
Careful with That Axe, Gene, Genome Perturbation after a PEG-Mediated Protoplast Transformation in Fusarium verticillioides

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Results
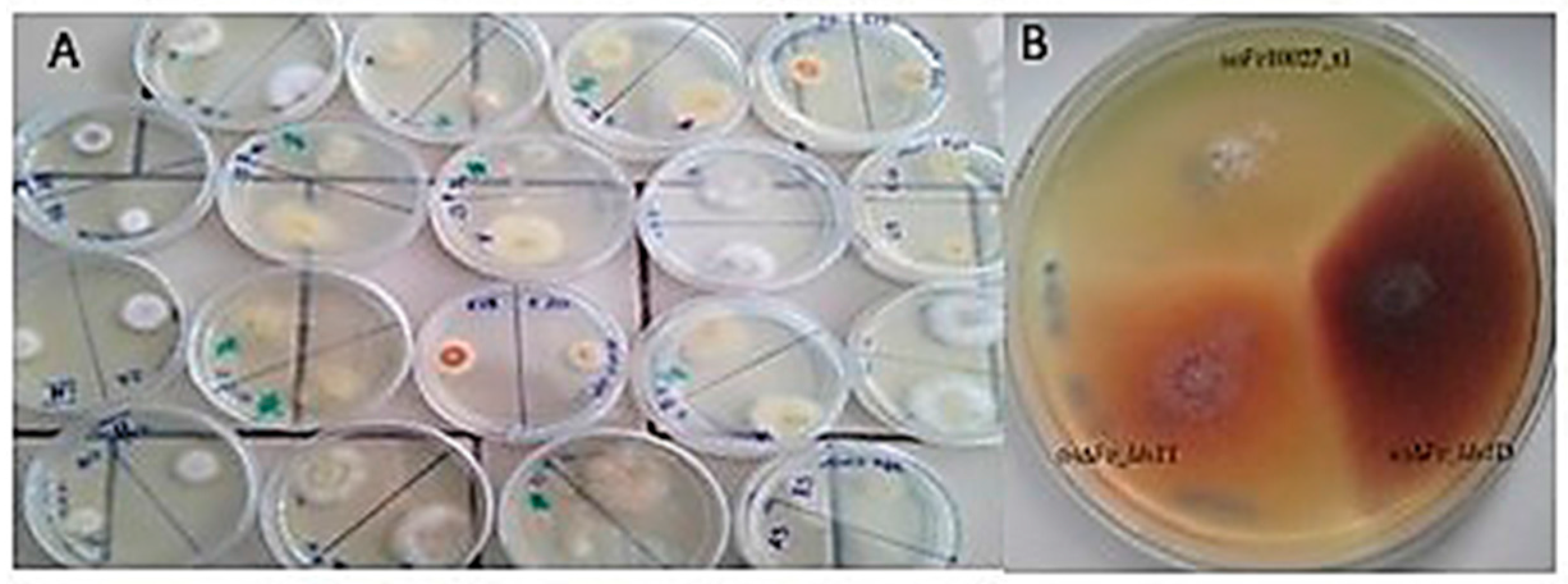
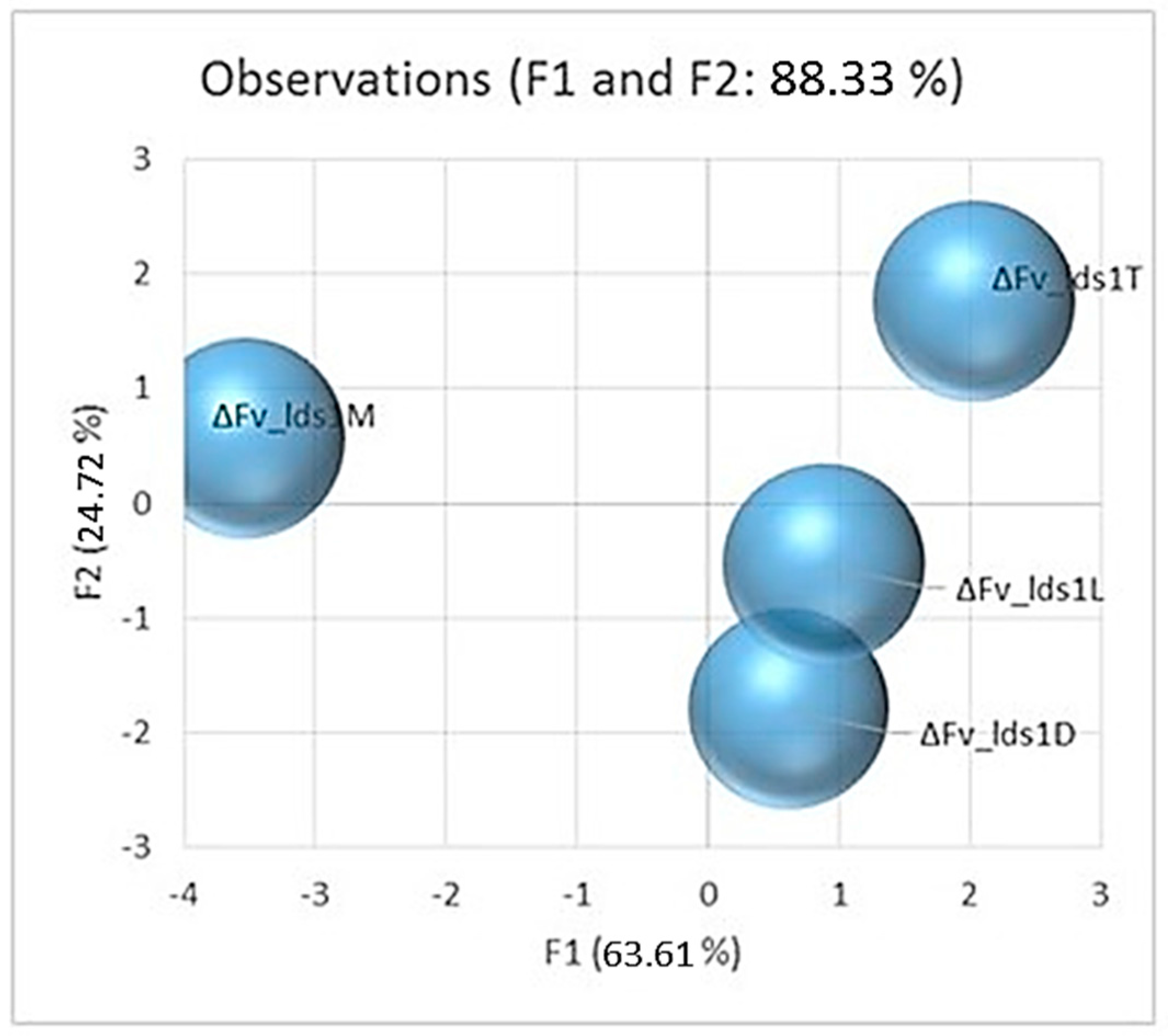
2.1. Phenotypic Variations Within lds1-Deleted Strains
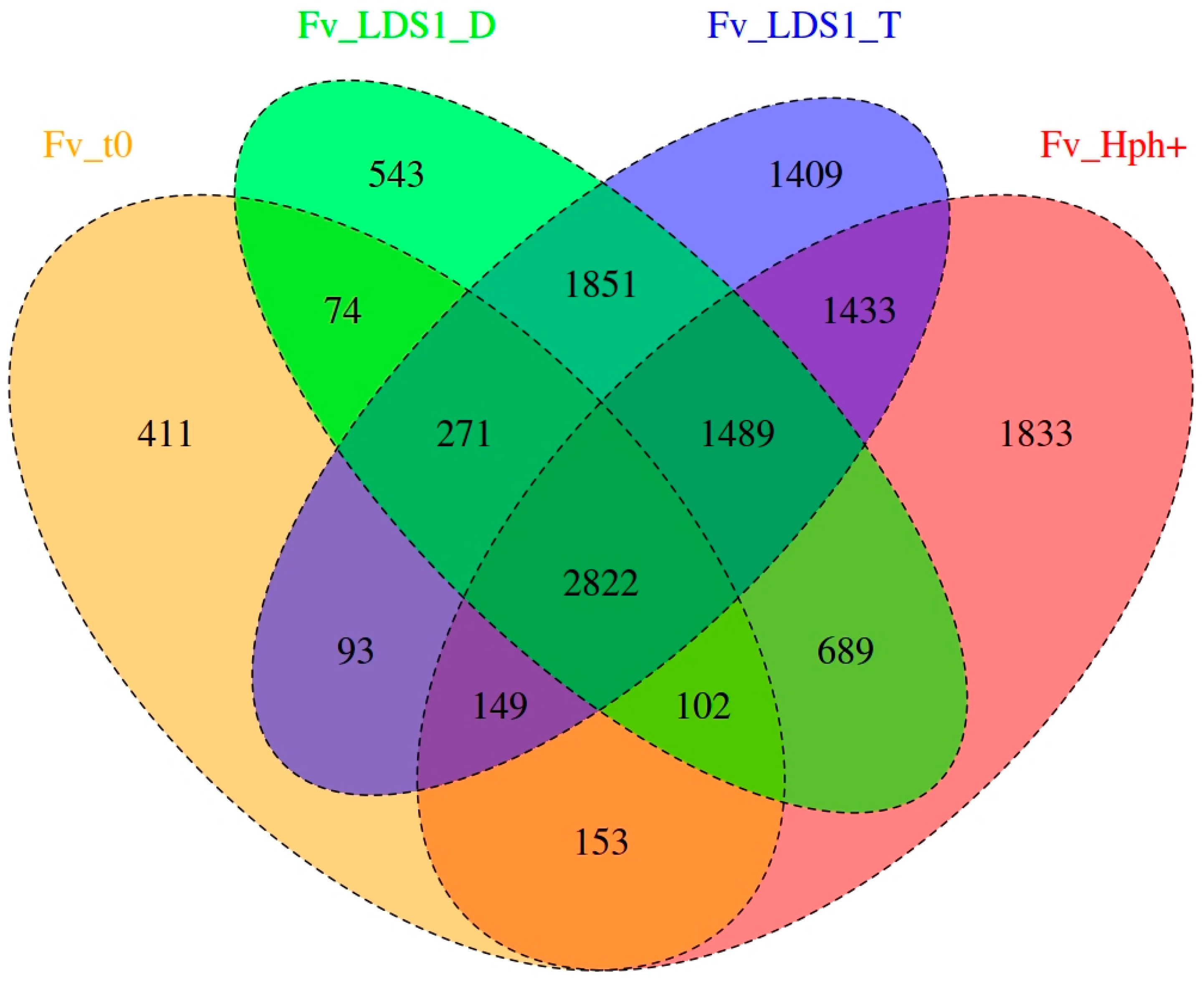
2.2. Variant Calling and Annotation
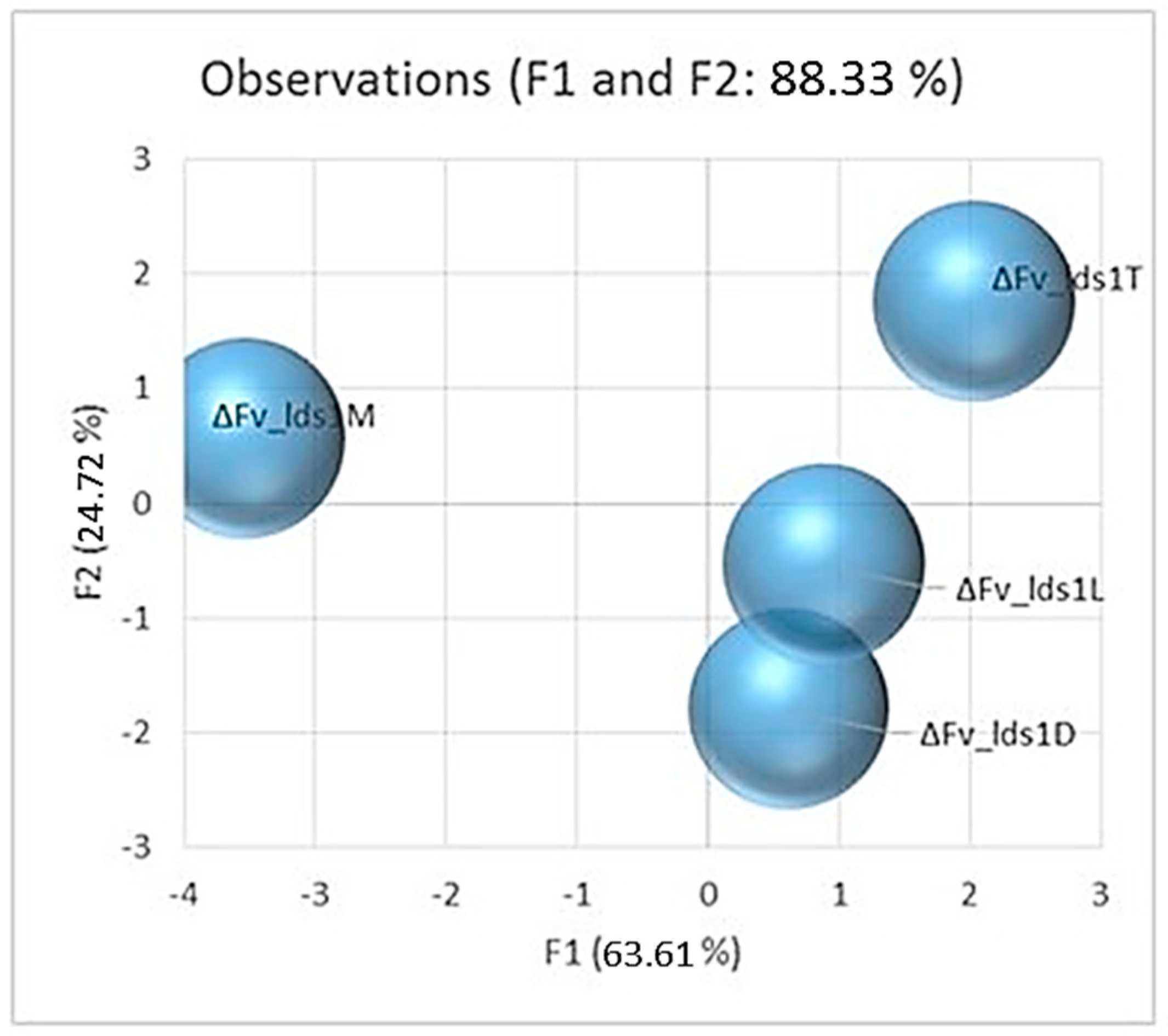
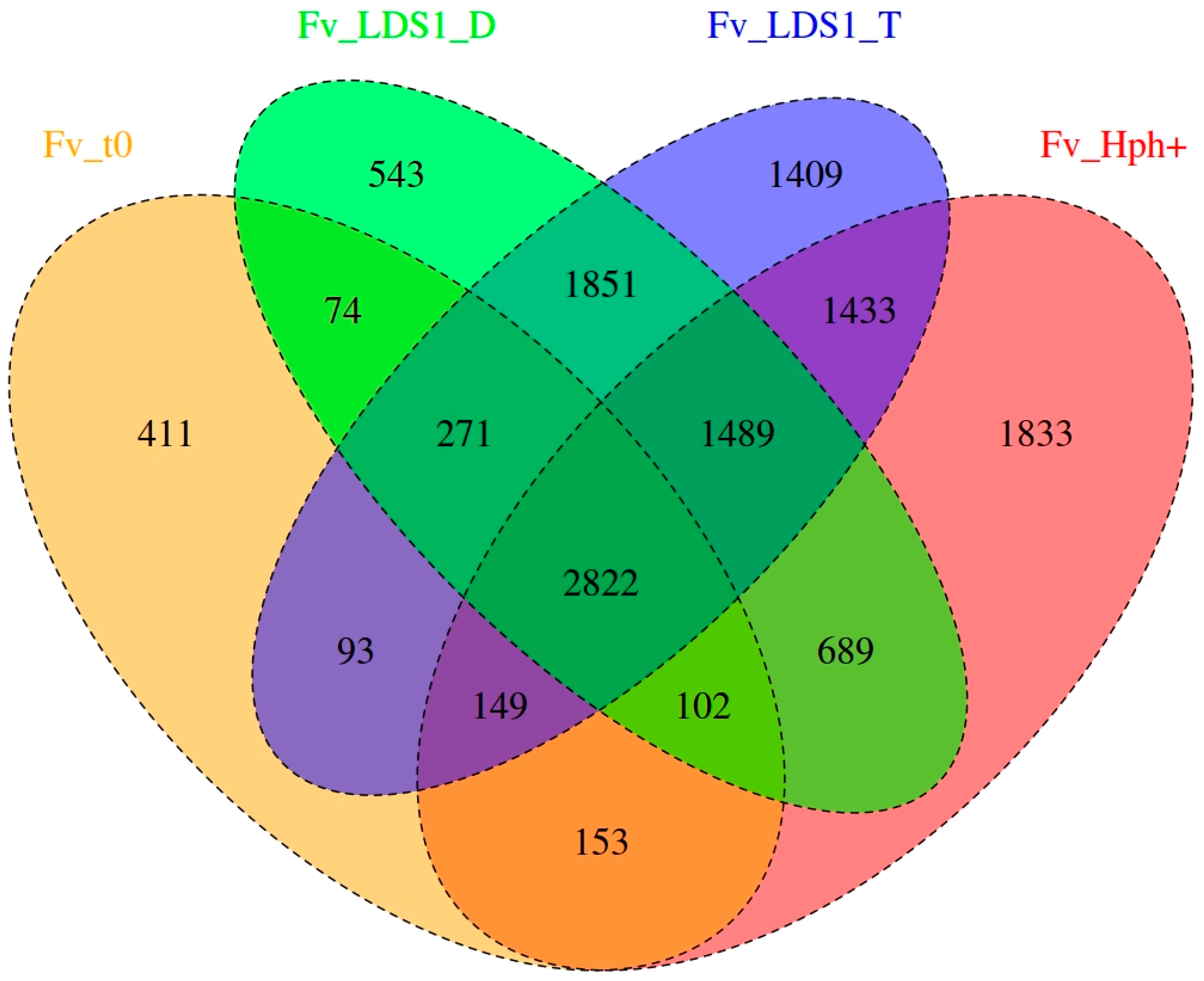
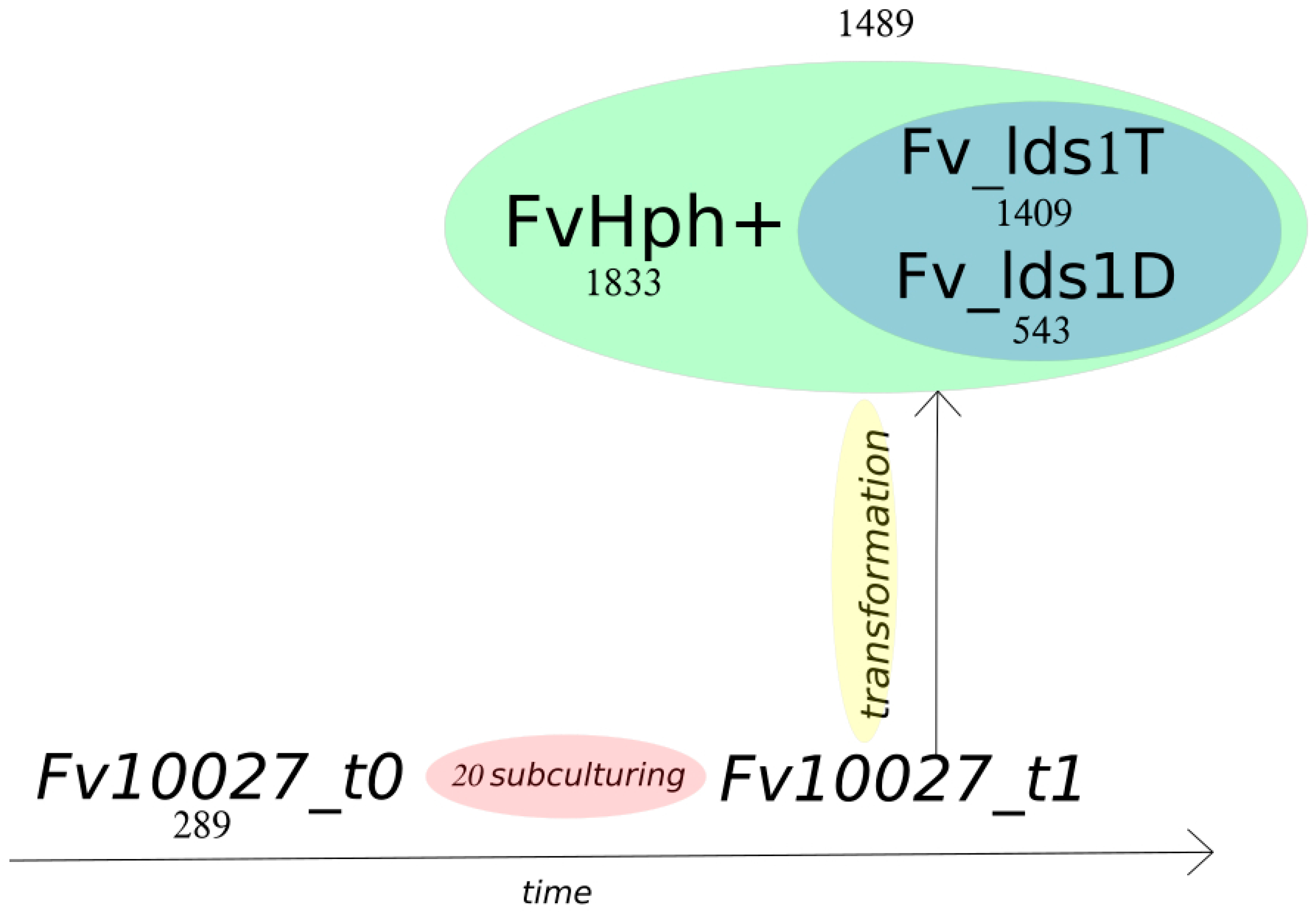
2.3. Significance of Variations in ΔFv_lds1 Mutant Strains
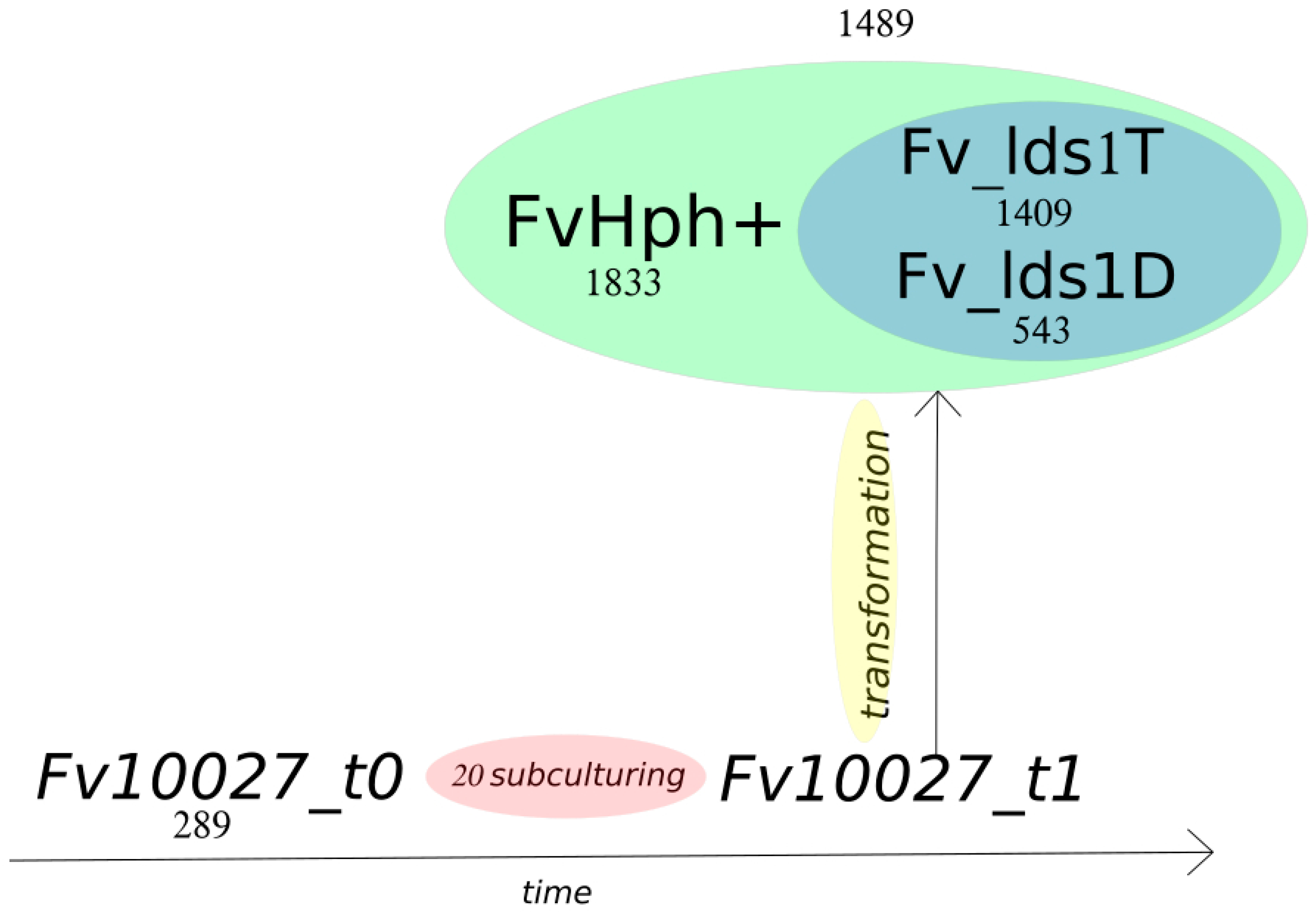
2.4. Multiple Nuclei as a Natural Source of Genome Variation
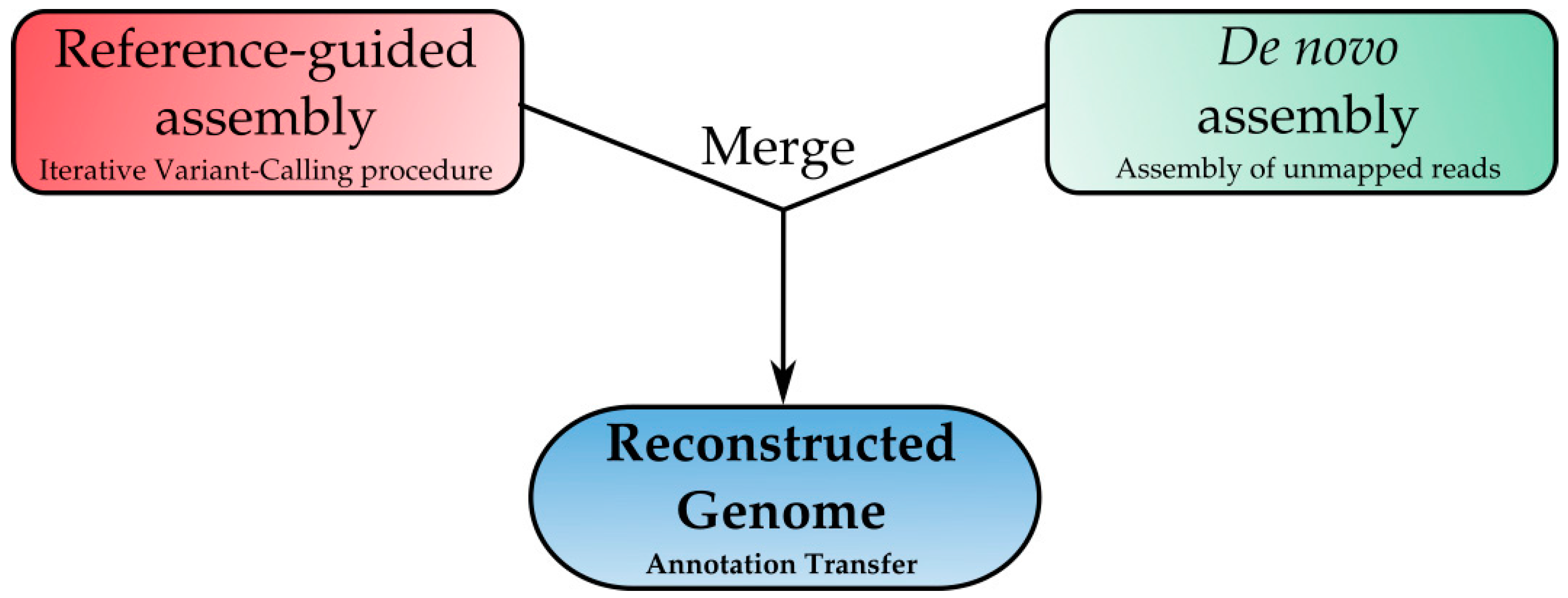
2.5. Genome Reconstruction: A New Frontier for Downstream Analysis
3. Discussion
4. Experimental Section
4.1. Fungal Strains and Media
4.2. Physiological Analyses
4.3. Sequencing
4.4. Bioinformatic Analysis
4.4.1. Filtering of Sequencing Data
4.4.2. SNP, DIP and SV Calling
4.4.3. Genome Reconstruction
4.5. Molecular Analysis
4.5.1. Nucleic Acid Extraction and cDNA Synthesis
4.5.2. Transcript Quantification Assay
4.5.3. Morphological Analyses by Fluorescence Microscopy
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Kerenyi, Z.; Moretti, A.; Waalwijk, C.; Olah, B.; Hornok, L. Mating Type Sequences in Asexually Reproducing Fusarium Species. Appl. Environ. Microbiol. 2004, 70, 4419–4423. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, S.; Kamoun, S. Genome evolution in filamentous plant pathogens: Why bigger can be better. Nat. Rev. Microbiol. 2012, 10, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Stukenbrock, E.H. Evolution, selection and isolation: A genomic view of speciation in fungal plant pathogens. New Phytol. 