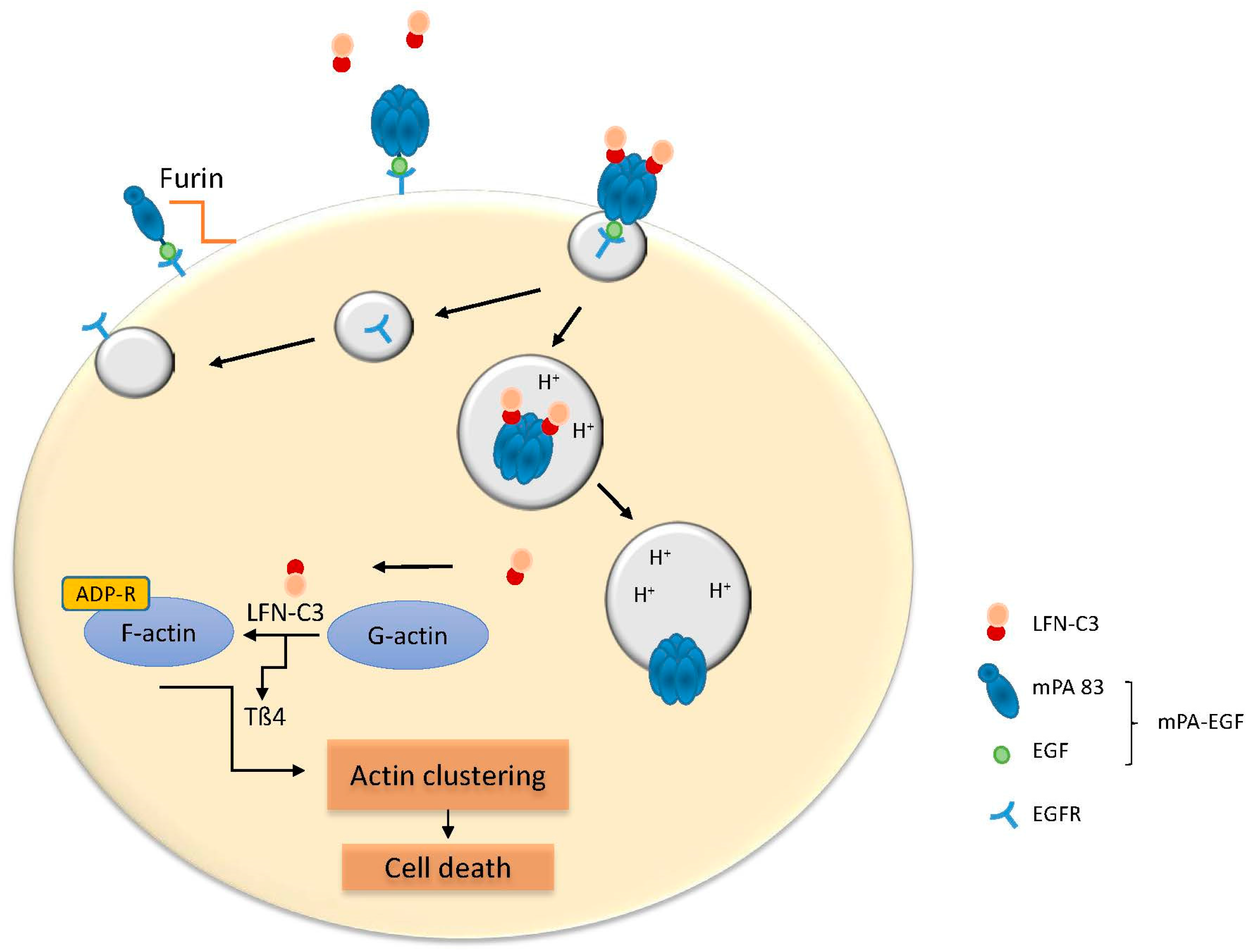
4. Anthrax Toxin as a Transporter
5. Testing Immunotoxins In Vivo
6. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thorpe, P.E.; Ross, W.C.; Cumber, A.J.; Hinson, C.A.; Edwards, D.C.; Davies, A.J. Toxicity of diphtheria toxin for lymphoblastoid cells is increased by conjugation to antilymphocytic globulin. Nature 1978, 271, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Nitta, H.; Kelly, B.D.; Jewell, S.; Banks, P.; Dennis, E.; Groqan, T.M. The assessment of HER2 status in breast cancer: The past, the present, and the future. Pathol. Int. 2016, 66, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Robak, T. Application of new drugs in chronic lymphocytic leukemia. Mediterr. J. Hematol. Infect. Dis. 2010, 2, e2010011. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J. Immunotoxins for targeted cancer therapy. AAPS J. 2006, 8, E532–E551. [Google Scholar] [CrossRef] [PubMed]
- Binz, T.; Rummel, A. Cell entry strategy of clostridial neurotoxins. J. Neurochem. 2009, 109, 1584–1595. [Google Scholar] [CrossRef] [PubMed]
- Jank, T.; Giesemann, T.; Aktories, K. Rho-glucosylating Clostridium difficile toxins A and B: New insights into structure and function. Glycobiology 2007, 17, 15R–22R. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.M.; Smith, D.C.; Roberts, L.M. Toxin entry: How bacterial proteins get into mammalian cells. Cell Microbiol. 1999, 1, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, L.C.; Liu, B.; Munker, R.; Andreeff, M.; Freireich, E.J.; Scheinberg, D.A.; Rosenblum, M.G. Humanized M195 monoclonal antibody conjugated to recombinant gelonin: An anti-CD33 immunotoxin with antileukemic activity. Clin. Cancer Res. 1998, 4, 1971–1976. [Google Scholar] [PubMed]
- Gill, D.M.; Pappenheimer, A.M., Jr.; Brown, R.; Kurnick, J.T. Studies on the mode of action of diphtheria toxin. VII.Toxin-stimulated hydrolysis of nicotinamide adenine dinucleotide in mammalian cell extracts. J. Exp. Med. 1969, 129, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J. Understanding the mode of action of diphtheria toxin: A perspective on progress during the 20th century. Toxicon 2001, 39, 1793–1803. [Google Scholar] [CrossRef]
- Murphy, J.R. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 2011, 3, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.; Kantardjieff, K.A.; Collier, R.J.; Eisenberg, D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Goor, R.S.; Pappenheimer, A.M., Jr.; Ames, E. Studies on the mode of action of diphtheria toxin. J. Exp. Med. 1964, 126, 923–939. [Google Scholar] [CrossRef]
- Strauss, N.; Hendee, E.D. The effect of the Diphtheria toxin on the metabolism of Hela cells. J. Exp. Med. 1959, 109, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Masaru Yamaizumi, E.M. One molecule of Diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar] [CrossRef]
