
HDAC8 Prevents Anthrax Lethal Toxin-induced Cell Cycle Arrest through Silencing PTEN in Human Monocytic THP-1 Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. HDAC8 Decreases Susceptibility to LeTx-Induced Cytotoxicity
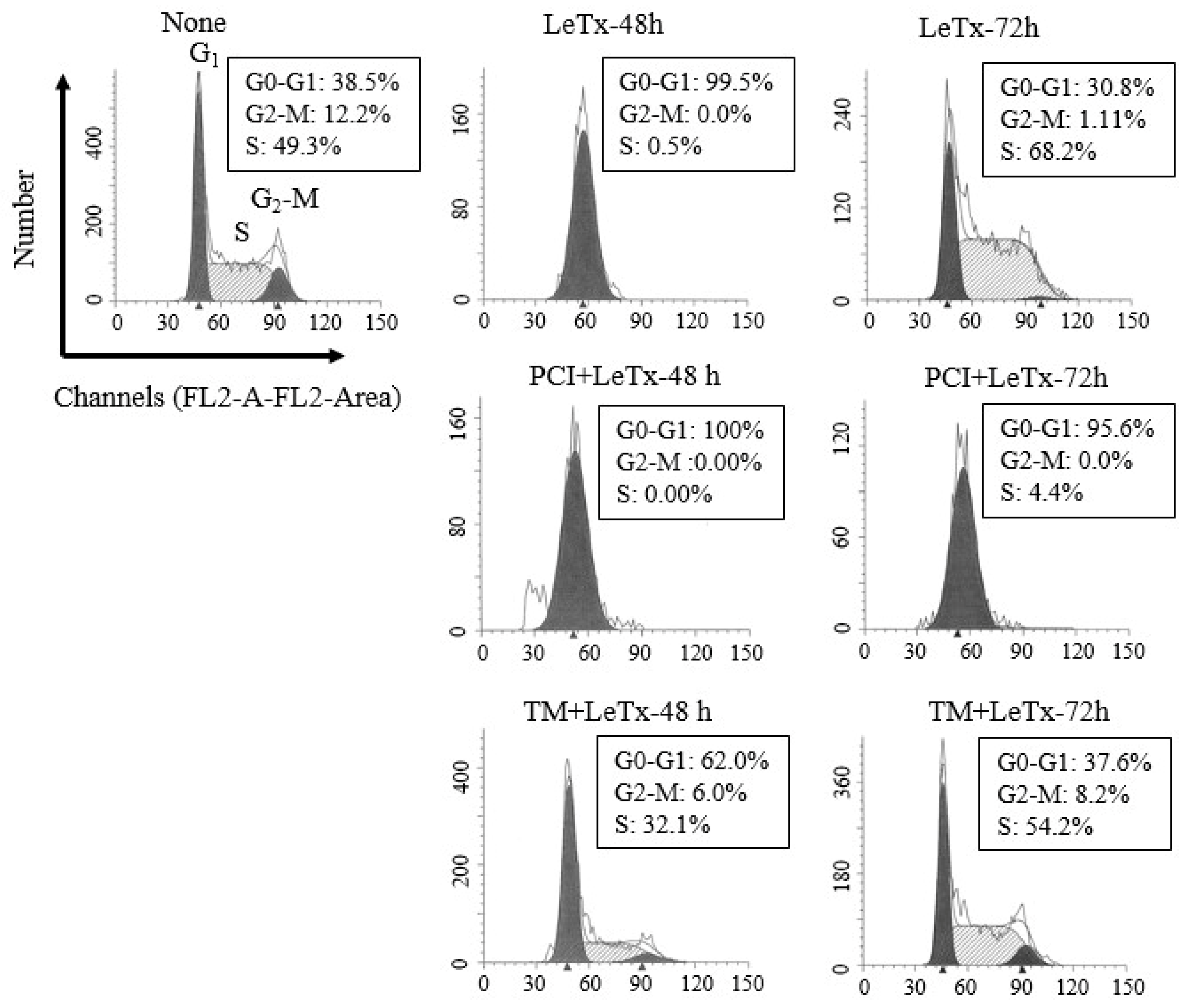
2.2. HDAC8 Prevents Cell Cycle Arrest Induced by LeTx
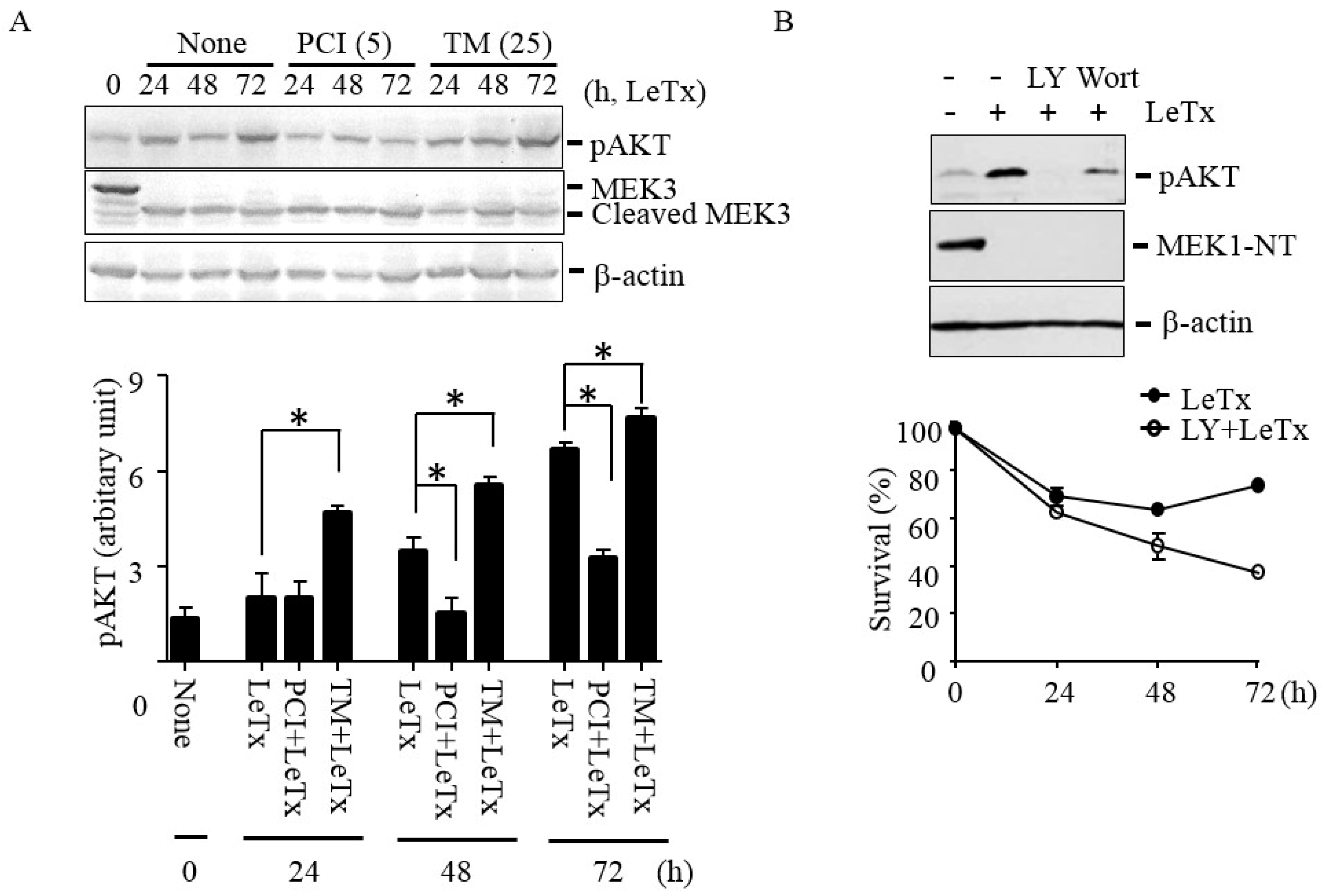
2.3. HDAC8 Activity is Required for Activating AKT in LeTx-Treated Cells
2.4. LeTx Suppresses PTEN Expression through HDAC8
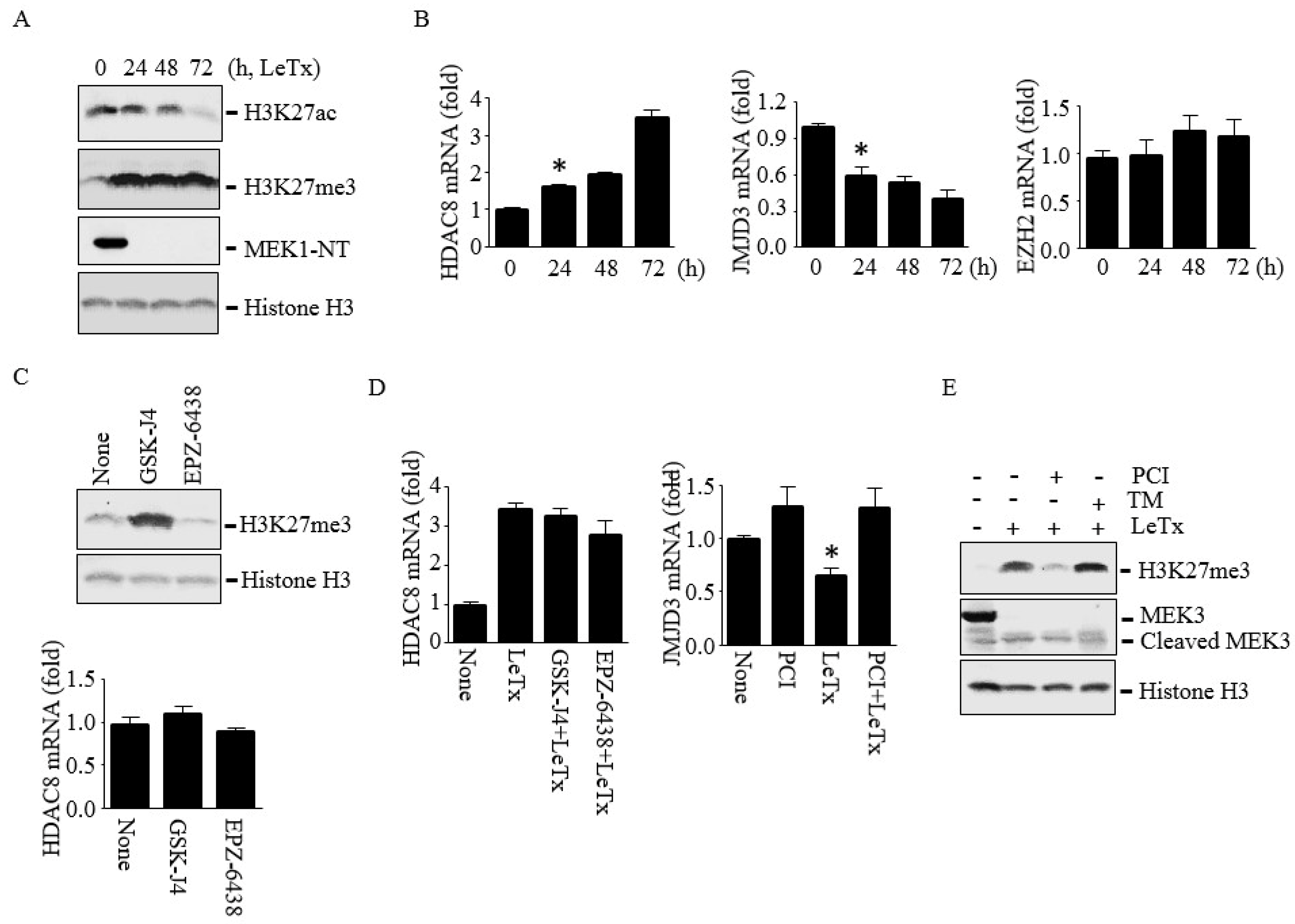
2.5. HDAC8 Induces Histone H3K27me3 by Silencing JMJD3
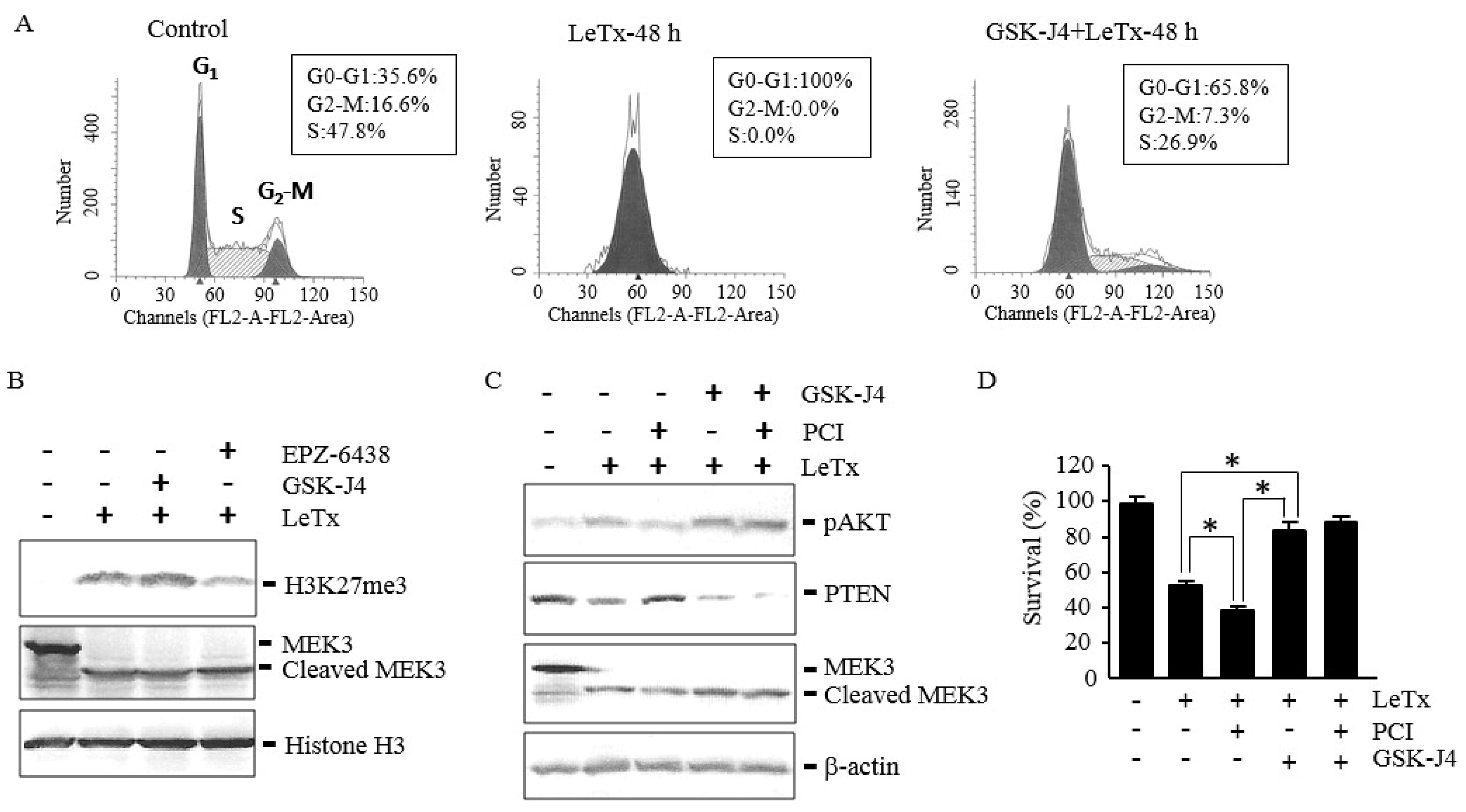
2.6. GSK-J4 Induces AKT Activation and Prevents Cell Cycle Arrest even in the Presence of PCI in LeTx-Treated Cells
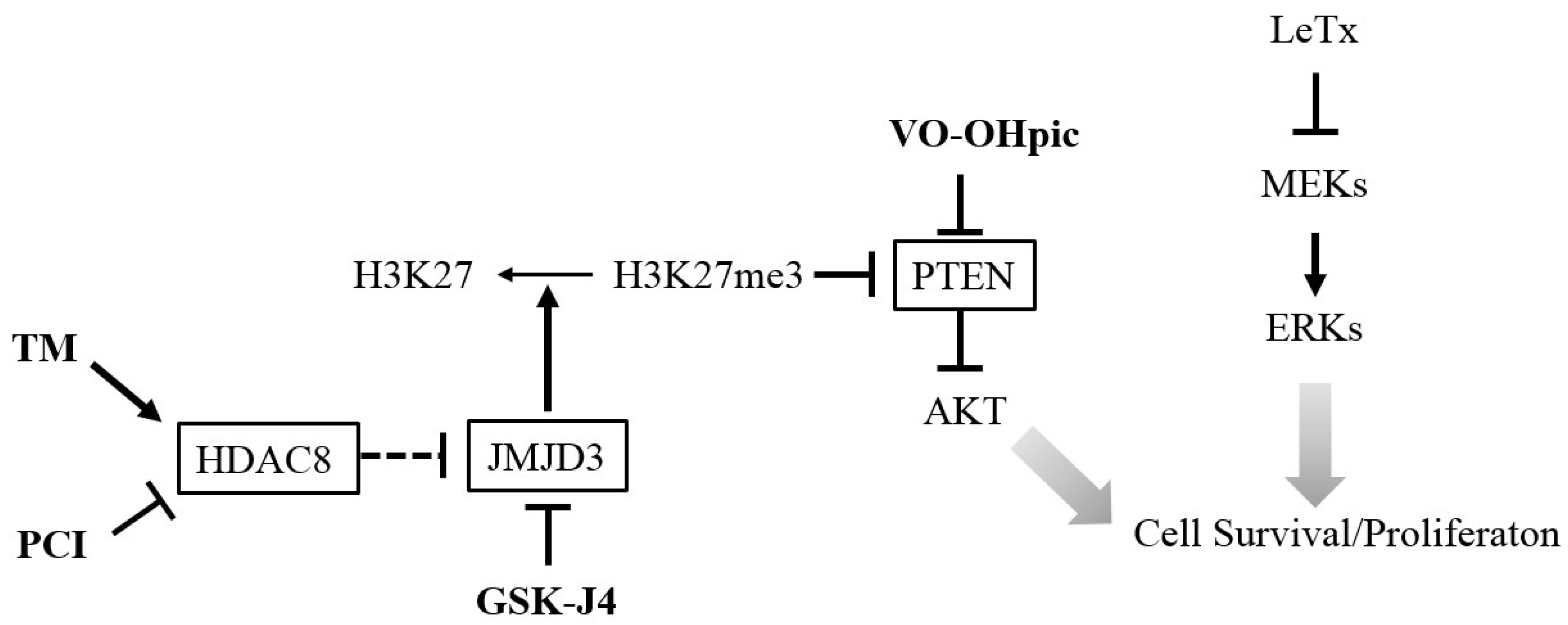
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Western Blot
4.5. Quantitative Real-Time PCR
4.6. Cell Cycle Analysis
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ERK | extracellular signal-regulated kinase |
| EZH2 | enhancer of zeste 2 |
| HDAC | histone deacetylase |
| H3K27Ac | histone H3 lysine 27 acetylation |
| H3K27me3 | histone H3 lysine 27 tri-methylation |
| JMJD3 | Jumonji Domain Containing 3 |
| LeTx | anthrax lethal toxin |
| LF | lethal factor |
| LY | LY942006 |
| MAPK | mitogen-activated protein kinase |
| MEK | MAPK kinase |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PA | protective antigen |
| PI3K | phosphoinositide 3-kinase |
| SMC3 | structural maintenance of chromosomes 3 |
| PCI | HDAC8 inhibitor PCI34051 |
| PTEN | phosphatase and tensin homolog 1 |
| TM | HDAC8 activator TM-2-51 |
References
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax pathogenesis. Annu. Rev. Microbiol. 2015, 69, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Fang, H.; Frucht, D.M. Anthrax lethal toxin increases superoxide production in murine neutrophils via differential effects on Mapk signaling pathways. J. Immunol. 2008, 180, 4139–4147. [Google Scholar] [CrossRef] [PubMed]
- Kassam, A.; Der, S.D.; Mogridge, J. Differentiation of human monocytic cell lines confers susceptibility to Bacillus anthracis lethal toxin. Cell Microbiol. 2005, 7, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Ng, D.; Lamothe, J.; Valvano, M.A.; Han, J.; Kim, S.O. Mitochondrial proteins Bnip3 and Bnip3L are involved in anthrax lethal toxin-induced macrophage cell death. J. Biol. Chem. 2007, 282, 26275–26283. [Google Scholar] [CrossRef] [PubMed]