2013, 199, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Agrios, G.N. Plant Pathology, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Runa, F.; Carbone, I.; Bhatnagar, D.; Payne, G.A. Nuclear heterogeneity in conidial populations of Aspergillus flavus. Fungal Genet. Biol. 2015, 84, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Pontecorvo, G. The Parasexual Cycle in Fungi. Annu. Rev. Microbiol. 1956, 10, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.F. Fungal Vegetative Compatibility. Annu. Rev. Phytopathol. 1993, 31, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Bourne, E.; Mina, D.; Gonçalves, S.C.; Loureiro, J.; Freitas, H.; Muller, L.A. Large and variable genome size unrelated to serpentine adaptation but supportive of cryptic sexuality in Cenococcum geophilum. Mycorrhiza 2014, 24, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Kvas, M.; Marasas, W.F.O.; Wingfield, B.D.; Wingfield, M.J.; Steenkamp, E.T. Diversity and evolution of Fusarium species in the Gibberella fujikuroi complex. Fungal Divers. 2009, 34, 1–21. [Google Scholar]
- Logrieco, A.; Bottalico, A.; Mulé, G.; Moretti, A.; Perrone, G. Epidemiology of toxigenic fungi and their associated mycotoxins for some Mediterranean crops. Eur. J. Plant Pathol. 2003, 109, 645. [Google Scholar] [CrossRef]
- Estrada, A.E.R.; Jonkers, W.; Kistler, H.C.; May, G. Interactions between Fusarium verticillioides, Ustilago maydis, and Zea mays: An endophyte, a pathogen, and their shared plant host. Fungal Genet. Biol. 2012, 49, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Falavigna, C.; Lazzaro, I.; Galaverna, G.; Dall’Asta, C.; Battilani, P. Oleoyl and linoleoyl esters of fumonisin B1 are differently produced by Fusarium verticillioides on maize and rice based media. Int. J. Food Microbiol. 2016, 217, 79. [Google Scholar] [CrossRef] [PubMed]
- Ochratoxin, A. Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 1993; Volume 56, pp. 489–521. [Google Scholar]
