1. Introduction
Nine voltage-gated sodium channel subtypes have been described to date (Na
V1.1–Na
V1.9), several of which are implicated as causative contributors to pain. Of particular interest is Na
V1.7, as loss-of-function mutations in humans lead to congenital insensitivity to pain, a rare condition that results in the inability to sense pain [
1,
2]. This makes pharmacological inhibition of Na
V1.7 an exciting therapeutic strategy for the treatment of a wide range of pain types, including inherited erythromelalgia and paroxysmal extreme pain disorder, two conditions whose pathophysiology arises from Na
V1.7 gain-of-function mutations. Accordingly, the development of Na
V1.7 inhibitors is being actively pursued by the pharmaceutical industry [
3]. Na
V1.7 selectivity is key to developing more effective analgesics as activity at major off-targets, including the skeletal muscle isoform Na
V1.4, the cardiac isoform Na
V1.5, as well as neuronal isoforms Na
V1.1, Na
V1.2 and Na
V1.6, is likely to impact the therapeutic window and cause dose-limiting adverse effects [
4,
5,
6]. However, achieving sufficient selectivity for Na
V1.7 over the other Na
V isoforms has proven challenging due to the high sequence homology within the Na
V family [
7]. Despite this challenge, several small molecules with reported Na
V1.7 selectivity are currently undergoing clinical development, including PF-05089771 (Pfizer) and CNV1014802 (Convergence Pharmaceuticals) [
8]. In addition to small molecules, venom-derived peptides are proving to be an alternate source for Na
V1.7 inhibitors, with several spider peptides, including GpTx-1, Huwentoxin-IV and ProTx-II, reported to have some Na
V1.7 selectivity [
9,
10,
11]. Evaluating the analgesic efficacy of this increasing number of Na
V1.7-selective compounds in relevant pre-clinical models of pain is a crucial translational step to clinical success. However, the pathophysiological basis of most commonly used animal models is multi-factorial, making selection of the best disease model for rapid efficacy profiling of Na
V1.7 inhibitors challenging. We thus sought to characterize a novel animal model of Na
V1.7-mediated pain that is suitable for the rapid
in vivo profiling of the analgesic efficacy of Na
V1.7 inhibitors. As gain-of-function mutations of Na
V1.7 in humans are associated with a range of painful syndromes [
12,
13], we hypothesized that intraplantar administration of the Na
V1.7 activator OD1 could be used as a pharmacological tool to establish a Na
V1.7-mediated mouse model of pain.
OD1 is a scorpion toxin isolated from the venom of the Iranian yellow scorpion (
Odontobuthus doriae) that potently enhances the activity of Na
V1.7 by inhibiting channel fast inactivation and increasing peak current with an EC
50 between 4 and 8 nM [
14,
15]. OD1 also has activity at Na
V1.4 (EC
50 = 10 nM) and Na
V1.6 (EC
50 = 30–47 nM); however, it is 1000-fold selective (EC
50 > 1 μM) over Na
V1.1, Na
V1.2, Na
V1.3, Na
V1.5 and Na
V1.8 [
14,
15]. Consistent with its
in vitro pharmacological activity, intraplantar administration of OD1 elicited pain behaviors, including licking and flinching of the hind paw, and we have previously used this model to assess the analgesic effects of ProTx-III [
16]. The aim of this study was to characterize the OD1 mouse model of pain and to validate the use of this model by testing the efficacy of several reported selective Na
V1.7 inhibitors, including the spider peptide GpTx-1, PF-04856264 (as the structure of clinical candidate PF-05089771 is not publicly available) [
17] and the clinical candidate CNV1014802 (raxatrigine). As the full pharmacological activity of these inhibitors is not reported, we determined their selectivity at Na
V1.1–Na
V1.8 and the mode of action at Na
V1.7 using functional assays.
3. Discussion
Choosing a relevant animal pain model to evaluate the analgesic efficacy of NaV1.7-selective inhibitors is an essential step in the pre-clinical development of these compounds. We therefore developed a novel mouse model of NaV1.7-mediated pain based on intraplantar injection of the scorpion toxin OD1 that allows rapid in vivo characterization of the analgesic efficacy of NaV1.7 inhibitors.
