
Sensitive Detection of α-Conotoxin GI in Human Plasma Using a Solid-Phase Extraction Column and LC-MS/MS


Abstract
:1. Introduction
2. Results
2.1. Sample Treatment
2.2. Liquid Chromatography
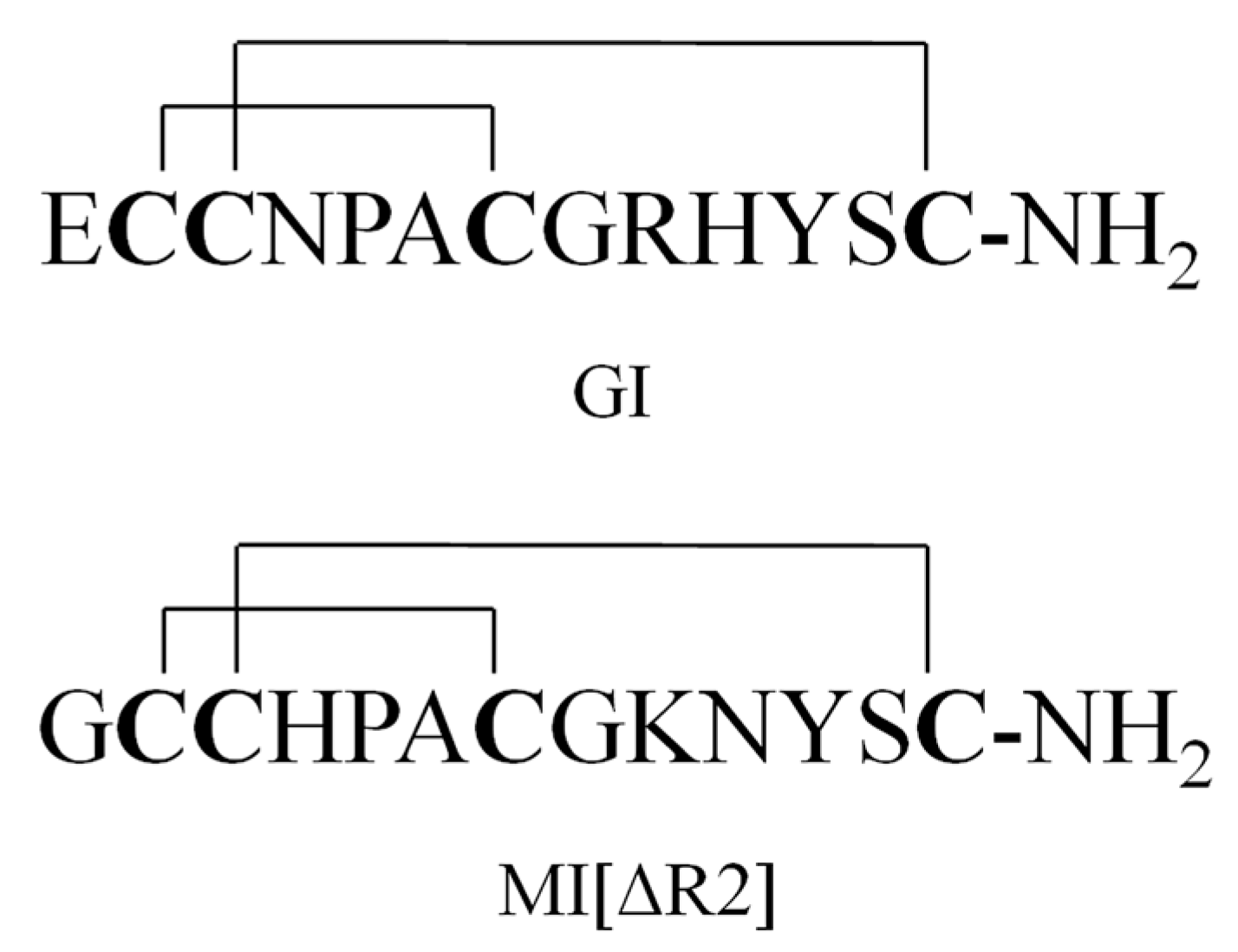
2.3. MS/MS
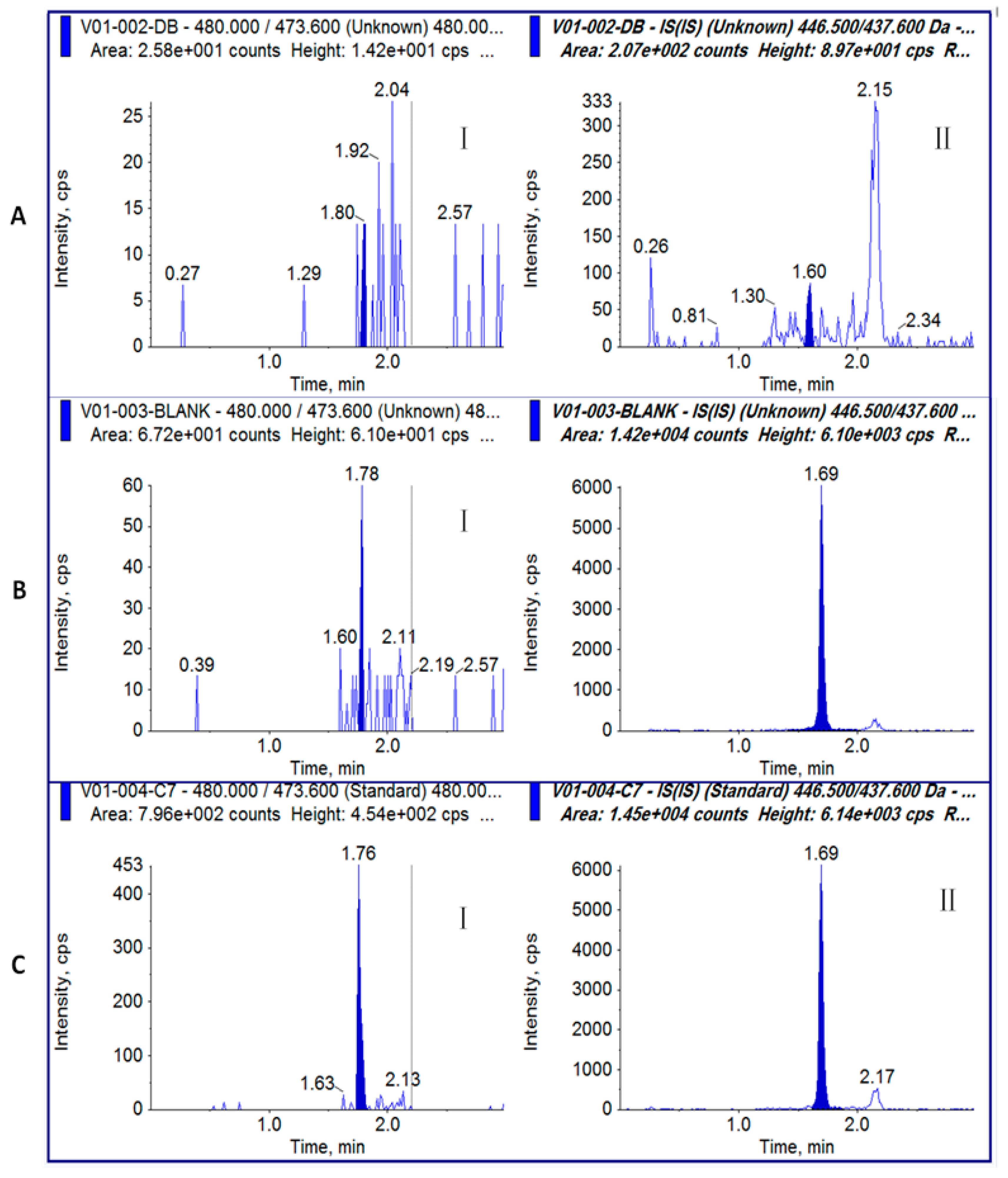
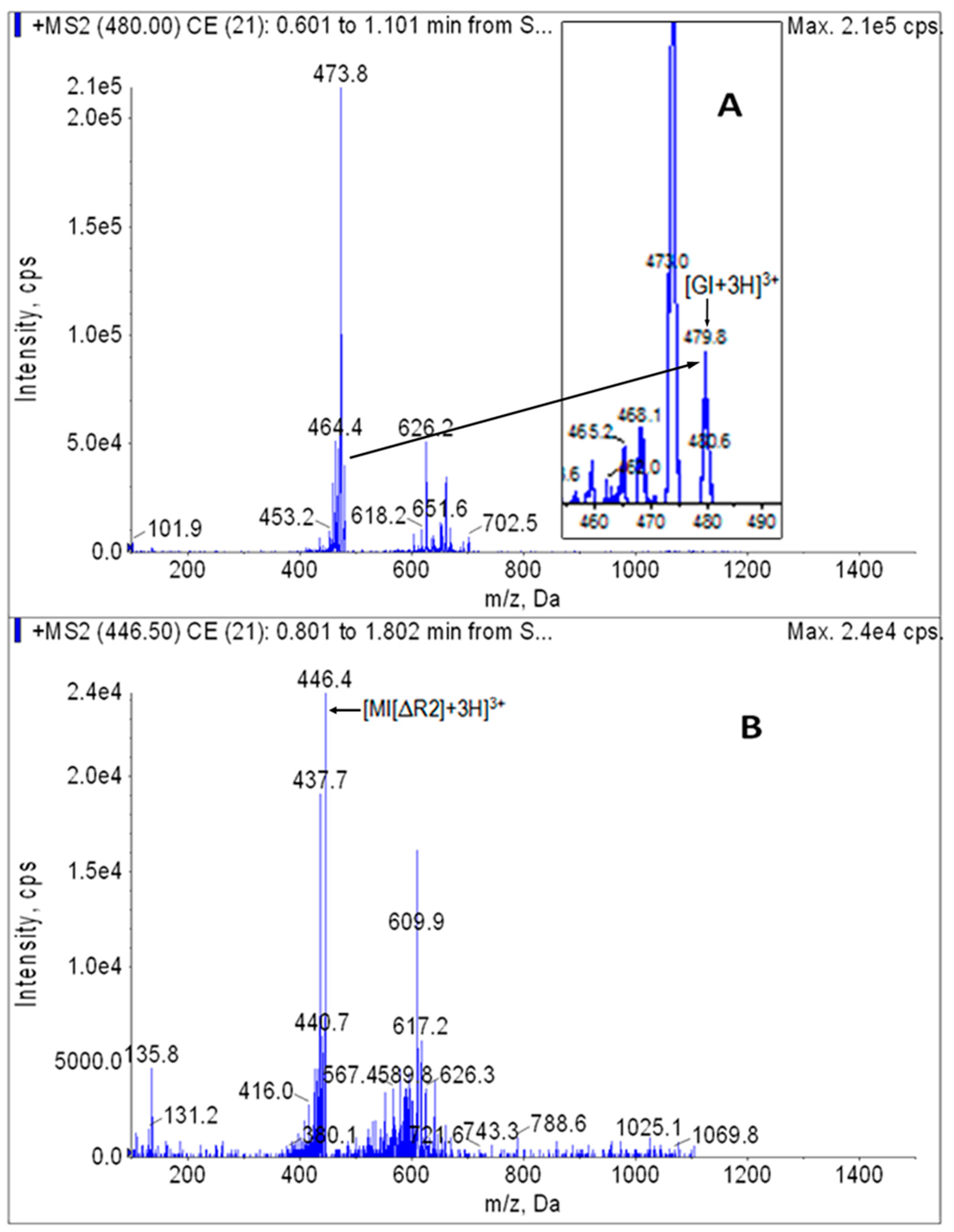
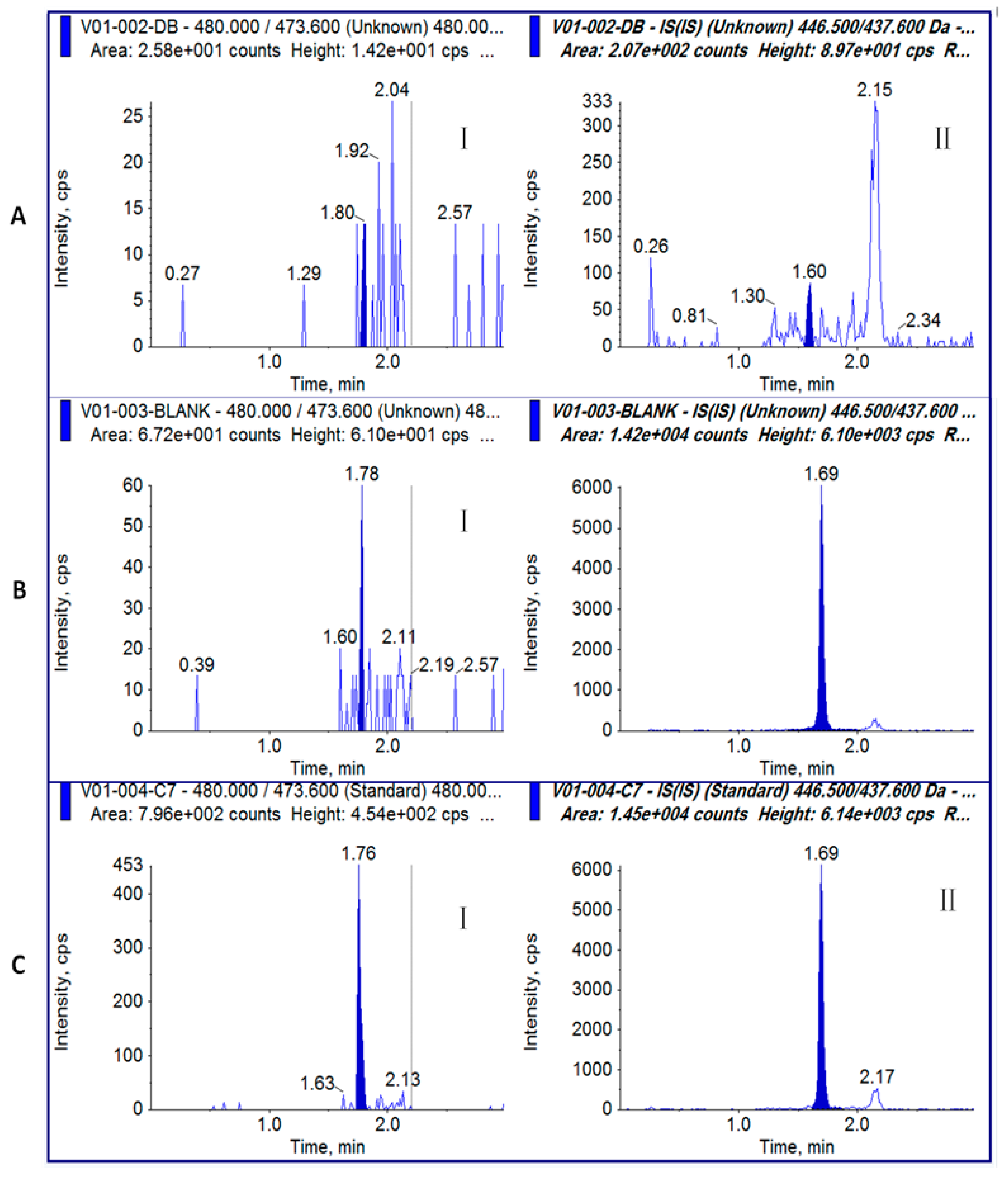
2.4. Specificity
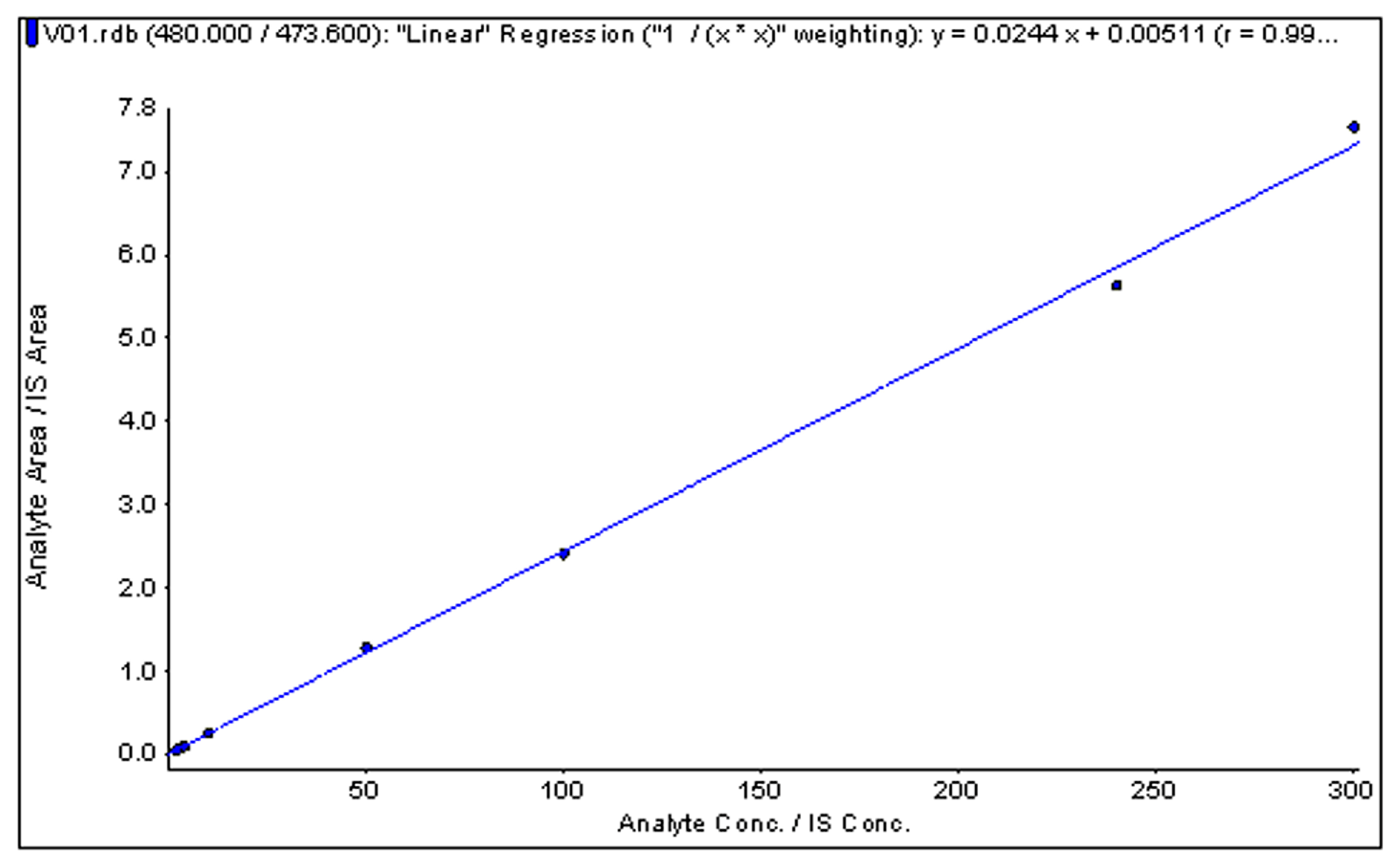
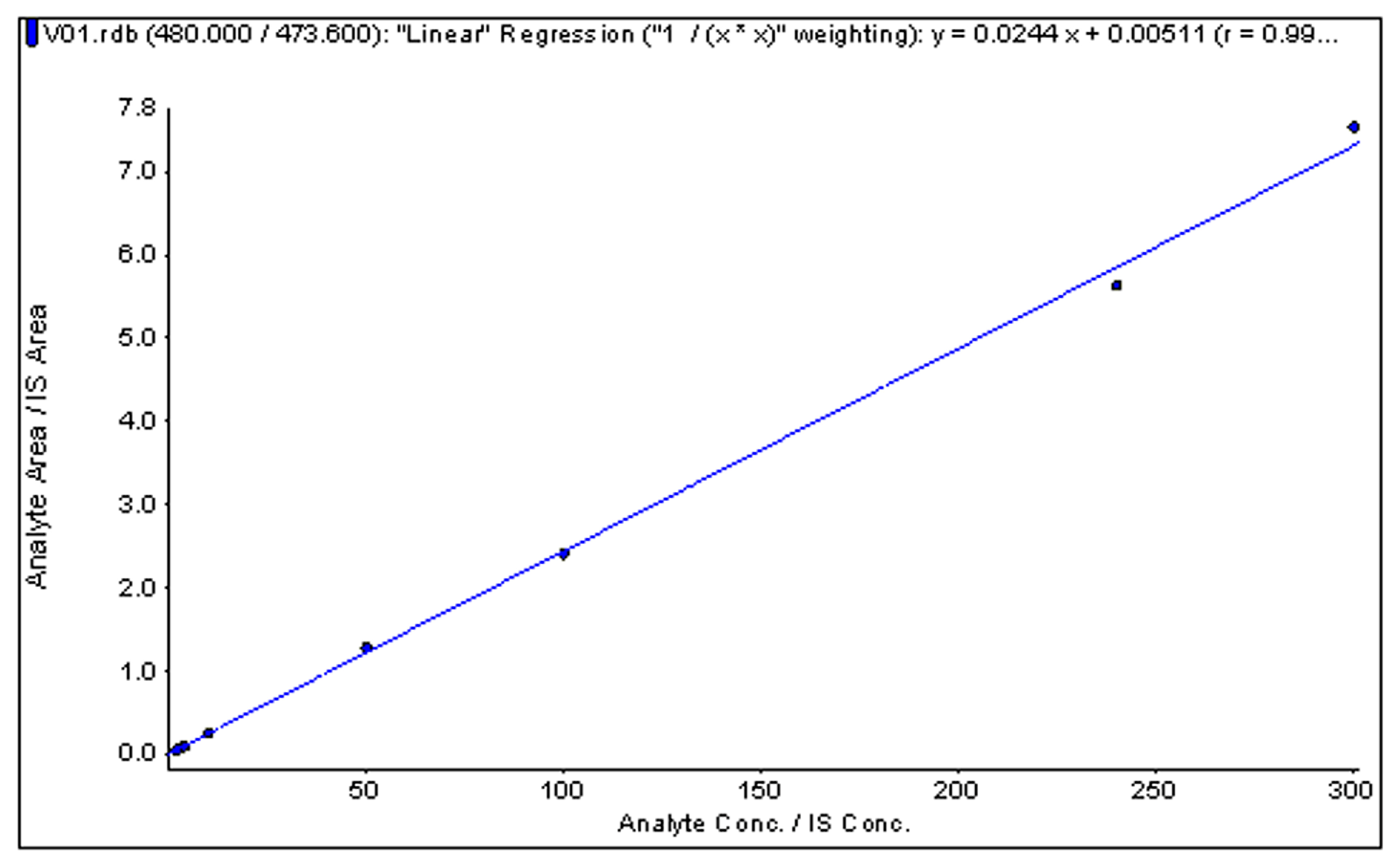
2.5. Linearity and Sensitivity
2.6. Assay Precision and Accuracy
2.7. Matrix Effect and Recovery
2.8. Stability
2.9. Analysis of GI in Human Blood
3. Discussion
4. Conclusions
5. Experimental Section
5.1. Chemicals and Reagents
5.2. Preparation of Standards
5.3. Sample Preparation and Extraction Procedure
5.4. LC-MS/MS Analysis
5.5. Method Validation
5.6. Analysis of GI in Human Blood
5.7. Data Acquisition and Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaas, Q.; Westermann, J.C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Duda, T.F., Jr.; Remigio, E.A. Variation and evolution of toxin gene expression patterns of six closely related venomous marine snails. Mol. Ecol. 2008, 17, 3018–3032. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.G.; Li, H.Y.; Liu, N.; Wu, C.X.; Jiang, J.; Yue, J.J.; Jing, Y.; Dai, Q.Y. Diversity and evolution of conotoxins in Conus virgo, Conus eburneus, Conus imperialis and Conus marmoreus from the South China Sea. Toxicon 2012, 60, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.J.; Perron, F.E. Life History and Biogeography Patterns in Conus; Clarendon Press VII: Oxford, UK, 1994; p. 106. [Google Scholar]