- Marasas, W.F.O. Discovery and occurrence of the fumonisins: A historical perspective. Environ. Health Perspect. 2001, 109, 239–243. [Google Scholar] [CrossRef] [PubMed]
- De Ruyck, K.; De Boevre, M.; Huybrechts, I.; De Saeger, S. Dietary mycotoxins, co-exposure, and carcinogenesis in humans: Short review. Mutat. Res. 2015, 766, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Scala, V.; Giorni, P.; Cirlini, M.; Ludovici, M.; Visentin, I.; Cardinale, F.; Fabbri, A.A.; Fanelli, C.; Reverberi, M.; Battilani, P.; et al. LDS1-produced oxylipins are negative regulators of growth, conidiation and fumonisin synthesis in the fungal maize pathogen Fusarium verticillioides. Front. Microbiol. 2014, 5, 669. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Sung, W.; Morris, K.; Coffey, N.; Landry, C.R.; Dopman, E.P.; Dickinson, W.J.; Okamoto, K.; Kulkarni, S.; Hartl, D.L.; et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl. Acad. Sci. USA 2008, 105, 9272–9277. [Google Scholar] [CrossRef] [PubMed]
- Denver, R.; Dolan, P.C.; Wilhelm, L.J.; Sung, W.; Lucas-Lledó, J.I.; Howe, D.K.; Lewis, S.C.; Okamoto, K.; Thomas, W.K.; Lynch, M.; et al. A genome-wide view of Caenorhabditis elegans base substitution mutation processes. Proc. Natl. Acad. Sci. USA 2009, 106, 16310–16314. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Halligan, D.L.; Keightley, P.D. Spontaneous mutation accumulation studies in evolutionary genetics. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 151–172. [Google Scholar] [CrossRef]
- Latham, J.R.; Wilson, A.K.; Steinbrecher, R.A. The mutational consequences of plant transformation. J. Biomed. Biotechnol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Fincham, J.R.S. Transformation in fungi. Microbiol. Rev. 1989, 53, 148–170. [Google Scholar] [PubMed]