- Kiyokawa, T.; Shirono, K.; Hattori, T.; Nishimura, H.; Yamaguchi, K.; Nichols, J.C.; Strom, T.B.; Murphy, J.R. Cytotoxicity of Interleukin 2-Toxin toward Lymphocytes from Patients with Adult T-Cell Leukemia. Cancer Res. 1989, 49, 4042–4046. [Google Scholar] [PubMed]
- Wasik, M.A.; Sioutos, N.; Tuttle, M.; Butmarc, J.R.; Kaplan, W.D.; Kadin, M.E. Constitutive secretion of soluble interleukin-2 receptor by human T cell lymphoma xenografted into SCID mice. Correlation of tumor volume with concentration of tumor-derived soluble interleukin-2 receptor in body fluids of the host mice. Am. J. Pathol. 1994, 144, 1089–1097. [Google Scholar] [PubMed]
- Waldmann, T.A. The Role of the Multichain IL-2 Receptor Complex in the Control of Normal and Malignant T-Cell Proliferation. Environ. Health Perspect. 1987, 75, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Hymes, K. Denileukin diftitox for the treatment of cutaneous T-cell lymphoma. Biologics 2008, 2, 717–724. [Google Scholar] [CrossRef]
- Avarbock, A.B.; Loren, A.W.; Park, J.Y.; Junkins-Hopkins, J.M.; Choi, J.; Litzky, L.A.; Rook, A.H. Lethal vascular leak syndrome after denileukin diftitox administration to a patient with cutaneous gamma/delta T-cell lymphoma and occult cirrhosis. Am. J. Hematol. 2008, 83, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Wamg, Z.; Zheng, Q.; Zhang, H.; Bronson, R.T.; Madsen, J.C.; Sachs, D.H.; Huang, C.A.; Wang, Z. Ontak-like human IL-2 fusion toxin. J. Immunol. Methods 2017. [Google Scholar] [CrossRef] [PubMed]
- Callahan, L.T., III. Purification and Characterization of Pseudomonas aeruginosa Exotoxin. Infect. Immun. 1974, 9, 113–118. [Google Scholar] [PubMed]
- Li, M.; Dtda, F.; Benhar, I.; Pastan, I.; Davies, D.R. The crystal structure of Pseudomonas aeruginosa exotoxin domain III with nicotinamide and AMP: Conformational differences with the intact exotoxin. Proc. Natl. Acad. Sci. USA 1995, 92, 9308–9312. [Google Scholar] [CrossRef] [PubMed]
- Schorch, B.; Song, S.; van Diemen, F.R.; Bock, H.H.; May, P.; Herz, J.; Brummelkamp, T.R.; Papatheodorou, P.; Aktories, K. LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc. Natl. Acad. Sci. USA 2014, 111, 6431–6436. [Google Scholar] [CrossRef] [PubMed]
- Iglewski, B.H.; Kabat, D. NAD-Dependent Inhibition of Protein Synthesis by Pseudomonas aeruginosa Toxin: (Inactivation of mammalian elongation factor 2/Pseudomonas aeruginosa exotoxin/ ADP-ribosyl transferases). Proc. Natl. Acad. Sci. USA 1975, 72, 2284–2288. [Google Scholar] [CrossRef] [PubMed]
- David, J.P.; Fitzgerald, I.S.T.; Pastan, I.; Willingham, M.C. Enhancement of toxicity of antitransferrin receptor antibody-Pseudomonas exotoxin conjugates by adenovirus. Proc. Natl. Acad. Sci. USA 1983, 80, 4134–4138. [Google Scholar]
- David, J.P.; FitzGerald, R.P.; Pastan, I.; Willingham, M.C. Adenovirus-Induced Release of Epidermal Growth Factor and Pseudomonas Toxin into the Cytosol of KB Cells during Receptor-Mediated Endocytosis. Cell 1983, 32, 607–617. [Google Scholar]