- Kohn, A.J. Human injuries and fatalities due to venomous marine snails of the family Conidae. Int. J. Clin. Pharmacol. Ther. 2016, 54, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L. Venomous snails: One slip, and you’re dead. Nature 2004, 429, 798–799. [Google Scholar] [CrossRef] [PubMed]
- Halford, Z.A.; Yu, P.Y.; Likeman, R.K.; Hawley-Molloy, J.S.; Thomas, C.; Bingham, J.P. Cone shell envenomation: Epidemiology, pharmacology and medical care. Diving Hyperb. Med. 2015, 45, 200–207. [Google Scholar] [PubMed]
- Yoshiba, S. An estimation of the most dangerous species of cone shell, Conus geographus venom’s lethal dose in humans. Nihon Eiseigaku Zasshi 1984, 39, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Fegan, D.; Andresen, D. Conus geographus envenomation. Lancet 1997, 349, 1672. [Google Scholar] [CrossRef]
- Dutertre, S.; Jin, A.H.; Alewood, P.F.; Lewis, R.J. Intraspecific variations in Conus geographus defence-evoked venom and estimation of the human lethal dose. Toxicon 2014, 91, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Mir, R.; Karim, S.; Kamal, M.A.; Wilson, C.M.; Mirza, Z. Conotoxins: Structure, therapeutic potential and pharmacological applications. Curr. Pharm. Des. 2016, 22, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Azam, L.; McIntosh, J.M. Alpha-conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol. Sin. 2009, 30, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Munasinghe, N.R.; Christie, M.J. Conotoxins that could provide analgesia through voltage gated sodium channel inhibition. Toxins 2015, 7, 5386–5407. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Berecki, G. Mechanisms of conotoxin inhibition of N-type (Cav2.2) calcium channels. Biochim. Biophys. Acta 2013, 1828, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Prorok, M.; Castellino, F.J. The molecular basis of conantokin antagonism of NMDA receptor function. Curr. Drug Targets 2007, 8, 633–642. [Google Scholar] [CrossRef] [PubMed]
- McManus, O.B.; Musick, J.R. Postsynaptic block of frog neuromuscular transmission by conotoxin GI. J. Neurosci. 1985, 5, 110–116. [Google Scholar] [PubMed]
- McManus, O.B.; Musick, J.R.; Gonzalez, C. Peptides isolated from the venom of Conus geographus block neuromuscular transmission. Neurosci. Lett. 1981, 25, 57–62. [Google Scholar] [CrossRef]
- Gray, W.R.; Luque, A.; Olivera, B.M.; Barrett, J.; Cruz, L.J. Peptide toxins from Conus geographus venom. J. Biol. Chem. 1981, 256, 4734–4740. [Google Scholar] [PubMed]