- Ma, L.J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Sanseverino, W.; Hénaff, E.; Vives, C.; Pinosio, S.; Burgos-Paz, W.; Morgante, M.; Ramos-Onsins, S.E.; Garcia-Mas, J.; Casacuberta, J.M. Transposon Insertions, Structural Variations, and SNPs contribute to the evolution of the melon genome. Mol. Biol. Evol. 2015, 32, 2760–2774. [Google Scholar] [CrossRef] [PubMed]
- Roper, M.; Ellison, C.; Taylor, J.W.; Glass, N.L. Nuclear and Genome Dynamics in Multinucleate Ascomycete Fungi. Curr. Biol. 2011, 21, R786–R793. [Google Scholar] [CrossRef] [PubMed]
- Slepecky, R.A.; Starmer, W.T. Phenotypic plasticity in fungi: a review with observations on Aureobasidium pullulans. Mycologia 2009, 101, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Reverberi, M.; Punelli, M.; Scala, V.; Scarpari, M.; Uva, P.; Mentzen, W.I.; Dolezal, A.L.; Woloshuk, C.; Pinzari, F.; Fabbri, A.A.; et al. Genotypic and phenotypic versatility of Aspergillus flavus during maize exploitation. PLoS ONE 2013, 8, e68735. [Google Scholar] [CrossRef] [PubMed]
- Savile, D.B.O. The Meaning of “Pleomorphism”. Mycologia 1969, 61, 1161–1162. [Google Scholar] [CrossRef] [PubMed]
- West-Eberhard, M.J. Phenotypic plasticity and the origins of diversity. Annu. Rev. Ecol. Syst. 1989, 20, 249–278. [Google Scholar] [CrossRef]
- Promislow, D. A Regulatory network analysis of phenotypic plasticity in yeast. Am. Nat. 2005, 165, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.W.; Proctor, R.H. Fusarium: Genomics, Molecular and Cellular Biology; Horizon Scientific Press: Haverhill, UK, 2013; ISBN 1908230258, 9781908230256. [Google Scholar]
- Gan, X.; Stegle, O.; Behr, J.; Steffen, J.G.; Drewe, P.; Hildebrand, K.L.; Lyngsoe, R.; Schultheiss, S.J.; Osborne, E.J.; Sreedharan, V.T.; et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 2011, 477, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, M.; Mateo, J.J.; Hinojo, M.J.; Mateo, R. Sugars and amino acids as factors affecting the synthesis of fumonisins in liquid cultures by isolates of the Gibberella fujikuroi complex. Int. J. Food Microbiol. 2003, 89, 185–193. [Google Scholar] [CrossRef]