- Kreitman, R.J.; Sieqall, C.B.; Chaudhary, V.K.; FitzGerald, D.J.; Pastan, I. Properties of Chimeric Toxins with Two Recognition Domains: Interleukin 6 and Transforming Growth Factor α at Different Locations in Pseudomonas Exotoxin. Bioconjug. Chem. 1992, 3, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin—recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Bruland, O.S.; Pastan, I. TP-3 immunotoxins improve antitumor activity in mice with osteosarcoma. Clin. Orthop. Relat. Res. 2005, 430, 142–148. [Google Scholar] [CrossRef]
- Kuan, C.T.; Wang, Q.; Pastan, I. Pseudomonas exotoxin A mutants. Replacement of surface exposed residues in domain II with cysteine residues that can be modified with polyethylene glycol in a site-specific manner. J. Biol. Chem. 1994, 269, 7610–7616. [Google Scholar] [PubMed]
- Alderson, R.F.; Kreitman, R.J.; Chen, T.; Yeung, P.; Herbst, R.; Fox, J.A.; Pastan, I. CAT-8015: A second-generation pseudomonas exotoxin A-based immunotherapy targeting CD22-expressing hematologic malignancies. Clin. Cancer Res. 2009, 15, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Kaplan, G.; Park, D.; Jang, Y.; Lee, F.; Kreitman, R.; Pastan, I. Rational design of low immunogenic anti CD25 recombinant immunotoxin for T cell malignancies by elimination of T cell epitopes in PE38. Cell Immunol. 2017, 313, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, Y.; Onda, M.; Nagata, S.; Lee, B.; Kreitman, R.J.; Pastan, I. Site-specific chemical modification with polyethylene glycol of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc. Natl. Acad. Sci. USA 2000, 97, 8548–8553. [Google Scholar] [CrossRef] [PubMed]
- Lesieur, C.; Vécsey-Semjén, B.; Abrami, L.; Fivaz, M.; Gisou van der Goot, F. Membrane insertion: The strategies of toxins (Review). Mol. Membr. Biol. 1997, 14, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Gatter, K.C.; Brown, G.; Trowbridge, I.S.; Woolston, R.E.; Mason, D.Y. Transferrin receptors in human tissues: Their distribution and possible clinical relevance. J. Clin. Pathol. 1983, 36, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Thorstensen, K.; Romslo, I. The role of transferrin in the mechanism of cellular iron uptake. Biochem. J. 1990, 271, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.; Delia, D.; Schneider, C.; Newman, R.; Kemshead, J.; Greaves, M. Ubiquitous cell-surface glycoprotein on tumor cells is proliferationassociated receptor for transferrin. Proc. Natl. Acad. Sci. USA 1981, 78, 4515–4519. [Google Scholar] [CrossRef] [PubMed]
- Pederzolli, C.; Belmonte, G.; Serra, M.D.; Macek, P.; Menestrina, G. Biochemical and Cytotoxic Properties of Conjugates of Transferrin with Equinatoxin 11, a Cytolysin from a Sea Anemone. Bioconjug. Chem. 1995, 6, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G. Novel Therapeutic Strategies to Selectively Kill Cancer Cells. Biochem. Pharmacol. 1998, 55, 247–252. [Google Scholar] [CrossRef]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Bubeck Wardenburg, J. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; Bayley, H. A pore-forming protein with a protease-activated trigger. Protein Eng. 1994, 7, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Pollack, M. The Role of Exotoxin A in Pseudomonas Disease and Immunity. Rev. Infect. Dis. 1983, 5, S979–S984. [Google Scholar] [CrossRef] [PubMed]