- Raymond, B.; Batsche, E.; Boutillon, F.; Wu, Y.Z.; Leduc, D.; Balloy, V.; Raoust, E.; Muchardt, C.; Goossens, P.L.; Touqui, L. Anthrax lethal toxin impairs IL-8 expression in epithelial cells through inhibition of histone H3 modification. PLoS Pathog. 2009, 5, e1000359. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Greten, F.R.; Li, Z.W.; Karin, M. Macrophage apoptosis by anthrax lethal factor through p38 Map kinase inhibition. Science 2002, 297, 2048–2051. [Google Scholar] [CrossRef] [PubMed]
- Comer, J.E.; Chopra, A.K.; Peterson, J.W.; Konig, R. Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infect. Immun. 2005, 73, 8275–8281. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.M.; VanBrocklin, M.; McWilliams, M.J.; Leppla, S.H.; Duesbery, N.S.; Woude, G.F. Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Popov, S.G.; Villasmil, R.; Bernardi, J.; Grene, E.; Cardwell, J.; Wu, A.; Alibek, D.; Bailey, C.; Alibek, K. Lethal toxin of Bacillus anthracis causes apoptosis of macrophages. Biochem. Biophys. Res. Commun. 2002, 293, 349–355. [Google Scholar] [CrossRef]
- Kirby, J.E. Anthrax lethal toxin induces human endothelial cell apoptosis. Infect. Immun. 2004, 72, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Xu, L.; Chen, T.Y.; Cyr, J.M.; Frucht, D.M. Anthrax lethal toxin has direct and potent inhibitory effects on B cell proliferation and immunoglobulin production. J. Immunol. 2006, 176, 6155–6161. [Google Scholar] [CrossRef] [PubMed]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D'Elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.Y.; Kang, C.I.; Hur, G.H.; Yang, J.M.; Shin, S. Bacillus anthracis lethal toxin induces cell-type-specific cytotoxicity in human lung cell lines. J. Appl. Microbiol. 2014, 116, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Ng, D.; Pelech, S.L.; Kim, S.O. Critical role of the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase-3 signaling pathway in recovery from anthrax lethal toxin-induced cell cycle arrest and Mek cleavage in macrophages. J. Biol. Chem. 2007, 282, 36230–36239. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Mehta, H.; Chakrabarty, K.; Booth, J.L.; Duggan, E.S.; Patel, K.B.; Ballard, J.D.; Coggeshall, K.M.; Metcalf, J.P. Resistance of human alveolar macrophages to Bacillus anthracis lethal toxin. J. Immunol. 2009, 183, 5799–5806. [Google Scholar] [CrossRef] [PubMed]