- López-Errasquín, E.; Vázquez, C.; Jiménez, M.; González-Jaén, M.T. Real-Time RT-PCR assay to quantify the expression of fum1 and fum19 genes from the Fumonisin-producing Fusarium verticillioides. J. Microbiol. Methods 2007, 68, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Ludovici, M.; Ialongo, C.; Reverberi, M.; Beccaccioli, M.; Scarpari, M.; Scala, V. Quantitative profiling of oxylipins through comprehensive LC-MS/MS analysis of Fusarium verticillioides and maize kernels. Food Addit. Contam. 2014, 31, 2026–2033. [Google Scholar] [CrossRef] [PubMed]
- Scala, V.; Camera, E.; Ludovici, M.; Dall’Asta, C.; Cirlini, M.; Giorni, P.; Battilani, P.; Bello, C.; Fabbri, A.A.; Fanelli, C.; et al. Fusarium verticillioides and maize interaction in vitro: relationship between oxylipin cross-talk and fumonisin synthesis. World Mycotoxin J. 2013, 6, 343–351. [Google Scholar] [CrossRef]
- Addinsoft. XLSTAT, Analyse de Données et Statistique Avec MS Excel, Addinsoft: New York, NY, USA, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Antioxidant Enzymes | Oxylipin Genes | Oxylipins | Growth | Conidia | Conidia Germination | FBs |
|---|---|---|---|---|---|---|---|
| In vitro | |||||||
| ΔFv_lds1D | 0.20 ± 0.02 | 29.9 ± 1.2 | 0.52 ± 0.02 | 0.95 ± 0.11 | 0.74 ± 0.08 | 0.72 ± 0.05 | 3.4 ± 0.2 |
| ΔFv_lds1T | 0.24 ± 0.04 | 63.5 ± 5.3 | 0.54 ± 0.05 | 0.88 ± 0.05 | 2.22 ± 0.15 | 0.99 ± 0.08 | 3.2 ± 0.2 |
| ΔFv_lds1M | 0.31 ± 0.02 | 2.9 ± 0.5 | 0.62 ± 0.04 | 0.97 ± 0.02 | 0.60 ± 0.04 | 1.21 ± 0.12 | 1.0 ± 0.1 |
| ΔFv_lds1L | 0.19 ± 0.04 | 30.7 ± 5.2 | 0.49 ± 0.03 | 0.92 ± 0.08 | 1.07 ± 0.14 | 1.21 ± 0.09 | 3.2 ± 0.2 |
| FvHph+ | -- | 17.3 ± 4.2 | -- | 0.68 ± 0.10 | 1.01 ± 0.07 | -- | 1.3 ± 0.1 |
| In vivo | |||||||
| ΔFv_lds1D | 9.9 ± 1.5 | 1.1 ± 0.2 | 0.76 ± 0.04 | 15.2 ± 3.2 | |||
| ΔFv_lds1T | 57.5 ± 5.1 | 1.3 ± 0.2 | 1.56 ± 0.22 | 139.4 ± 16.3 | |||