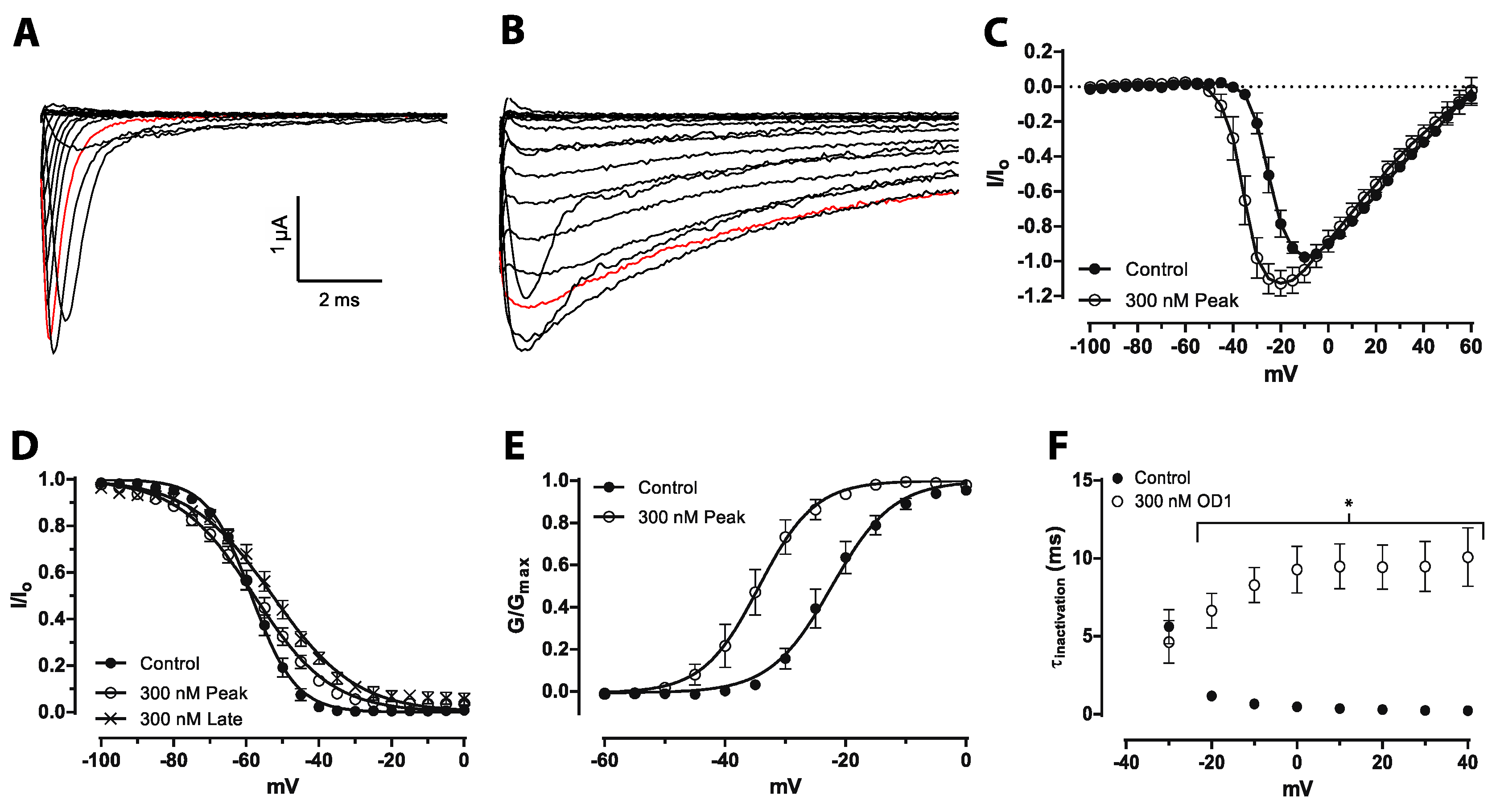
OD1 was initially classed as a α-scorpion toxin that increases peak current by delaying fast inactivation, but has little effect on the voltage dependence of activation at Na
V1.7 expressed in oocytes at 50 nM. [
14,
22]. Similarly, a small, albeit statistically-significant shift in the voltage dependence of activation was recently reported at Na
V1.7 expressed in mammalian cells [
23]. Our results confirm that this shift in the voltage dependence of activation is further enhanced at higher concentrations. Thus, OD1 has mixed α/β-scorpion toxin effects at Na
V1.7, not only increasing peak current and delaying inactivation, but also shifting the voltage dependence of activation to more hyperpolarized potentials. Thus, the mixed functional activity of OD1, which was also previously reported for Na
V1.4 and Na
V1.6 [
15], appears to be concentration dependent, suggesting that OD1 preferentially interacts with the α-scorpion toxin-binding site with additional interaction with the β-scorpion toxin-binding site at higher concentrations. Both pharmacological effects are consistent with the ability of OD1 to induce spontaneous action potential firing in sensory neurons. While pure α-toxins would be expected to amplify supra-threshold responses due to the inhibition of channel inactivation and would lead to amplification of small generator potentials, a shift in the voltage dependence of activation would additionally result in channel opening closer to resting membrane potential, resulting in greater amplification of sub-threshold responses and induction of spontaneous action potential firing.