- Green, B.R.; Bulaj, G.; Norton, R.S. Structure and function of μ-conotoxins, peptide-based sodium channel blockers with analgesic activity. Future Med. Chem. 2014, 6, 1677–1698. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Yamaguchi, Y.; Ishida, Y.; Ohizumi, Y. Roles of basic amino acid residues in the activity of μ-conotoxin GIIIA and GIIIB, peptide blockers of muscle sodium channels. Chem. Biol. Drug Des. 2015, 85, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Ishida, Y.; Wakamatsu, K.; Kato, R.; Honda, H.; Ohizumi, Y.; Nakamura, H.; Ohya, M.; Lancelin, J.M.; Kohda, D.; et al. Active site of mu-conotoxin GIIIA, a peptide blocker of muscle sodium channels. J. Biol. Chem. 1991, 266, 16989–16991. [Google Scholar] [PubMed]
- Hannon, H.E.; Atchison, W.D. Omega-conotoxins as experimental tools and therapeutics in pain management. Mar. Drugs 2013, 11, 680–699. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.J.; Schroeder, T.; Lewis, R. Structure-activity relationships of omega-conotoxins at N-type voltage-sensitive calcium channels. J. Mol. Recognit. 2000, 13, 55–70. [Google Scholar] [CrossRef]
- Wang, F.; Yan, Z.; Liu, Z.; Wang, S.; Wu, Q.; Yu, S.; Ding, J.; Dai, Q. Molecular basis of toxicity of N-type calcium channel inhibitor MVIIA. Neuropharmacology 2016, 101, 137–145. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, M.; Cruz, L.J.; Hunkapiller, M.W.; Gray, W.R.; Olivera, B.M. Isolation and structure of a peptide toxin from the marine snail Conus magus. Arch. Biochem. Biophys. 1982, 218, 329–334. [Google Scholar] [CrossRef]