- Meloche, S.; Pouyssegur, J. The Erk1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, N.; Fukui, Y.; Takuwa, Y. Cyclin D1 expression mediated by phosphatidylinositol 3-kinase through mTor-p70(s6k)-independent signaling in growth factor-stimulated NIH 3T3 fibroblasts. Mol. Cell. Biol. 1999, 19, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Radeff-Huang, J.; Seasholtz, T.M.; Chang, J.W.; Smith, J.M.; Walsh, C.T.; Brown, J.H. Tumor necrosis factor-alpha-stimulated cell proliferation is mediated through sphingosine kinase-dependent Akt activation and cyclin D expression. J. Biol. Chem. 2007, 282, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Salles, I.I.; Tucker, A.E.; Voth, D.E.; Ballard, J.D. Toxin-induced resistance in Bacillus anthracis lethal toxin-treated macrophages. Proc. Natl. Acad. Sci. USA 2003, 100, 12426–12431. [Google Scholar] [CrossRef] [PubMed]
- Das, N.D.; Jung, K.H.; Chai, Y.G. The role of NF-kappaB and H3K27me3 demethylase, JMJD3, on the anthrax lethal toxin tolerance of Raw 264.7 cells. PLoS ONE 2010, 5, e9913. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Han, C.Y.; Reid, C.; Kim, S.O. HDAC8-mediated epigenetic reprogramming plays a key role in resistance to anthrax lethal toxin-induced pyroptosis in macrophages. J. Immunol. 2014, 193, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Park, S.; Han, C.Y.; Nguyen, M.L.; Kim, S.O. Cellular adaptation to anthrax lethal toxin-induced mitochondrial cholesterol enrichment, hyperpolarization, and reactive oxygen species generation through downregulating MLN64 in macrophages. Mol. Cell. Biol. 2012, 32, 4846–4860. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Mandal, T.; Balsubramanian, N.; Viaene, T.; Leedahl, T.; Sule, N.; Cook, G.; Srivastava, D.K. Histone deacetylase activators: N-acetylthioureas serve as highly potent and isozyme selective activators for human histone deacetylase-8 on a fluorescent substrate. Bioorg. Med. Chem. Lett. 2011, 21, 5920–5923. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Worby, C.A.; Dixon, J.E. Pten. Annu. Rev. Biochem. 2014, 83, 641–669. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Reid, C.; Meshkibaf, S.; Kim, S.O. Inhibition of IL-1β expression by anthrax lethal toxin is reversed by HDAC8 inhibition in murine macrophages. J. Biol. Chem. 2016, 291. [Google Scholar] [CrossRef]