| ALL Variants | Raw Variant | Quality Filtered and Background Removed | Referenced to Fv_10027_t1 |
|---|---|---|---|
| Total variants | 285,588 | 18,964 | 13,202 |
| Total SNPs | 273,744 | 18,802 | 13,086 |
| Total DIPs | 11,844 | 162 | 116 |
| Name of Variant | Position | Gene Name | Type of Variant | Description of Gene | Predicted Function |
|---|---|---|---|---|---|
| SNP1 | Scaffold_2:1748174 | FVEG_14979 | stop_gained | hypothetical protein | |
| SNP2 | Scaffold_3:383277 | FVEG_02691 | stop_gained | amidohydrolase ytcJ-like | |
| SNP3 | Scaffold_3:985831 | FVEG_02859 | stop_gained | hypothetical protein | |
| SNP4 | Scaffold_4:2239365 | FVEG_04184 | stop_gained | hypothetical protein | GPI-anchor transamidase |
| SNP5 | Scaffold_5:2242191 | FVEG_05070 | stop_gained | hypothetical protein | THUMP domain, involved in RNA metabolism |
| SNP6 | Scaffold_9:1378671 | FVEG_07628 | stop_gained | hypothetical protein | |
| SNP7 | Scaffold_12:878190 | FVEG_09192 | stop_gained | hypothetical protein | Myc-type, basic helix-loop-helix DNA binding protein |
| SNP8 | Scaffold_13:426900 | FVEG_09582 | stop_gained | hypothetical protein | |
| SNP9 | Scaffold_13:1337782 | FVEG_09864 | stop_gained | hypothetical protein | AMP-dependent synthetase/ligase; phosphopantetheine binding ACP domain |
| SNP10 | Scaffold_15:1311389 | FVEG_10951 | stop_gained | hypothetical protein | Proton-dependent oligopeptide transporter family |
| SNP11 | Scaffold_17:682448 | FVEG_11590 | stop_gained | iron-sulfur clusters transporter ATM1 | |
| SNP12 | Scaffold_18:160090 | FVEG_11865 | stop_gained | hypothetical protein | Zn(2)-C6 fungal-type DNA-binding domain; transcription factor domain, fungi |
| SNP13 | Scaffold_19:97648 | FVEG_12210 | stop_gained | hypothetical protein | |
| SNP14 | Scaffold_19:261693 | FVEG_12278 | stop_gained | hypothetical protein | |
| SNP15 | Scaffold_19:418919 | FVEG_17230 | stop_gained | hypothetical protein | |