The functional effects of OD1 on Na
V1.7 can be correlated to the functional consequences of Na
V1.7 gain-of-function mutations in the painful syndromes inherited erythromelalgia and paroxysmal extreme pain disorder. Typically, mutations present in inherited erythromelalgia patients cause a voltage-dependent shift of activation of Na
V1.7 to more hyperpolarized potentials [
24,
25,
26,
27], corresponding to the β-scorpion toxin-like effects of OD1, whereas paroxysmal extreme pain disorder mutations generally delay the fast inactivation [
28,
29,
30], corresponding to the α-scorpion toxin activity of OD1. This indicates that the OD1 mouse model may be used to identify new analgesics with the potential to treat the poorly managed conditions of inherited erythromelalgia and paroxysmal extreme pain disorder.
Na
V1.7 is preferentially expressed in the majority of peripheral sensory neurons that have been functionally characterized as nociceptive A- and C-fibers [
31]. Specifically, it is present on the peripheral terminals of nociceptive neurons, where it contributes to the amplification of sub-threshold generator potentials by producing ramp currents that depolarize the membrane to the threshold required to generate an action potential [
32,
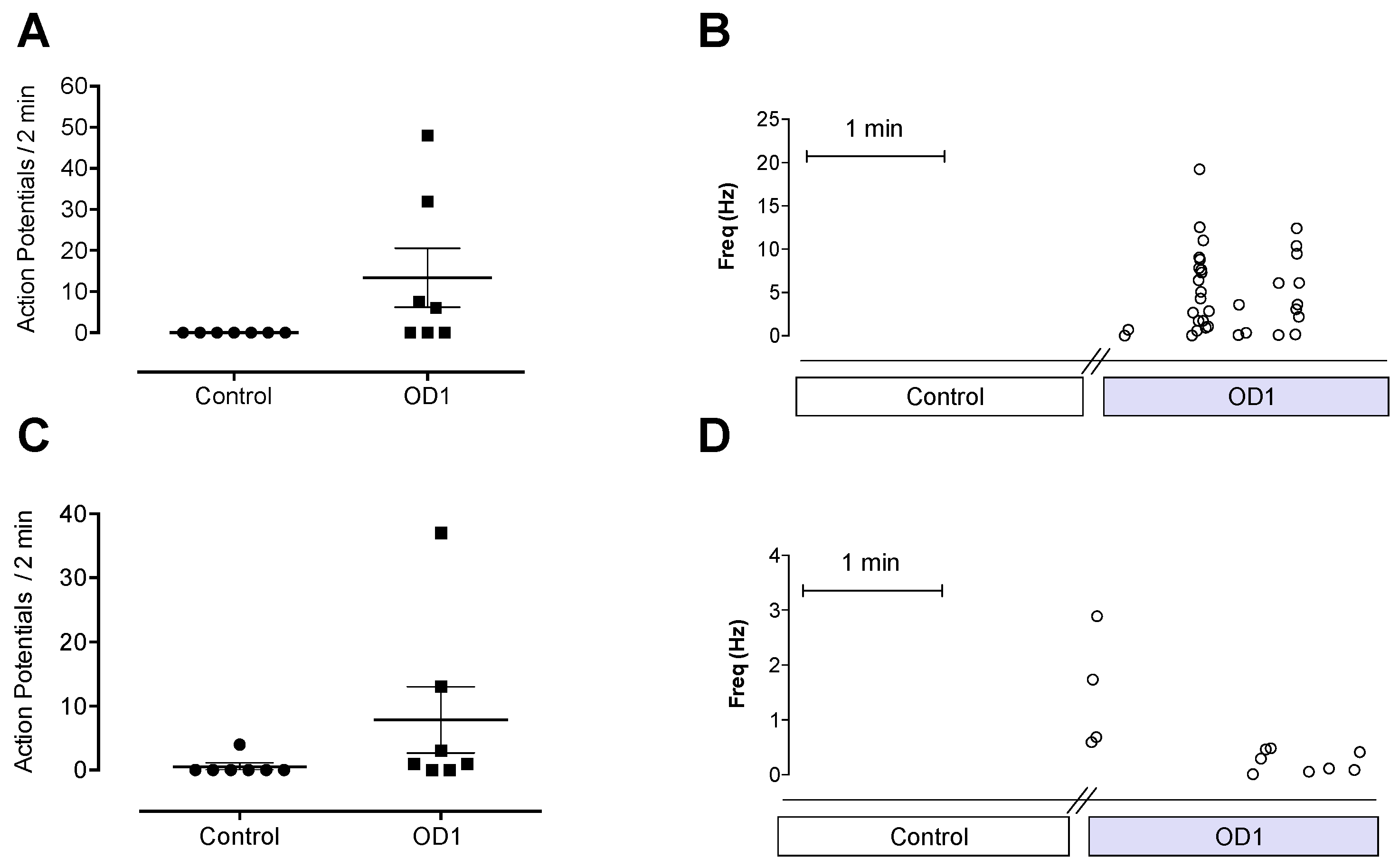
33]. Application of OD1 to the receptive fields of A- and C-fibers caused spontaneous action potential firing, consistent with the effect of OD1 on the biophysical properties of Na
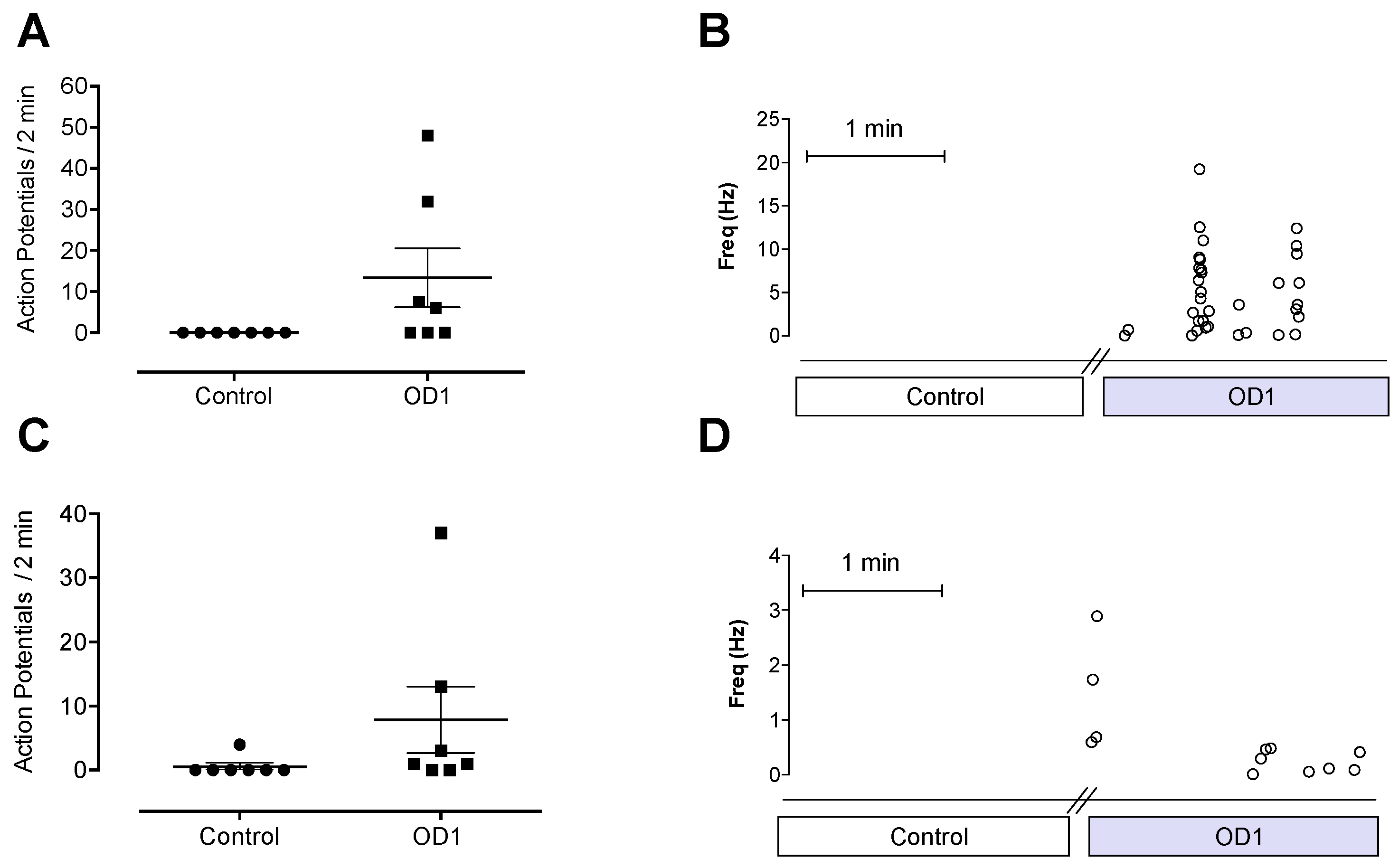
V1.7, which would result in depolarization closer to resting membrane potential, leading to spontaneous action potential firing. The spontaneous pain behaviors induced by intraplantar injection of OD1 are consistent with the spontaneous action potential firing seen in A- and C-fibers in the skin-nerve preparation.