- Pasini, D.; Malatesta, M.; Jung, H.R.; Walfridsson, J.; Willer, A.; Olsson, L.; Skotte, J.; Wutz, A.; Porse, B.; Jensen, O.N.; et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of polycomb group target genes. Nucleic. Acids. Res. 2010, 38, 4958–4969. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.; Salmon-Divon, M.; Dvinge, H.; Hynes-Allen, A.; Balasooriya, G.; Leaford, D.; Behrens, A.; Bertone, P.; Hendrich, B. Nurd-mediated deacetylation of H3K27 facilitates recruitment of polycomb repressive complex 2 to direct gene repression. EMBO J. 2012, 31, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Tie, F.; Banerjee, R.; Stratton, C.A.; Prasad-Sinha, J.; Stepanik, V.; Zlobin, A.; Diaz, M.O.; Scacheri, P.C.; Harte, P.J. Cbp-mediated acetylation of histone H3 lysine 27 antagonizes drosophila polycomb silencing. Development 2009, 136, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Xu, J.F.; Chang, R.M.; Fang, F.; Zuo, C.H.; Yang, L.Y. Jarid2 promotes invasion and metastasis of hepatocellular carcinoma by facilitating epithelial-mesenchymal transition through PTEN/Akt signaling. Oncotarget 2016, 7, 40266–40284. [Google Scholar] [CrossRef] [PubMed]
- Gall Troselj, K.; Novak Kujundzic, R.; Ugarkovic, D. Polycomb repressive complex's evolutionary conserved function: The role of EZH2 status and cellular background. Clin. Epigenetics 2016, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase JMJD3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in hox gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Risso, G.; Blaustein, M.; Pozzi, B.; Mammi, P.; Srebrow, A. Akt/PKB: One kinase, many modifications. Biochem. J. 2015, 468, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Hart, J.R.; Vogt, P.K. Phosphorylation of Akt: A mutational analysis. Oncotarget 2011, 2, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Fayard, E.; Xue, G.; Parcellier, A.; Bozulic, L.; Hemmings, B.A. Protein kinase b (Pkb/Akt), a key mediator of the pi3k signaling pathway. Curr. Top. Microbiol. Immunol. 2010, 346, 31–56. [Google Scholar] [PubMed]
- Mahajan, K.; Mahajan, N.P. Pi3k-independent AKT activation in cancers: A treasure trove for novel therapeutics. J. Cell. Physiol. 2012, 227, 3178–3184. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Dormond, O. PI3K and AKT: Unfaithful partners in cancer. Int. J. Mol. Sci. 2015, 16, 21138–21152. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Molina, A.; Efeyan, A.; Lopez-Guadamillas, E.; Munoz-Martin, M.; Gomez-Lopez, G.; Canamero, M.; Mulero, F.; Pastor, J.; Martinez, S.; Romanos, E.; et al. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab. 2012, 15, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Correia, N.C.; Girio, A.; Antunes, I.; Martins, L.R.; Barata, J.T. The multiple layers of non-genetic regulation of pten tumour suppressor activity. Eur. J. Cancer 2014, 50, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lei, C.; Fan, J.; Wang, J. Mir-18a promotes cell proliferation of esophageal squamous cell carcinoma cells by increasing cylin D1 via regulating PTEN-PI3K-ALT-mTor signaling axis. Biochem. Biophys. Res. Commun. 2016, 477, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Virolle, T.; Adamson, E.D.; Baron, V.; Birle, D.; Mercola, D.; Mustelin, T.; de Belle, I. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat. Cell Biol. 2001, 3, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Pass, I.; Coxon, P.; Downes, C.P.; Smith, S.A.; Macphee, C.H. Tumor suppressor and anti-inflammatory actions of ppargamma agonists are mediated via upregulation of pten. Curr. Biol. 2001, 11, 764–768. [Google Scholar] [CrossRef]
- Moorehead, R.A.; Hojilla, C.V.; De Belle, I.; Wood, G.A.; Fata, J.E.; Adamson, E.D.; Watson, K.L.; Edwards, D.R.; Khokha, R. Insulin-like growth factor-II regulates PTEN expression in the mammary gland. J. Biol. Chem. 2003, 278, 50422–50427. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; Zhang, L.; Gan, Y.; Wang, X.; Wang, J.; LeMaire, S.A.; Coselli, J.S.; Wang, X.L. Up-regulation of PTEN (phosphatase and tensin homolog deleted on chromosome ten) mediates p38 MAPK stress signal-induced inhibition of insulin signaling. A cross-talk between stress signaling and insulin signaling in resistin-treated human endothelial cells. J. Biol. Chem. 2006, 281, 7727–7736. [Google Scholar] [PubMed]
- Jiang, X.; Li, H. Overexpression of LRG1 regulates PTEN via mapk/mek signaling pathway in esophageal squamous cell carcinoma. Exp. Ther. Med. 2016, 12, 2045–2052. [Google Scholar] [PubMed]
- Wu, Q.X.; Yuan, S.X.; Ren, C.M.; Yu, Y.; Sun, W.J.; He, B.C.; Wu, K. Oridonin upregulates PTEN through activating p38 MAPK and inhibits proliferation in human colon cancer cells. Oncol. Rep. 2016, 35, 3341–3348. [Google Scholar] [PubMed]