- Gray, W.R.; Rivier, J.E.; Galyean, R.; Cruz, L.J.; Olivera, B.M.; Conotoxin, M.I. Disulfide bonding and conformational states. J. Biol. Chem. 1983, 258, 12247–12251. [Google Scholar] [PubMed]
- Anderson, P.D. Bioterrorism: Toxins as weapons. J. Pharm. Pract. 2012, 25, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, X.; Dai, S.; Ou, L.; Sun, X.; Zhu, B.; Chen, F.; Shang, M.; Song, H. Quantification of puerarin in plasma by on-line solid-phase extraction column switching liquid chromatography-tandem mass spectrometry and its applications to a pharmacokinetic study. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 863, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Meng, Q.; Chen, Z.; Hou, Y.; Shan, C.; Cheng, Y. Quantitative analysis of a novel HIV fusion inhibitor (sifuvirtide) in HIV infected human plasma using high-performance liquid chromatography-electrospray ionization tandem mass spectrometry. J. Pharm. Biomed. Anal. 2010, 51, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, D.; Hong, Z. A rapid LC-HRMS method for the determination of domoic acid in urine using a self-assembly pipette tip solid-phase extraction. Toxins 2016, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Belén Serrano, A.; Capriotti, A.L.; Cavaliere, C.; Piovesana, S.; Samperi, R.; Ventura, S.; Laganà, A. Development of a rapid LC-MS/MS method for the determination of emerging fusarium mycotoxins enniatins and beauvericin in human biological fluids. Toxins 2015, 7, 3554–3571. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, M.; Tsilia, V.; Rajkovic, A.; De Cremer, K.; Van Loco, J. Application of LC-MS/MS MRM to determine staphylococcal enterotoxins (SEB and SEA) in Milk. Toxins 2016, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Dong, F.; Wang, N.; He, K.; Liu, B.; Wu, S.; Li, A.; Zhang, X. Rapid detection of conotoxin SO-3 in serum using Cu-chelated magnetic beads coupled with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J. Anal. Toxicol. 2009, 33, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, C.; Liu, Z.; Wang, X.; Liu, N.; Du, W.; Dai, Q. Structural and Functional Characterization of a Novel α-Conotoxin Mr1.7 from conus marmoreus targeting neuronal nAChR α3β2, α9α10 and α6/α3β2β3 Subtypes. Mar. Drugs 2015, 13, 3259–3275. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Guidance for Industry Bioanalytical Method Validation; Food and Drug Administration: Silver Spring, MD, USA, 2001.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample No. | Concentration (ng/mL) |
|---|---|
| 2.0 | |
| 1 | 1.83 |
| 2 | 2.27 |
| 3 | 2.28 |
| 4 | 2.32 |
| 5 | 2.41 |
| 6 | 1.77 |
| Mean (ng/mL) | 2.15 |
| SD | 0.27 |
| RSD% | 12.70 |
| RE% | 7.27 |
| Assay | Concentration (ng/mL) | ||
|---|---|---|---|
| 6.0 | 40.0 | 225.0 | |
| Intra-batch (n = 6) | |||
| Mean ± SD (ng/mL) | 5.7 ± 0.3 | 40.5 ± 2.6 | 223.3 ± 9.1 |
| RSD% | 5.72 | 6.31 | 4.08 |
| RE% | −4.80 | 1.25 | −0.76 |