- Tobkes, N.; Wallace, B.A.; Bayley, H. Secondary structure and assembly mechanism of an oligomeric channel protein. Biochemistry 1985, 24, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Blakey, D.C.; Skilleter, D.N.; Price, R.J.; Thorpe, P.E. Uptake of native and deglycosylated ricin A-chain immunotoxins by mouse liver parenchymai and non-parenchymal cells in vitro and in vivo. Biochim. Biophys. Acta 1988, 968, 172. [Google Scholar] [CrossRef]
- Mason-Osann, E.; Hollevoet, K.; Niederfellner, G.; Pastan, I. Quantification of recombinant immunotoxin delivery to solid tumors allows for direct comparison of in vivo and in vitro results. Sci. Rep. 2015, 5, 10832. [Google Scholar] [CrossRef] [PubMed]
- Skilleter, D.N.; Skilleter, D.N.; Price, R.J.; Parnell, G.D.; Cumber, A.J. The low uptake of an abrin A-chain immunotoxin by rat hepatic cells in vivo and in vitro. Cancer Lett. 1989, 46, 161–166. [Google Scholar] [CrossRef]
- Ravel, S.; Colombatti, M.; Casellas, P. Internalization and Intracellular Fate of Anti-CDS Monoclonal Antibody and Anti-CDS Ricin A-Chain Immunotoxin in Human Leukemic T Cells. Blood 1992, 796, 1511–1517. [Google Scholar]
- Young, J.A.; Collier, R.J. Anthrax Toxin: Receptor-Binding, Internalization, Pore Formation, and Translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Kunz, B.; van der Goot, F.G. Anthrax toxin triggers the activation of src-like kinases to mediate its own uptake. Proc. Natl. Acad. Sci. USA 2010, 107, 1420–1424. [Google Scholar] [CrossRef] [PubMed]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic Inactivation of MAP-Kinase-Kinase by Anthrax Lethal Factor. Science 1998, 280, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Pellizzari, R.; Recchi, C.; Napolitani, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves the N-terminus of MAPKKS and induces tyrosine/threonine phosphorylation of MAPKS in cultured macrophages. J. Appl. Microbiol. 1999, 87, 288. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations in eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Netzel-Arnett, S.; Birkedal-Hansen, H.; Leppla, S.H. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000, 60, 6061–6067. [Google Scholar] [PubMed]
