
RnlB Antitoxin of the Escherichia coli RnlA-RnlB Toxin–Antitoxin Module Requires RNase HI for Inhibition of RnlA Toxin Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
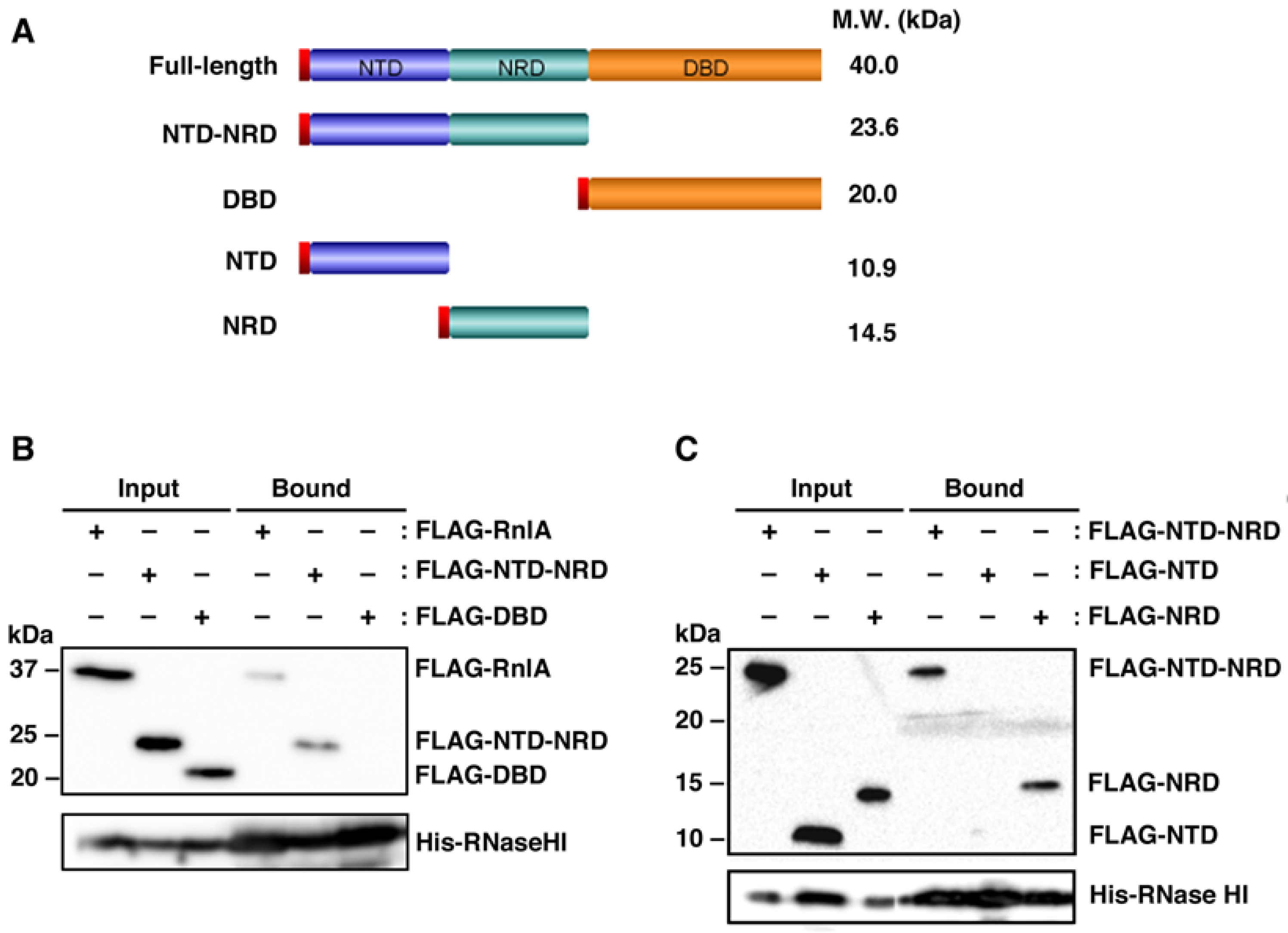
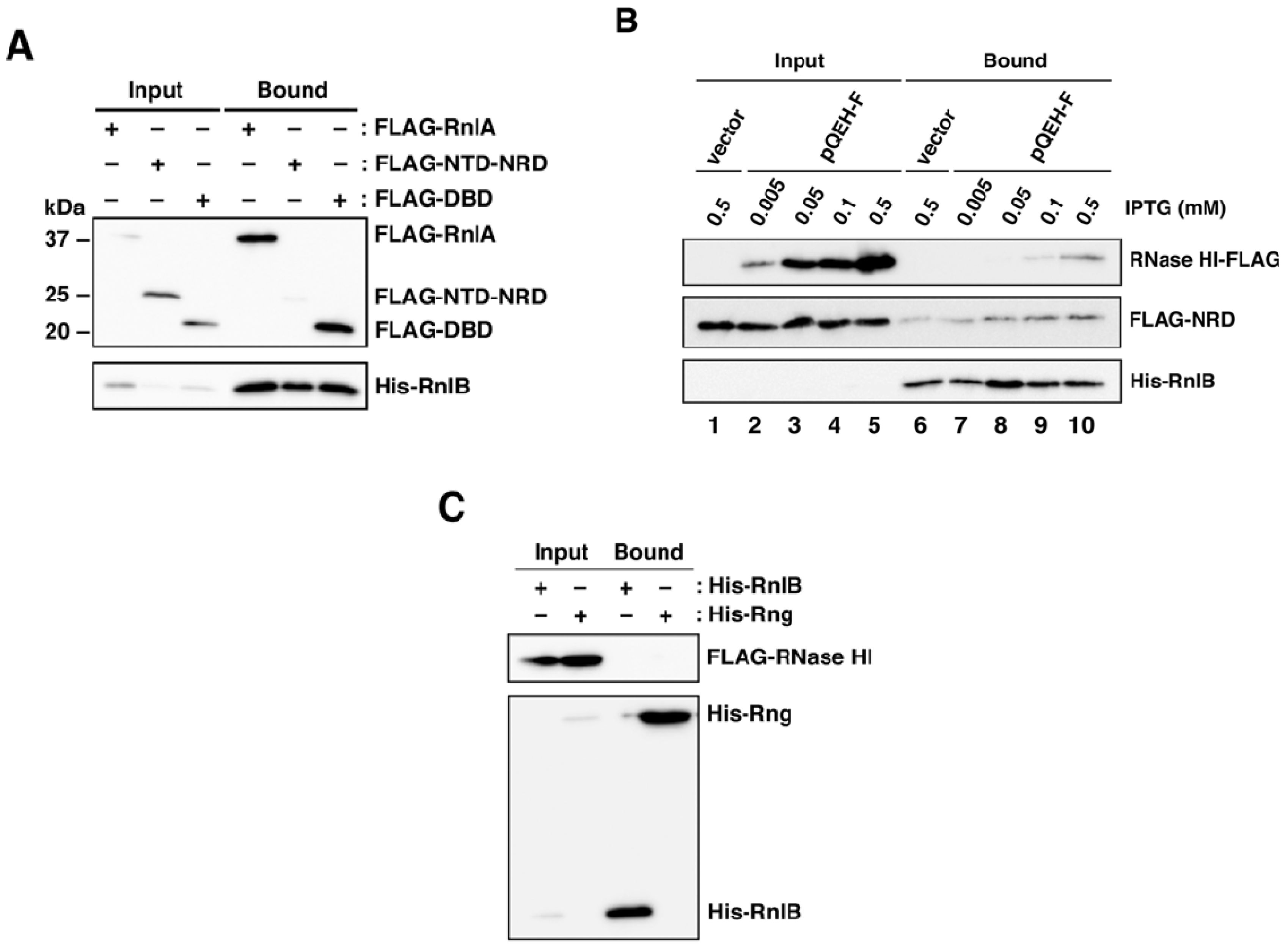
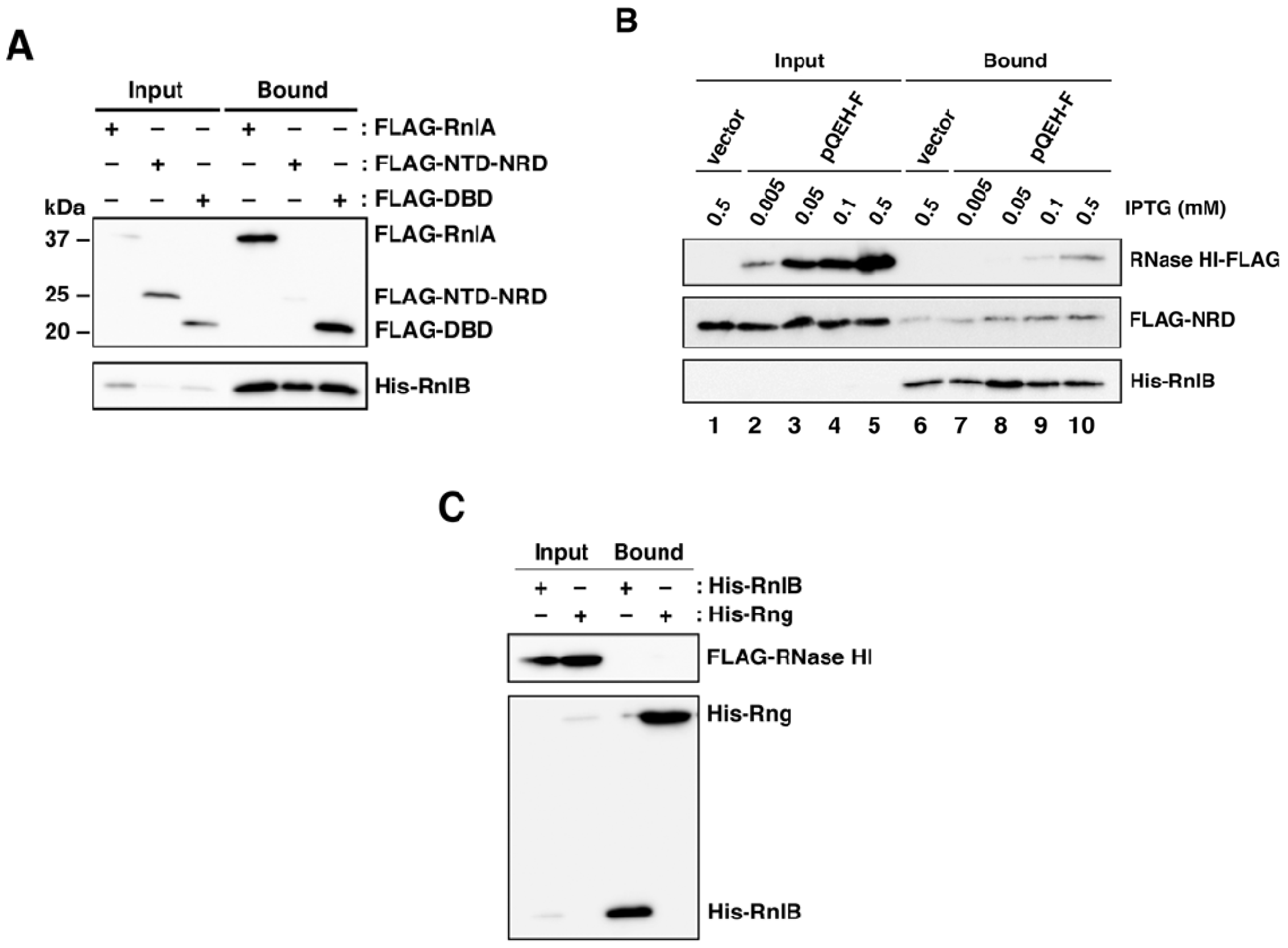
2.1. RNase HI Interacts with NRD of RnlA
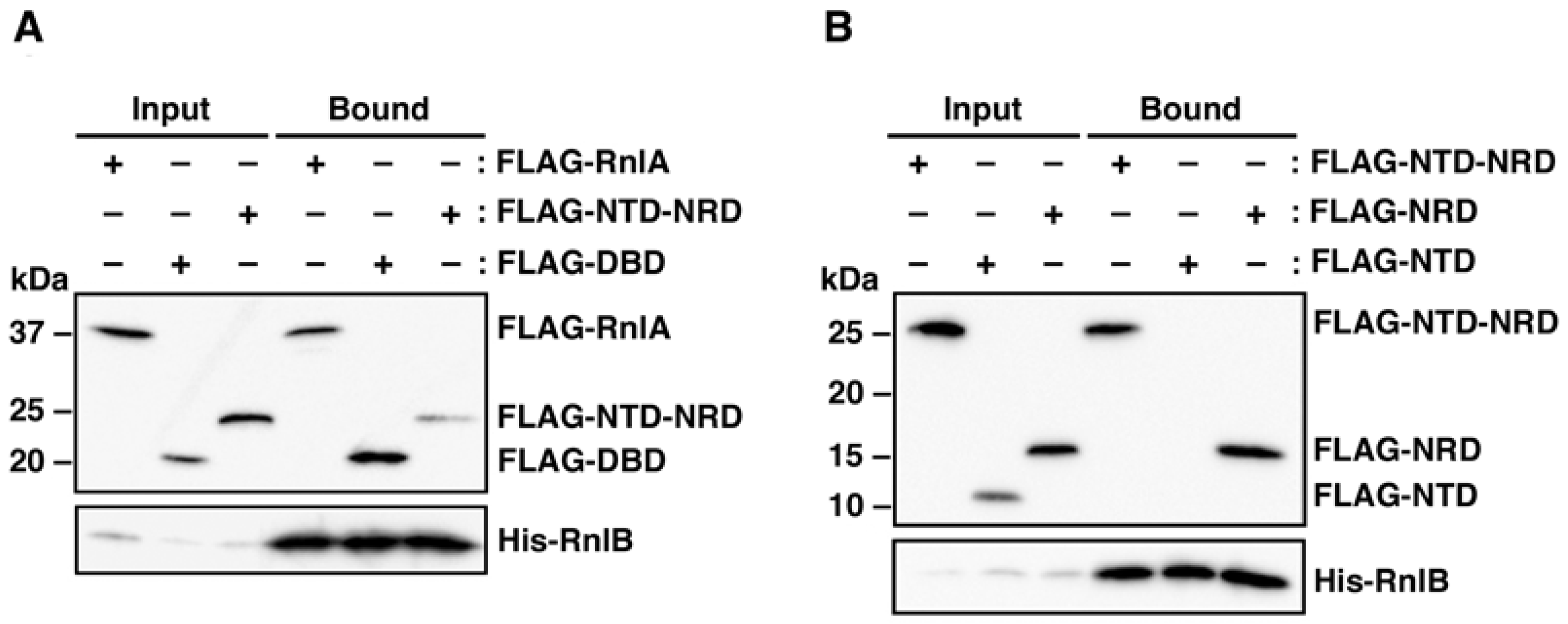
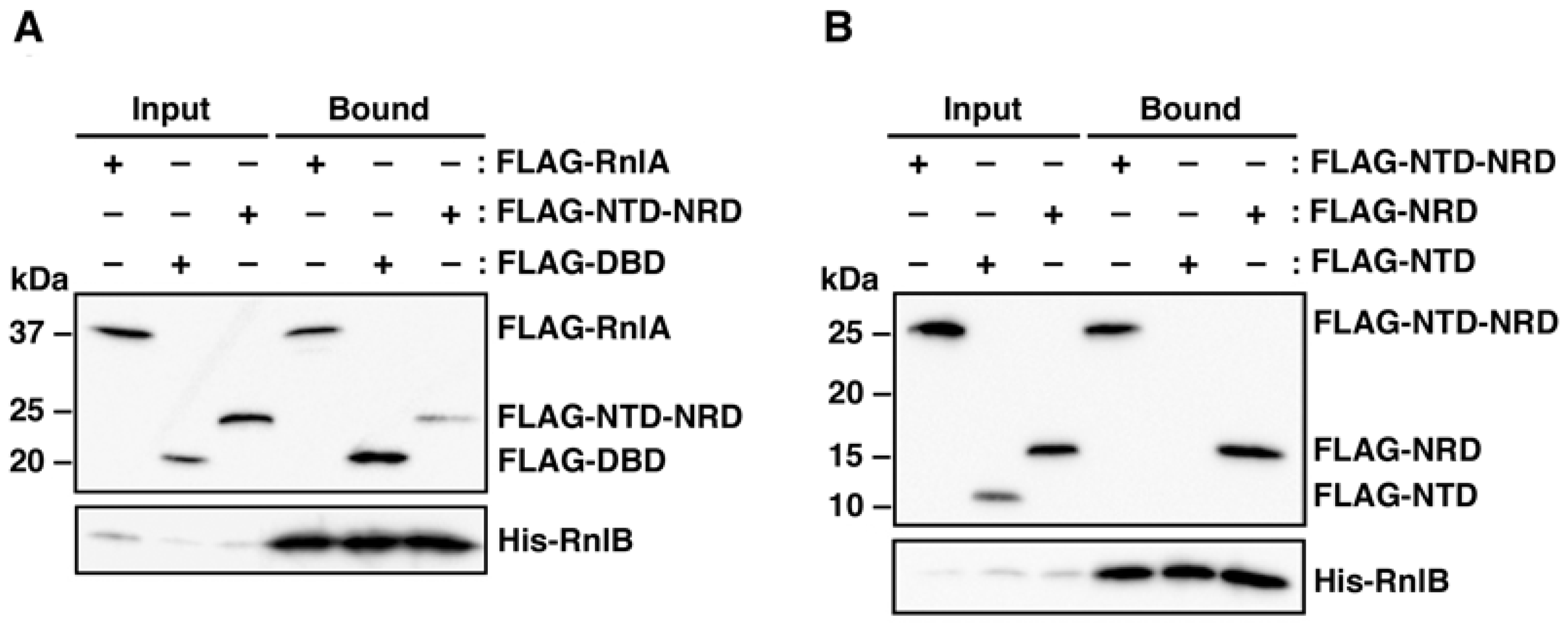
2.2. RnlB Interacts with NRD and DBD of RnlA
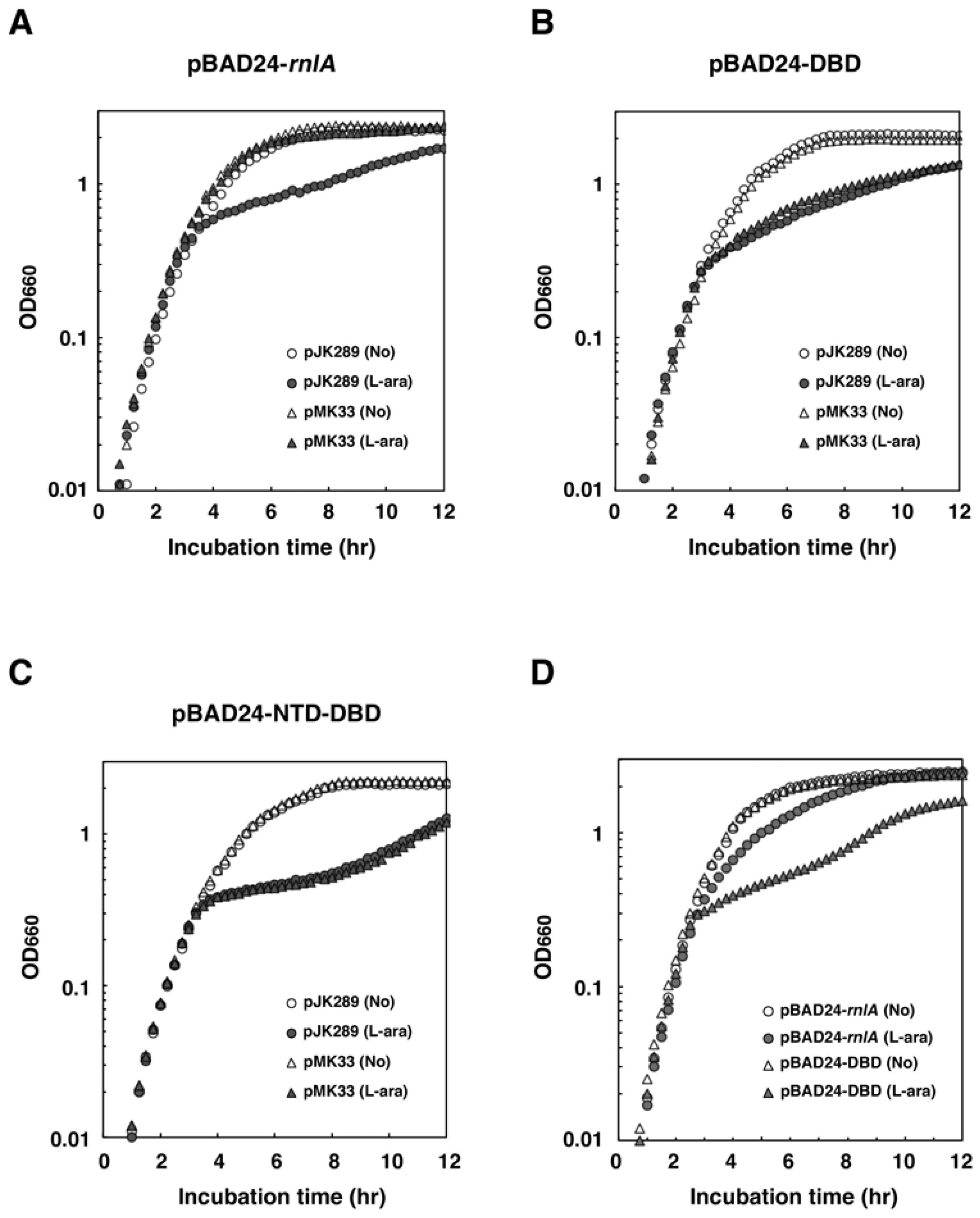
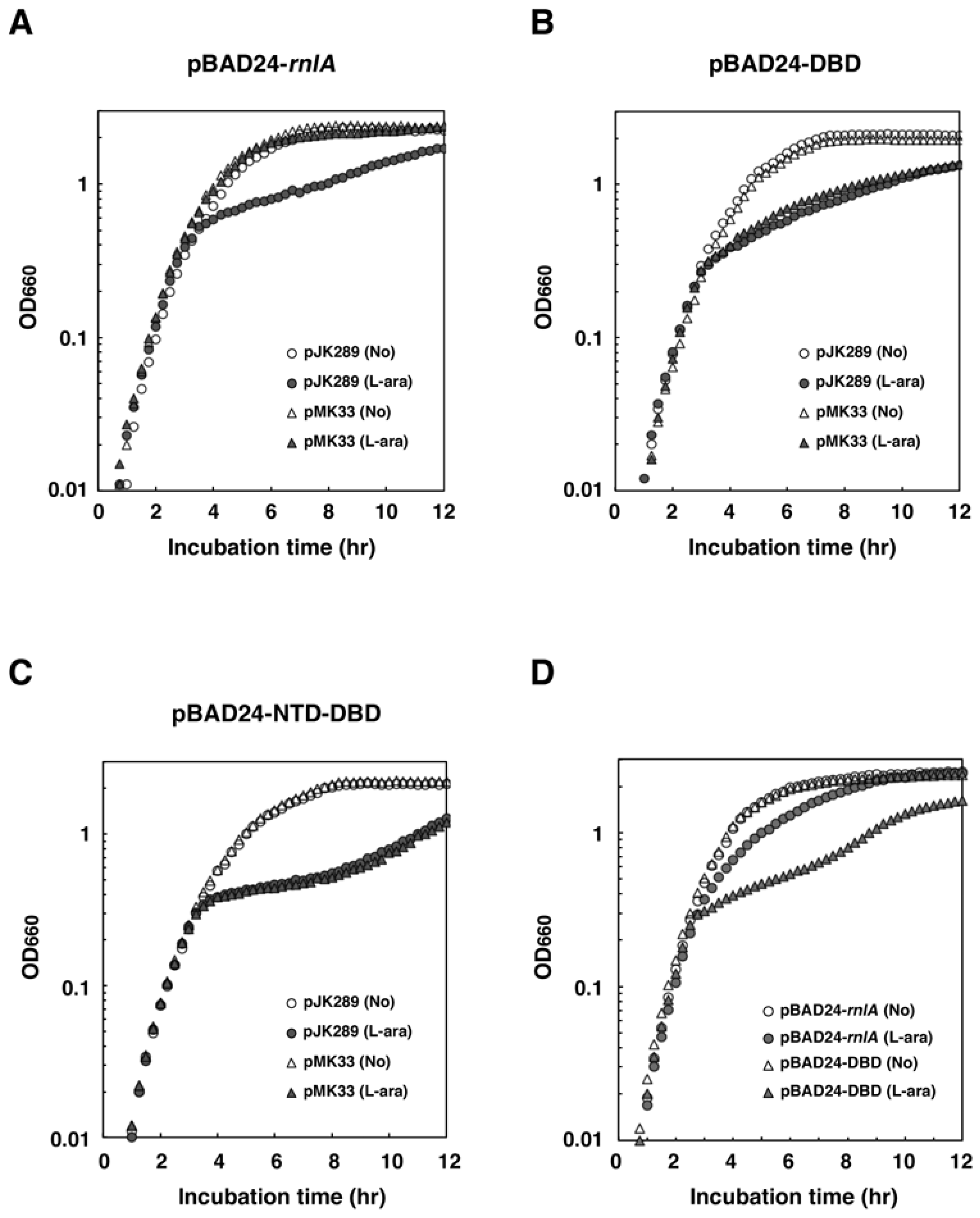
2.3. NRD is Required for the Inhibition of RnlA Toxicity by RnlB
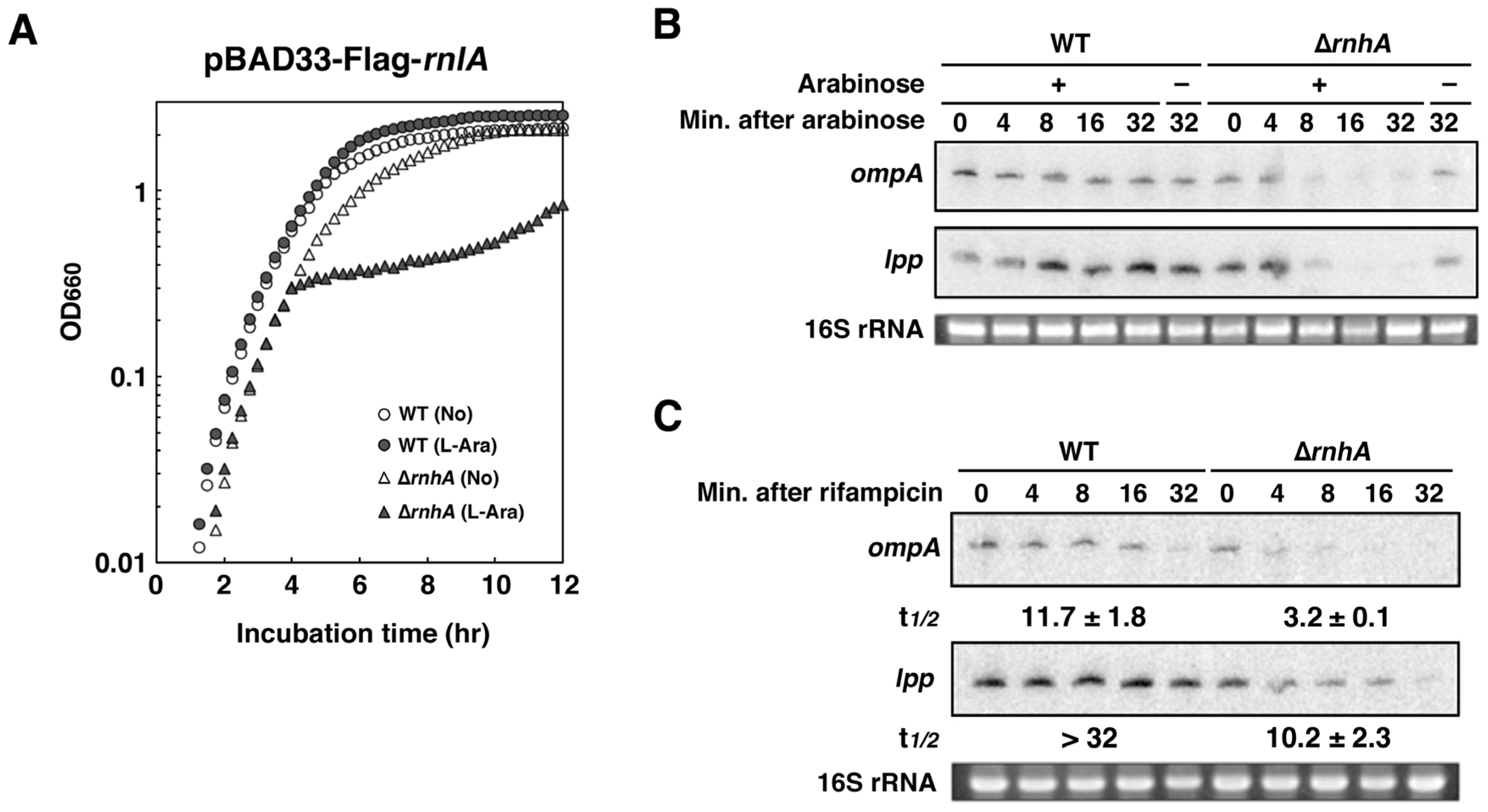
2.4. RNase HI is Required for the Inhibition of RnlA Activity by RnlB
2.5. The Recruitment of RnlB to NRD of RnlA Depends on RNase HI
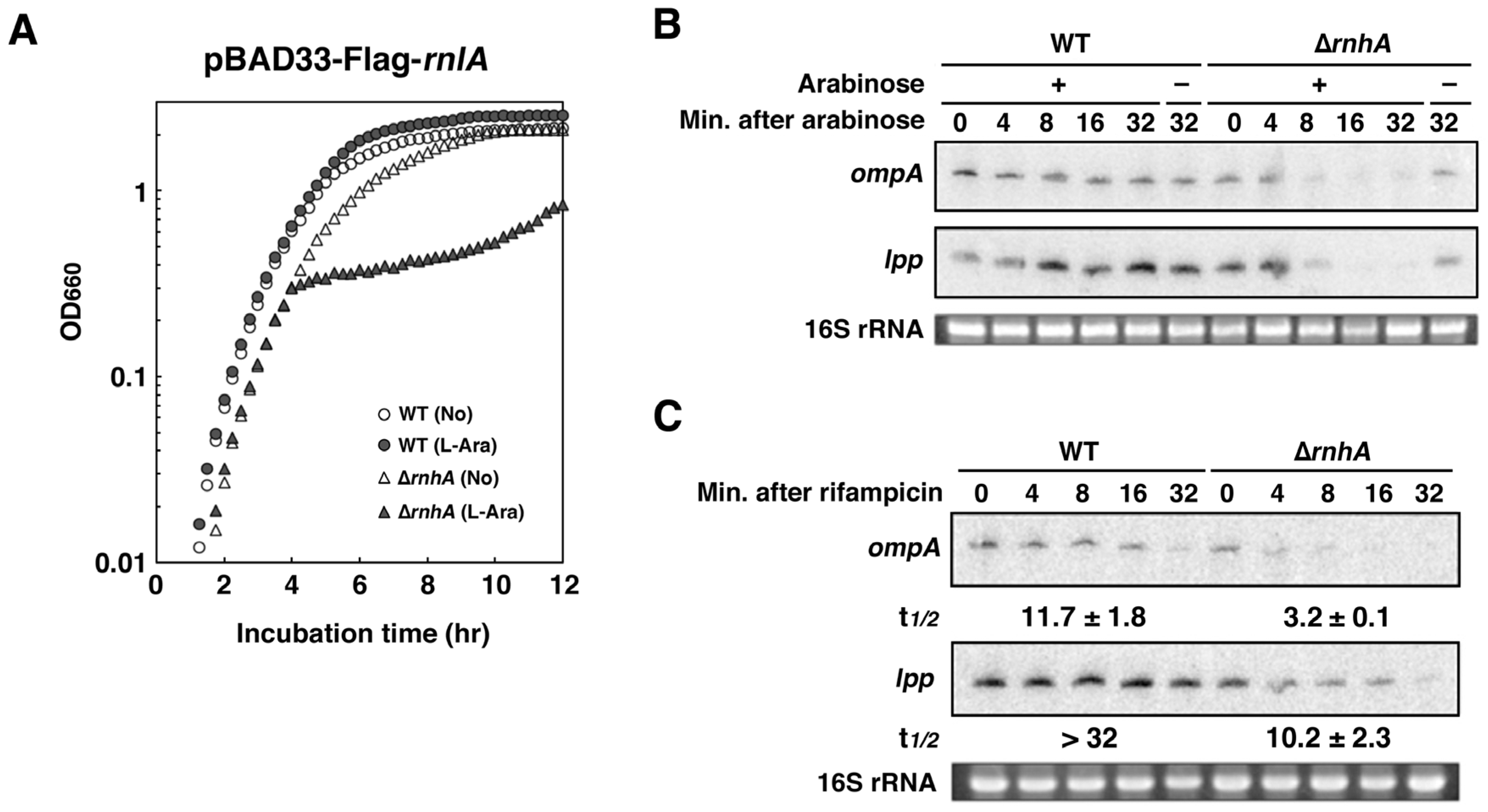
2.6. Depletion of RNase HI Leads to the Inactivation of RnlA in the Absence of RnlB
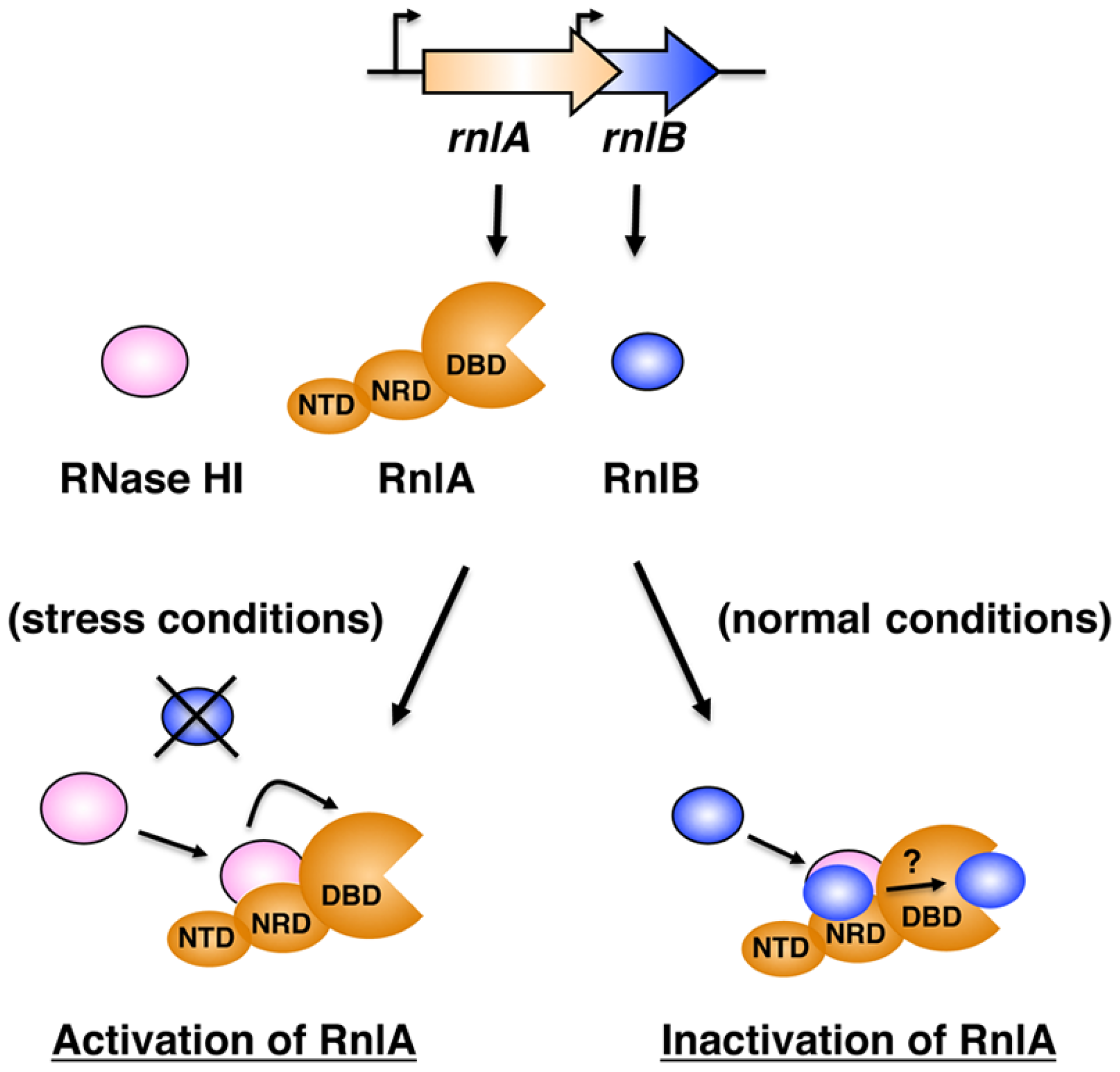
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Construction of Plasmids
4.3. Pull-Down and Western Blotting Analyses
4.4. RNA Purification and Northern Blotting Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gerdes, K.; Christensen, S.K.; Løbner-Olesen, A. Prokaryotic toxin–antitoxin stress response loci. Nat. Rev. Microbiol. 2005, 3, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Pogliano, J.; Helinski, D.R.; Konieczny, I. ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol. Microbiol. 2002, 44, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Inouye, M. mRNA interferases, sequence-specific endoribonucleases from the toxin–antitoxin systems. Prog. Mol. Biol. Transl. Sci. 2009, 85, 467–500. [Google Scholar] [PubMed]
- Zhang, Y.; Inouye, M. RatA (YfjG), an Escherichia coli toxin, inhibits 70S ribosome association to block translation initiation. Mol. Microbiol. 2011, 79, 1418–1429. [Google Scholar] [CrossRef] [PubMed]
- Mutschler, H.; Gebhardt, M.; Shoeman, R.L.; Meinhart, A. A novel mechanism of programmed cell death in bacteria by toxin–antitoxin systems corrupts peptidoglycan synthesis. PLoS Biol. 2011, 9, e1001033. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Awano, N.; Inouye, M. YeeV is an Escherichia coli toxin that inhibits cell division by targeting the cytoskeleton proteins, FtsZ and MreB. Mol. Microbiol. 2011, 79, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Buts, L.; Lah, J.; Dao-Thi, M.-H.; Wyns, L.; Loris, R. Toxin–antitoxin modules as bacterial metabolic stress managers. Trends Biochem. Sci. 2005, 30, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.F.; Bertram, R. Toxin–antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 2013, 340, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Yarmolinsky, M.B. Programmed cell death in bacterial populations. Science 1995, 267, 836–837. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Hiraga, S. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc. Natl. Acad. Sci. USA 1983, 80, 4784–4788. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Bedzyk, L.A.; Thomas, S.M.; Ye, R.W.; Wood, T.K. Gene expression in Escherichia coli biofilms. Appl. Microbiol. Biotechnol. 2004, 64, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Wang, X.; Ma, Q.; Zhang, X.S.; Wood, T.K. Toxin–antitoxin systems in Escherichia coli influence biofilm formation through YjgK (TabA) and fimbriae. J. Bacteriol. 2009, 191, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Shakespeare, L.J.; Jørgensen, M.G.; Gerdes, K. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. USA 2011, 108, 13206–13211. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Castro-Camargo, M.; Gerdes, K. (p)ppGpp controls bacterial persistence by stochastic induction of toxin–antitoxin activity. Cell 2013, 154, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Amato, S.M.; Orman, M.A.; Brynildsen, M.P. Metabolic control of persister formation in Escherichia coli. Mol. Cell 2013, 50, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Germain, E.; Castro-Roa, D.; Zenkin, N.; Gerdes, K. Molecular mechanism of bacterial persistence by HipA. Mol. Cell 2013, 52, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, N.; Knapen, W.J.; Kint, C.I.; Liebens, V.; Bergh, B.V.; Dewachter, L.; Michiels, J.E.; Fu, Q.; David, C.C.; Fierro, A.C.; et al. Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol. Cell 2015, 59, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kim, Y.; Hong, S.H.; Ma, Q.; Brown, B.L.; Pu, M.; Tarone, A.M.; Benedik, M.J.; Peti, W.; Page, R.; et al. Antitoxin MqsA helps mediate the bacterial general stress response. Nat. Chem. Biol. 2011, 7, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Benedik, M.J.; Wood, T.K. Antitoxin DinJ influences the general stress response through transcript stabilizer CspE. Environ. Microbiol. 2012, 14, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Pecota, D.C.; Wood, T.K. Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J. Bacteriol. 1996, 178, 2044–2050. [Google Scholar] [CrossRef] [PubMed]
- Hazan, R.; Engelberg-Kulka, H. Escherichia coli mazEF-mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol. Genet. Genom. 2004, 272, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Fineran, P.C.; Blower, T.R.; Foulds, I.J.; Humphreys, D.P.; Lilley, K.S.; Salmond, G.P.C. The phage abortive infection system, ToxIN, functions as a protein–RNA toxin–antitoxin pair. Proc. Natl. Acad. Sci. USA 2009, 106, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Dy, R.L.; Przybilski, R.; Semeijn, K.; Salmond, G.P.; Fineran, P.C. A widespread bacteriophage abortive infection system functions through a Type IV toxin–antitoxin mechanism. Nucleic Acids Res. 2014, 42, 4590–4605. [Google Scholar] [CrossRef] [PubMed]
- Alawneh, A.M.; Qi, D.; Yonesaki, T.; Otsuka, Y. An ADP-ribosyltransferase Alt of bacteriophage T4 negatively regulates the Escherichia coli MazF toxin of a toxin–antitoxin module. Mol. Microbiol. 2016, 99, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Otsuka, Y.; Lemire, S.; Yonesaki, T. Escherichia coli rnlA and rnlB compose a novel toxin–antitoxin systems. Genetics 2011, 187, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, Y.; Yonesaki, T. Dmd of bacteriophage T4 functions as an antitoxin against Escherichia coli LsoA and RnlA toxins. Mol. Microbiol. 2012, 83, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E.; White, T.; Kashlev, M.; Uzan, M.; Mckinney, J.; Guttman, B. Effects on host genome structure and expression. In Molecular Biology of Bacteriophage T4; Karam, J.D., Drake, J.W., Kreuzer, K.N., Mosig, G., Hall, D.H., Eiserling, F.A., Black, L.W., Spicer, E.K., Kutter, E., Carlson, K., et al., Eds.; American Society for Microbiology Press: Washington, DC, USA, 1994; pp. 357–368. [Google Scholar]
- Kai, T.; Selick, H.E.; Yonesaki, T. Destabilization of bacteriophage T4 mRNAs by a mutation of gene 61.5. Genetics 1996, 144, 7–14. [Google Scholar] [PubMed]