| Inter-batch (n = 7) | |||
| Mean ± SD (ng/mL) | 6.1 ± 0.5 | 40.6 ± 2.9 | 227.9 ± 18.6 |
| RSD% | 8.61 | 7.02 | 8.16 |
| RE% | 1.47 | 1.50 | 1.27 |
| Concentration (ng/mL) | The Ratio of Peak Area b (A) | The Ratio of Peak Area c (B) | The Ratio of Peak Area d (C) | Matrix Effect e (%A) | Recovery f (%) |
|---|---|---|---|---|---|
| 6.0 | 0.129 | 0.146 | 0.077 | 113.32(RSD%7.66) | 52.81(RSD%2.53) |
| 40.0 | 0.853 | 0.955 | 0.583 | 111.96(RSD%3.06) | 61.07(RSD%9.19) |
| 225.0 | 5.512 | 5.462 | 3.216 | 99.10(RSD%6.83) | 58.88(RSD%3.90) |
| IS | 1.173 | 1.048 | 0.700 | 89.36(RSD%2.36) | 66.76(RSD%2.36) |
| Sample | QC1 | QC2 | QC3 |
|---|---|---|---|
| Concentration | 6.0 ng/mL | 40.0 ng/mL | 225.0 ng/mL |
| Room temperature (5.25 h) | |||
| Mean concentration founded (n = 6) | 5.9 ± 0.4 | 35.8 ± 2.5 | 200.5 ± 5.6 |
| RSD% | 6.58 | 7.00 | 2.81 |
| RE% | −1.77 | −10.54 | −10.87 |
| Autosampler(55 h) | |||
| Mean concentration founded (n = 6) | 6.2 ± 0.6 | 37.8 ± 4.3 | 203.5 ± 15.4 |
| RSD% | 9.11 | 11.37 | 7.54 |
| RE% | 3.13 | −5.49 | −9.56 |
| Freeze-thaw (3-cycles) | |||
| Mean concentration founded (n = 6) | 5.6 ± 0.4 | 38.2 ± 4.0 | 205.7 ± 16.9 |
| RSD% | 6.77 | 10.38 | 8.21 |
| RE% | −7.31 | −4.53 | −8.58 |
| Long-term (14 days at −20 °C) | |||
| Mean concentration founded (n = 6) | 6.2 ± 0.6 | 42.0 ± 3.4 | 221.9 ± 12.6 |
| RSD% | 9.86 | 8.14 | 5.67 |
| RE% | 3.76 | 4.95 | −1.38 |
| Matrix | Blood | Plasma |