- Mechaly, A.; McCluskey, A.J.; Collier, R.J. Changing the receptor specificity of anthrax toxin. mBio 2012, 3, e00088-12. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.I.; Gee, J.M.W.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- English, D.P.; Roque, D.M.; Santin, A.D. HER2 expression beyond breast cancer: Therapeutic implications for gynecologic malignancies. Mol. Diagn. Ther. 2013, 17, 85–99. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.J.; Olive, A.J.; Starnbach, M.N.; Collier, R.J. Targeting HER2-positive cancer cells with receptor-redirected anthrax protective antigen. Mol. Oncol. 2013, 7, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Zahaf, N.I.; Lang, A.E.; Kaiser, L.; Fichter, C.D.; Lassmann, S.; McCluskey, A.; Augspach, A.; Aktories, K.; Schmidt, G. Targeted delivery of an ADP-ribosylating bacterial toxin into cancer cells. Sci. Rep. 2017, 7, 41252. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, D.J.; Willingham, M.C.; Pastan, I. Antitumor effects of an immunotoxin made with Pseudomonas exotoxin in a nude mouse model of human ovarian cancer. Proc. Natl. Acad. Sci. USA 1986, 83, 6627–6630. [Google Scholar] [CrossRef] [PubMed]
- Bumol, T.F.; Wang, Q.C.; Reisfeld, R.A.; Kaplan, N.O. Monoclonal antibody and an antibody-toxin conjugate to a cell surface proteoglycan of melanoma cells suppress in vivo tumor growth. Proc. Natl. Acad. Sci. USA 1983, 80, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, U.K.; Moore, T.T.; Joo, H.G.; Tanaka, Y.; Herrmann, V.; Doherty, G.; Drebin, J.A.; Strasberg, S.M.; Eberlein, T.J.; Goedegebuure, P.S.; et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 2002, 169, 2756–2761. [Google Scholar] [CrossRef] [PubMed]
- Litzinger, M.T.; Fernando, R.; Curiel, T.J.; Grosenbach, D.W.; Schlom, J.; Palena, C. IL-2 immunotoxin denileukin diftitox reduces regulatory T cells and enhances vaccine-mediated T-cell immunity. Blood 2007, 110, 3192–3201. [Google Scholar] [CrossRef] [PubMed]
- Attia, P.; Maker, A.V.; Haworth, L.R.; Rogers-Freezer, L.; Rosenberg, S.A. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin Diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J. Immunother. 2005, 28, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Aoyama, A.; Tocco, G.; Boskovic, S.; Nadazdin, O.; Alessandrini, A.; Madsen, J.C.; Cosimi, A.B.; Benichou, G.; Kawai, T. Differential effects of denileukin diftitox IL-2 immunotoxin on NK and regulatory T cells in nonhuman primates. J. Immunol. 2012, 188, 6063–6070. [Google Scholar] [CrossRef] [PubMed]
- Galan, J.E.; Lara-Tejero, M.; Marlovits, T.C.; Wagner, S. Bacterial Type III Secretion Systems: Specialized Nanomachines for Protein Delivery into Target Cells. Annu. Rev. Microbiol. 2014, 68, 415–438. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahaf, N.-I.; Schmidt, G. Bacterial Toxins for Cancer Therapy. Toxins 2017, 9, 236. https://doi.org/10.3390/toxins9080236
Zahaf N-I, Schmidt G. Bacterial Toxins for Cancer Therapy. Toxins. 2017; 9(8):236. https://doi.org/10.3390/toxins9080236
Chicago/Turabian StyleZahaf, Nour-Imene, and Gudula Schmidt. 2017. "Bacterial Toxins for Cancer Therapy" Toxins 9, no. 8: 236. https://doi.org/10.3390/toxins9080236
APA StyleZahaf, N.-I., & Schmidt, G. (2017). Bacterial Toxins for Cancer Therapy. Toxins, 9(8), 236. https://doi.org/10.3390/toxins9080236