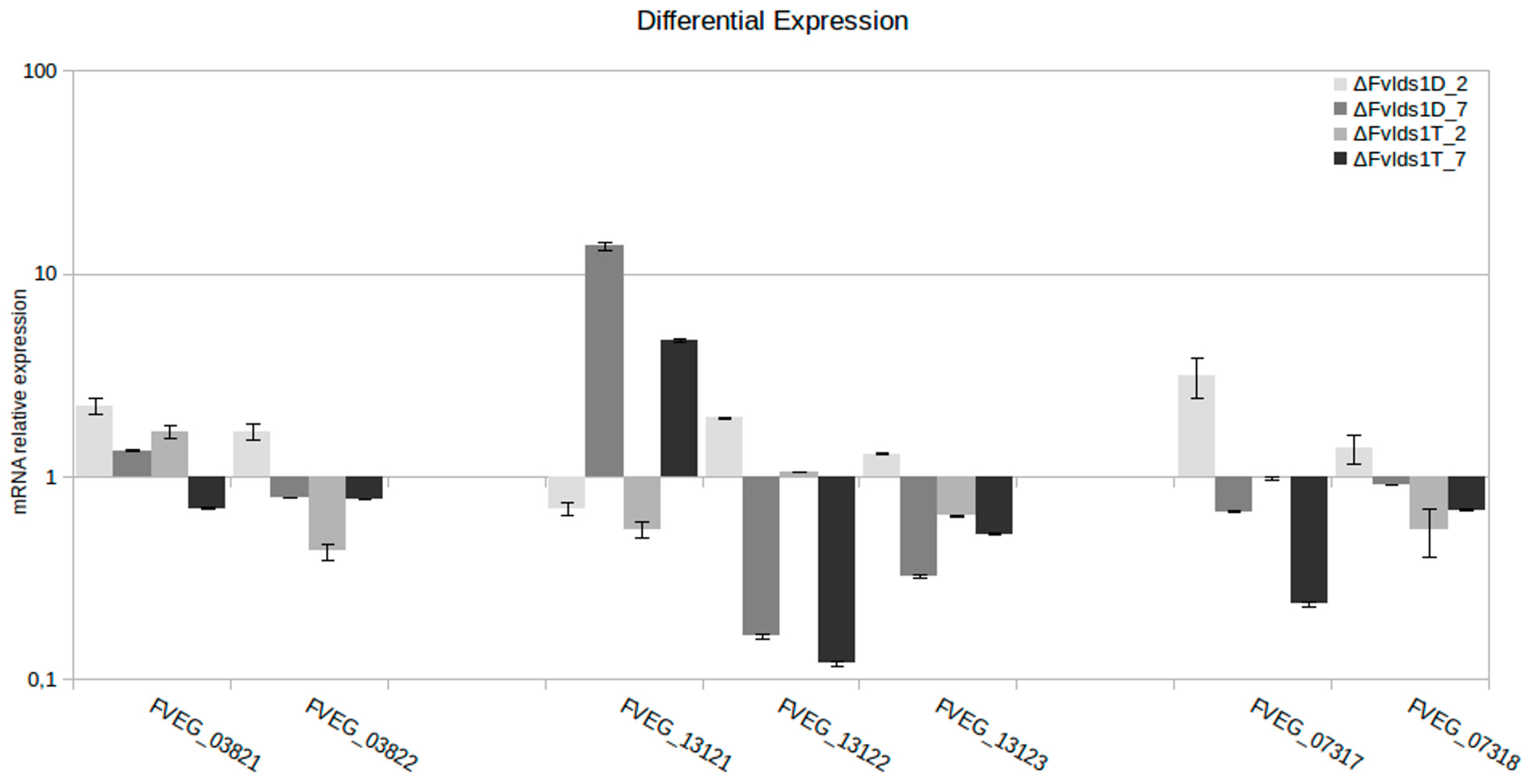
| DIP1 | Scaffold_4:1048999 | FVEG_03822 | frameshift | hypothetical protein | Basic region leucine zipper |
| DIP2 | Scaffold_1:2640553 | FVEG_00886 | frameshift | hypothetical protein | Leucine-rich repeat domain |
| DIP3 | Scaffold_4:2461229 | FVEG_04252 | frameshift | hypothetical protein | Metallo-beta-lactamase |
| DIP4 | Scaffold_22:131177 | FVEG_13121 | Upstream gene variant | hypothetical protein | Sensitivity to red light reduced-like, SRR1 |
| Scaffold_22:131177 | FVEG_13122 | Disruptive in-frame deletion | hypothetical protein | ||
| Scaffold_22:131177 | FVEG_13123 | Downstream gene variant | hypothetical protein | WD40-repeat-containing domain | |
| DIP5 | Scaffold_5:1387592 | FVEG_04792 | frameshift | hypothetical protein | Ribosome biogenesis protein Nop16 |
| DIP6 | Scaffold_9:391617 | FVEG_07297 | frameshift | hypothetical protein | MIF4G-like, type 3 domain; armadillo-type fold domain |
| DIP7 | Scaffold_12:1355894 | FVEG_09359 | frameshift | hypothetical protein | transmembrane transport |
| DIP8 | Scaffold_17:18487 | FVEG_11383 | frameshift | calcium binding protein 39 | |
| DIP9 | Scaffold_17:18487 | FVEG_11384 | frameshift | hypothetical protein | SWIB domain |
| DIP10 | Scaffold_18:298218 | FVEG_11924 | frameshift | hypothetical protein | |
| DIP11 | Scaffold_23:269982 | FVEG_13429 | frameshift | hypothetical protein | S-adenosyl-l-methionine-dependent methyltransferase |
| Structural Variants present only in ΔFvl_ds1T and ΔFv_lds1D stains | |||||
| SV1 | Scaffold_9:449257 | FVEG_07317 | stop lost, in-frame deletion, splice region variant | adenosine triphosphatase | ATPase family associated with various cellular activities |
| Scaffold_9:449257 | FVEG_07318 | Upstream gene variant | cystathionine beta-lyase | Cys/Met metabolism, pyridoxal phosphate-dependent enzyme | |