- Otsuka, Y.; Yonesaki, T. A novel endoribonuclease, RNase LS, in Escherichia coli. Genetics 2005, 169, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Gao, Z.Q.; Otsuka, Y.; Naka, K.; Yonesaki, T.; Zhang, H.; Dong, Y.H. Structure-function studies of Escherichia coli RnlA reveal a novel toxin structure involved in bacteriophage resistance. Mol. Microbiol. 2013, 90, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Koga, M.; Yonesaki, T.; Otsuka, Y. RNase HI stimulates the activity of RnlA toxin in Escherichia coli. Mol. Microbiol. 2014, 91, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.I.; Riggs, A.D.; Gill, G.N. Ribonuclease H (Hybrid) in Escherichia coli. Identification and characterization. J. Biol. Chem. 1973, 248, 2621–2624. [Google Scholar] [PubMed]
- Itoh, T.; Tomizawa, J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proc. Natl. Acad. Sci. USA 1980, 77, 2450–2454. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Otsuka, Y.; Gao, Z.Q.; Wei, Y.; Chen, Z.; Masuda, M.; Yonesaki, T.; Zhang, H.; Dong, Y.H. Structural insights into the inhibition mechanism of bacterial toxin LsoA by bacteriophage antitoxin Dmd. Mol. Microbiol. 2016, 101, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Kato, L.; Ikeda, H. Construction of mini-F plasmid vectors for plasmid shuffling in Escherichia coli. Gene 1996, 170, 141–142. [Google Scholar] [CrossRef]