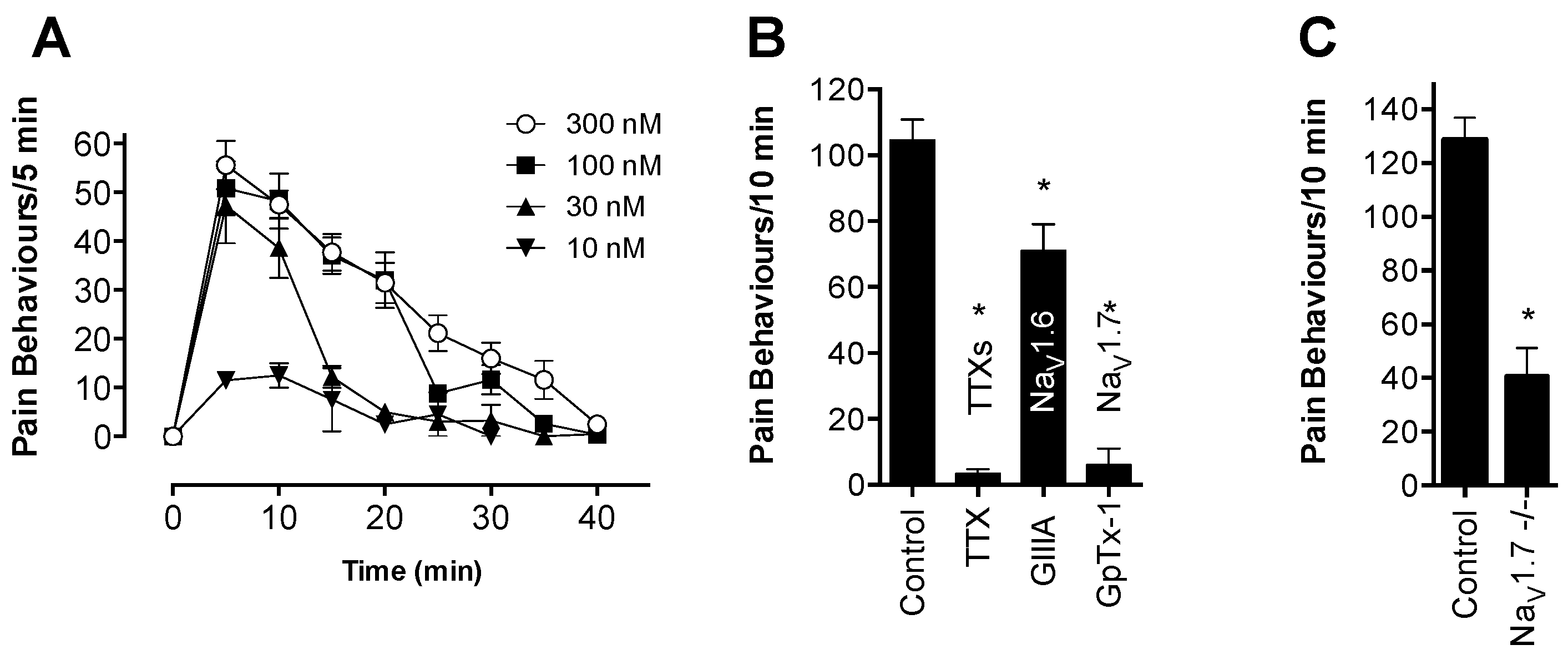
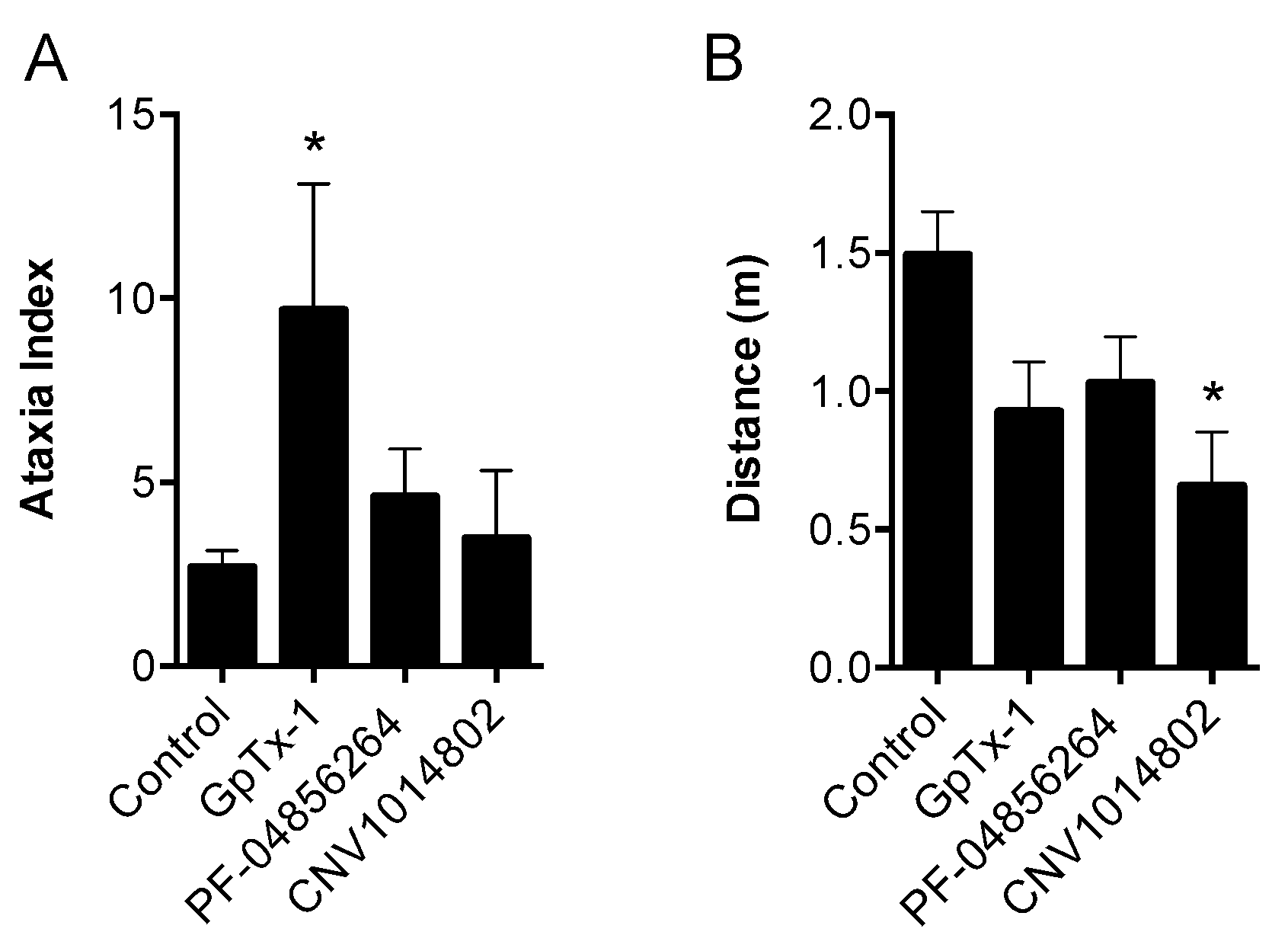
Although OD1 is a selective Na
V1.7 activator, it also has activity at Na
V1.6, albeit at higher concentrations. Given that intraplantar administration of the Na
V1.6 activator Cn2 also causes spontaneous pain behaviors in mice [
18], we assessed the relative contribution of Na
V1.7 and Na
V1.6 to spontaneous pain behaviors induced by OD1. While the Na
V1.6 inhibitor GIIIA partially reversed OD1-induced spontaneous pain by 33%, the Na
V1.7 selective inhibitors GpTx-1 and PF-04856264 were effective after intraplantar injection at concentrations unlikely to significantly inhibit Na
V1.6
in vivo, suggesting that the majority of spontaneous pain behaviors induced by OD1 are mediated through Na
V1.7. This was further confirmed in Na
V1.7
−/− mice, where the behavioral phenotype was strongly attenuated, with residual responses likely attributable to the Na
V1.6 activity of OD1. Thus, OD1 elicited a robust and long-lasting behavioral response that was mediated predominantly through Na
V1.7, allowing the efficacy of Na
V1.7 inhibitors to be readily assessed for their potential in the treatment of Na
V1.7-mediated pain.