|---|---|---|
| Mean volume founded (mL) | ||
| n = 4 | 2.0 | 1.0 |
| SD | 0.00 | 0.07 |
| RSD% | 0.00 | 6.90 |
| Mean concentration of GI (ng/mL) | ||
| n = 4 | 100.0 | 141.5 |
| SD | 0.00 | 14.57 |
| RSD% | 0.00 | 10.30 |
| Mean content of GI (ng) | ||
| n = 4 | 200.0 | 147.0 |
| SD | 0.00 | 19.11 |
| RSD% | 0.00 | 13.00 |
| The relative recovery of human blood (%) a | ||
| n = 4 | 73.48 | |
| SD | 9.55 | |
| RSD% | 13.00 | |
| The absolute recovery of human blood (%) b | ||
| n = 4 | 42.32 | |
| SD | 5.50 | |
| RSD% | 13.00 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, S.; Yang, B.; Yan, L.; Dai, Q. Sensitive Detection of α-Conotoxin GI in Human Plasma Using a Solid-Phase Extraction Column and LC-MS/MS. Toxins 2017, 9, 235. https://doi.org/10.3390/toxins9080235
Yu S, Yang B, Yan L, Dai Q. Sensitive Detection of α-Conotoxin GI in Human Plasma Using a Solid-Phase Extraction Column and LC-MS/MS. Toxins. 2017; 9(8):235. https://doi.org/10.3390/toxins9080235
Chicago/Turabian StyleYu, Shuo, Bo Yang, Liangping Yan, and Qiuyun Dai. 2017. "Sensitive Detection of α-Conotoxin GI in Human Plasma Using a Solid-Phase Extraction Column and LC-MS/MS" Toxins 9, no. 8: 235. https://doi.org/10.3390/toxins9080235
APA StyleYu, S., Yang, B., Yan, L., & Dai, Q. (2017). Sensitive Detection of α-Conotoxin GI in Human Plasma Using a Solid-Phase Extraction Column and LC-MS/MS. Toxins, 9(8), 235. https://doi.org/10.3390/toxins9080235