- Kogoma, T. Absence of RNase H allows replication of pBR322 in Escherichia coli mutants lacking DNA polymerase I. Proc. Natl. Acad. Sci. USA 1984, 81, 7845–7849. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Inouye, M. Regulation of growth and death in Escherichia coli by toxin–antitoxin systems. Nat. Rev. Microbiol. 2011, 9, 779–790. [Google Scholar] [CrossRef]
- Bordes, P.; Cirinesi, A.M.; Ummels, R.; Sala, A.; Sakr, S.; Bitter, W.; Genevaux, P. SecB-like chaperone controls a toxin–antitoxin stress-responsive system in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2011, 108, 8438–8443. [Google Scholar] [CrossRef] [PubMed]
- Sala, A.; Calderon, V.; Bordes, P.; Genevaux, P. TAC from Mycobacterium tuberculosis: A paradigm for stress-responsive toxin–antitoxin systems controlled by Sec-like chaperones. Cell Stress Chaperones 2013, 18, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.S.; Rawlings, D.E. The poison-antidote stability system of the broad-host-range Thiobacillus ferrooxidans plasmid pTF-FC2. Mol. Microbiol. 1997, 26, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Zielenkiewicz, U.; Ceglowski, P. The toxin-antitoxin system of the streptococcal plasmid pSM19035. J. Bacteriol. 2005, 187, 6094–6105. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, Y.; Koga, M.; Iwamoto, A.; Yonesaki, T. A role of RnlA in the RNase LS activity from Escherichia coli. Genes Genet. Syst. 2007, 82, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the Arabinose PBAD Promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Yonesaki, T. Phage-induced change in the stability of mRNAs. Virology 2004, 329, 134–141. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naka, K.; Qi, D.; Yonesaki, T.; Otsuka, Y. RnlB Antitoxin of the Escherichia coli RnlA-RnlB Toxin–Antitoxin Module Requires RNase HI for Inhibition of RnlA Toxin Activity. Toxins 2017, 9, 29. https://doi.org/10.3390/toxins9010029
Naka K, Qi D, Yonesaki T, Otsuka Y. RnlB Antitoxin of the Escherichia coli RnlA-RnlB Toxin–Antitoxin Module Requires RNase HI for Inhibition of RnlA Toxin Activity. Toxins. 2017; 9(1):29. https://doi.org/10.3390/toxins9010029
Chicago/Turabian StyleNaka, Kenta, Dan Qi, Tetsuro Yonesaki, and Yuichi Otsuka. 2017. "RnlB Antitoxin of the Escherichia coli RnlA-RnlB Toxin–Antitoxin Module Requires RNase HI for Inhibition of RnlA Toxin Activity" Toxins 9, no. 1: 29. https://doi.org/10.3390/toxins9010029
APA StyleNaka, K., Qi, D., Yonesaki, T., & Otsuka, Y. (2017). RnlB Antitoxin of the Escherichia coli RnlA-RnlB Toxin–Antitoxin Module Requires RNase HI for Inhibition of RnlA Toxin Activity. Toxins, 9(1), 29. https://doi.org/10.3390/toxins9010029