- Mak, L.H.; Vilar, R.; Woscholski, R. Characterisation of the pten inhibitor Vo-Ohpic. J. Chem. Biol. 2010, 3, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. Hdac8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Targeting class i histone deacetylases in cancer therapy. Expert Opin. Ther. Targets 2013, 17, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Gligoris, T.G.; Scheinost, J.C.; Burmann, F.; Petela, N.; Chan, K.L.; Uluocak, P.; Beckouet, F.; Gruber, S.; Nasmyth, K.; Lowe, J. Closing the cohesin ring: Structure and function of its SMC3-kleisin interface. Science 2014, 346, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Mannini, L.; Lamaze, F.C.; Cucco, F.; Amato, C.; Quarantotti, V.; Rizzo, I.M.; Krantz, I.D.; Bilodeau, S.; Musio, A. Mutant cohesin affects rna polymerase II regulation in Cornelia de Lange syndrome. Sci. Rep. 2015, 5, 16803. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, T.; Antony, J.; Braithwaite, A.W.; Horsfield, J.A. HDAC8 inhibition blocks SMC3 deacetylation and delays cell cycle progression without affecting cohesin-dependent transcription in MCF7 cancer cells. J. Biol. Chem. 2016, 291, 12761–12770. [Google Scholar] [CrossRef] [PubMed]
- Fazio, G.; Gaston-Massuet, C.; Bettini, L.R.; Graziola, F.; Scagliotti, V.; Cereda, A.; Ferrari, L.; Mazzola, M.; Cazzaniga, G.; Giordano, A.; et al. Cyclin D1 down-regulation and increased apoptosis are common features of cohesinopathies. J. Cell. Physiol. 2016, 231, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, J.; Han, Q.; Sun, Z.; Wang, J.; Wang, S.; Zhao, R.C. Enhancer of zeste homolog 2 is overexpressed and contributes to epigenetic inactivation of p21 and phosphatase and tensin homolog in B-cell acute lymphoblastic leukemia. Exp. Biol. Med. (Maywood) 2012, 237, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Cole, M.D. MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the Ezh2 methyltransferase. Cancer Res. 2013, 73, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.C.; Yang, Z.L. Overexpression of EZH2 and loss of expression of PTEN is associated with invasion, metastasis, and poor progression of gallbladder adenocarcinoma. Pathol. Res. Pract. 2011, 207, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Udaka, K.; Yokoyama, A. Imatinib causes epigenetic alterations of PTEN gene via upregulation of DNA methyltransferases and polycomb group proteins. Blood Cancer J. 2011, 1, e48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, F.; Liu, Y.; Lei, X.; Li, H.; Ji, G.; Yuan, Z.; Jiao, J. Ezh2 regulates adult hippocampal neurogenesis and memory. J. Neurosci. 