| ALL Variants | Fv10027_t0 | Fv_Hph+ | ΔFv_lds1D | ΔFv_lds1T |
|---|---|---|---|---|
| Total variants | 289 | 1833 | 542 | 1409 |
| Heterozygous variants | 289 | 1825 | 541 | 1408 |
| Homozygous variants | 0 | 8 | 1 | 1 |
| Number of Scaffolds | 36 |
| Total size | 41,749,604 bp |
| Longest sequence | 4,625,635 bp |
| Shortest sequence | 14,062 bp |
| Mean sequence size | 1,159,711 bp |
| N50 | 1,960,296 bp |
| Number of genes | 15,867 |
| Number of transcripts | 20,551 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scala, V.; Grottoli, A.; Aiese Cigliano, R.; Anzar, I.; Beccaccioli, M.; Fanelli, C.; Dall’Asta, C.; Battilani, P.; Reverberi, M.; Sanseverino, W. Careful with That Axe, Gene, Genome Perturbation after a PEG-Mediated Protoplast Transformation in Fusarium verticillioides. Toxins 2017, 9, 183. https://doi.org/10.3390/toxins9060183
Scala V, Grottoli A, Aiese Cigliano R, Anzar I, Beccaccioli M, Fanelli C, Dall’Asta C, Battilani P, Reverberi M, Sanseverino W. Careful with That Axe, Gene, Genome Perturbation after a PEG-Mediated Protoplast Transformation in Fusarium verticillioides. Toxins. 2017; 9(6):183. https://doi.org/10.3390/toxins9060183
Chicago/Turabian StyleScala, Valeria, Alessandro Grottoli, Riccardo Aiese Cigliano, Irantzu Anzar, Marzia Beccaccioli, Corrado Fanelli, Chiara Dall’Asta, Paola Battilani, Massimo Reverberi, and Walter Sanseverino. 2017. "Careful with That Axe, Gene, Genome Perturbation after a PEG-Mediated Protoplast Transformation in Fusarium verticillioides" Toxins 9, no. 6: 183. https://doi.org/10.3390/toxins9060183
APA StyleScala, V., Grottoli, A., Aiese Cigliano, R., Anzar, I., Beccaccioli, M., Fanelli, C., Dall’Asta, C., Battilani, P., Reverberi, M., & Sanseverino, W. (2017). Careful with That Axe, Gene, Genome Perturbation after a PEG-Mediated Protoplast Transformation in Fusarium verticillioides. Toxins, 9(6), 183. https://doi.org/10.3390/toxins9060183