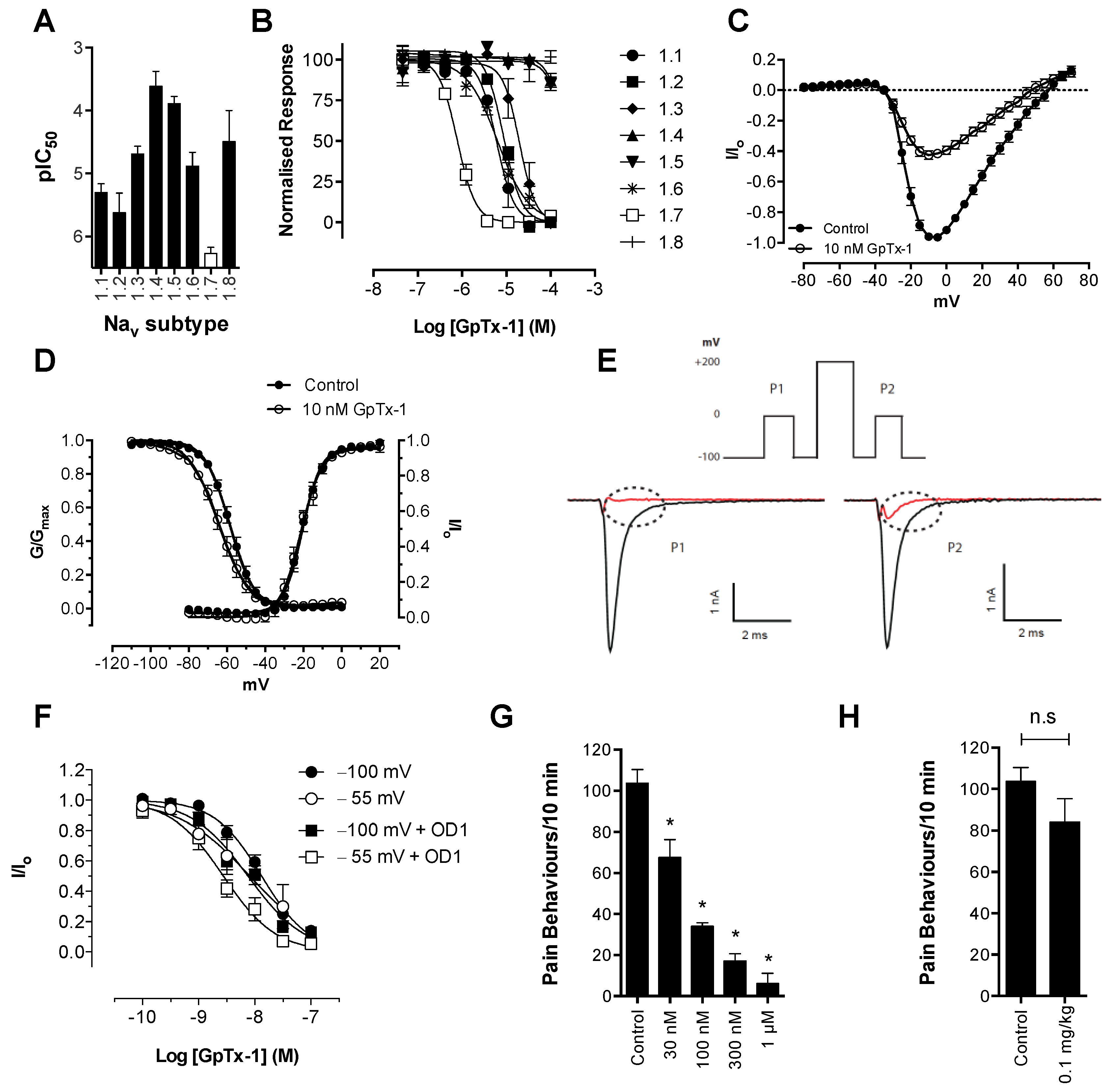