2014, 34, 5184–5199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zang, X.; Ponnusamy, M.; Masucci, M.V.; Tolbert, E.; Gong, R.; Zhao, T.C.; Liu, N.; Bayliss, G.; Dworkin, L.D.; et al. Enhancer of Zeste Homolog 2 inhibition attenuates renal fibrosis by maintaining Smad7 and phosphatase and tensin homolog expression. J. Am. Soc. Nephrol. 2016, 27, 2092–2108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ricciardi, R.P. E1A is the component of the MHC class І enhancer complex that mediates HDAC chromatin repression in adenovirus-12 tumorigenic cells. Virology. 2006, 352, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Durst, K.L.; Lutterbach, B.; Kummalue, T.; Friedman, A.D.; Hiebert, S.W. The inv(16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol. Cell. Biol. 2003, 23, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Singh, S.; Hua, W.K.; Cai, Q.; Chao, S.W.; Li, L.; Liu, H.; Ho, Y.; McDonald, T.; Lin, A.; et al. HDAC8 inhibition specifically targets inv(16) acute myeloid leukemic stem cells by restoring p53 acetylation. Cell Stem Cell 2015, 17, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Canettieri, G.; Morantte, I.; Guzman, E.; Asahara, H.; Herzig, S.; Anderson, S.D.; Yates, J.R., 3rd; Montminy, M. Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat. Struct. Biol. 2003, 10, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Karolczak-Bayatti, M.; Sweeney, M.; Cheng, J.; Edey, L.; Robson, S.C.; Ulrich, S.M.; Treumann, A.; Taggart, M.J.; Europe-Finner, G.N. Acetylation of heat shock protein 20 (hsp20) regulates human myometrial activity. J. Biol. Chem. 2011, 286, 34346–34355. [Google Scholar] [CrossRef] [PubMed]
- Waltregny, D.; Glenisson, W.; Tran, S.L.; North, B.J.; Verdin, E.; Colige, A.; Castronovo, V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005, 19, 966–968. [Google Scholar] [PubMed]
- Lee, H.; Sengupta, N.; Villagra, A.; Rezai-Zadeh, N.; Seto, E. Histone deacetylase 8 safeguards the human ever-shorter telomeres 1B (HEST1b) protein from ubiquitin-mediated degradation. Mol. Cell. Biol. 2006, 26, 5259–5269. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.J.; Tremblay, A.M.; Deblois, G.; Sylvain-Drolet, G.; Giguere, V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol. Endocrinol. 2010, 24, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.E.; Udeshi, N.D.; Wolfson, N.A.; Pitcairn, C.A.; Sullivan, E.D.; Jaffe, J.D.; Svinkina, T.; Natoli, T.; Lu, X.; Paulk, J.; et al. An unbiased approach to identify endogenous substrates of "Histone" Deacetylase 8. ACS Chem. Biol. 2014, 9, 2210–2216. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Bill, K.L.; Bid, H.K.; Braggio, D.; Constantino, D.; Prudner, B.; Zewdu, A.; Batte, K.; Lev, D.; Pollock, R.E. Hdac8, a potential therapeutic target for the treatment of malignant peripheral nerve sheath tumors (mpnst). PLoS ONE 2015, 10, e0133302. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Tournier, J.N.; Quesnel-Hellmann, A.; Cleret, A.; Vidal, D.R. Contribution of toxins to the pathogenesis of inhalational anthrax. Cell Microbiol. 2007, 9, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Cote, C.K.; Welkos, S.L. Anthrax toxins in context of Bacillus anthracis spores and spore germination. Toxins (Basel) 2015, 7, 3167–3178. [Google Scholar] [CrossRef] [PubMed]
- Banks, D.J.; Barnajian, M.; Maldonado-Arocho, F.J.; Sanchez, A.M.; Bradley, K.A. Anthrax toxin receptor 2 mediates Bacillus anthracis killing of macrophages following spore challenge. Cell Microbiol. 2005, 7, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Guidi-Rontani, C.; Levy, M.; Ohayon, H.; Mock, M. Fate of germinated Bacillus anthracis spores in primary murine macrophages. Mol. Microbiol. 2001, 42, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Cote, C.K.; Van Rooijen, N.; Welkos, S.L. Roles of macrophages and neutrophils in the early host response to Bacillus anthracis spores in a mouse model of infection. Infect. Immun. 2006, 74, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Bachran, C.; Leppla, S.H. Tumor targeting and drug delivery by anthrax toxin. Toxins (Basel) 2016, 8, 197. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, S.-D.; Cho, W.; Kim, S.O. HDAC8 Prevents Anthrax Lethal Toxin-induced Cell Cycle Arrest through Silencing PTEN in Human Monocytic THP-1 Cells. Toxins 2017, 9, 162. https://doi.org/10.3390/toxins9050162
Ha S-D, Cho W, Kim SO. HDAC8 Prevents Anthrax Lethal Toxin-induced Cell Cycle Arrest through Silencing PTEN in Human Monocytic THP-1 Cells. Toxins. 2017; 9(5):162. https://doi.org/10.3390/toxins9050162
Chicago/Turabian StyleHa, Soon-Duck, Woohyun Cho, and Sung Ouk Kim. 2017. "HDAC8 Prevents Anthrax Lethal Toxin-induced Cell Cycle Arrest through Silencing PTEN in Human Monocytic THP-1 Cells" Toxins 9, no. 5: 162. https://doi.org/10.3390/toxins9050162
APA StyleHa, S.-D., Cho, W., & Kim, S. O. (2017). HDAC8 Prevents Anthrax Lethal Toxin-induced Cell Cycle Arrest through Silencing PTEN in Human Monocytic THP-1 Cells. Toxins, 9(5), 162. https://doi.org/10.3390/toxins9050162