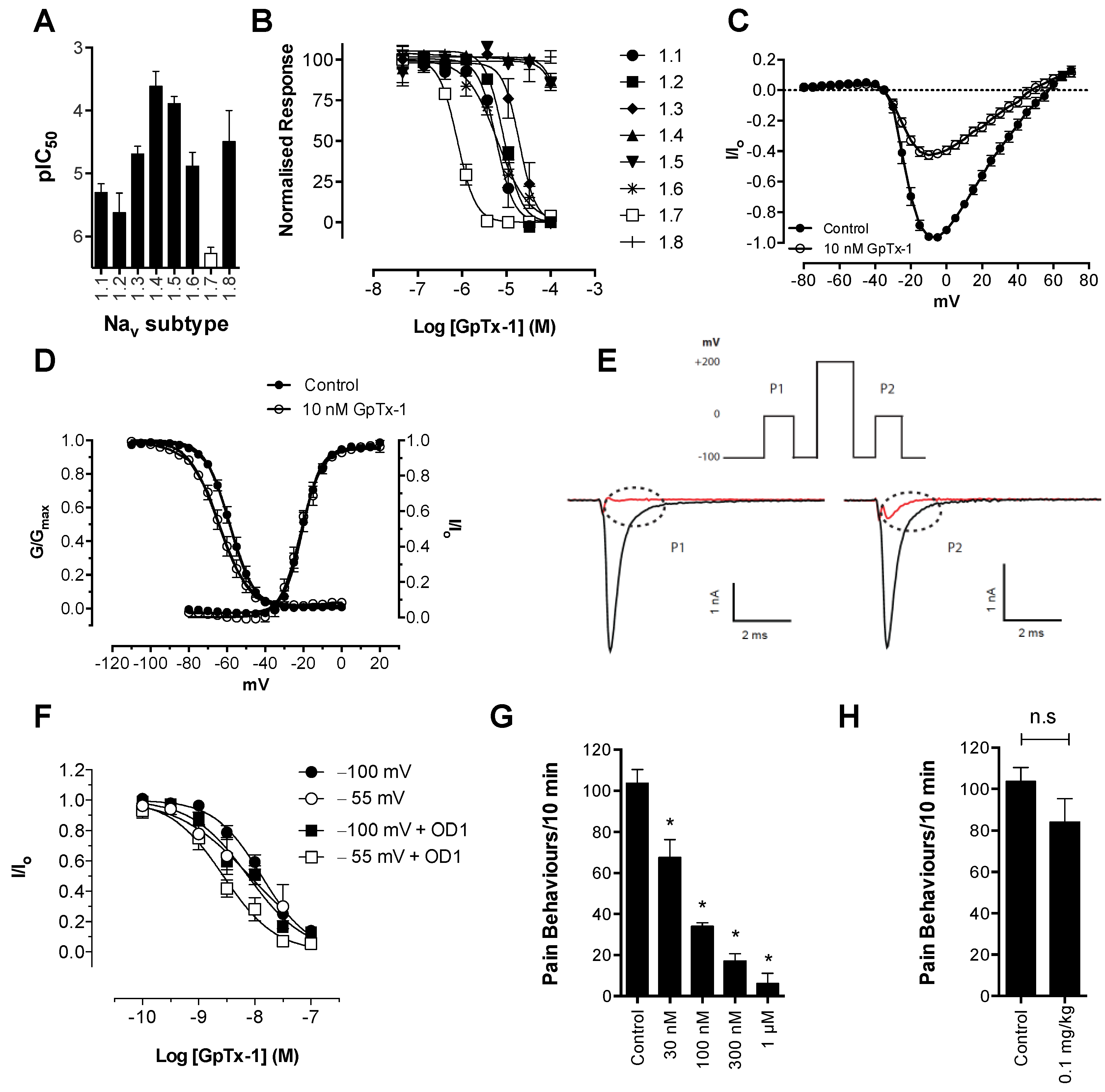
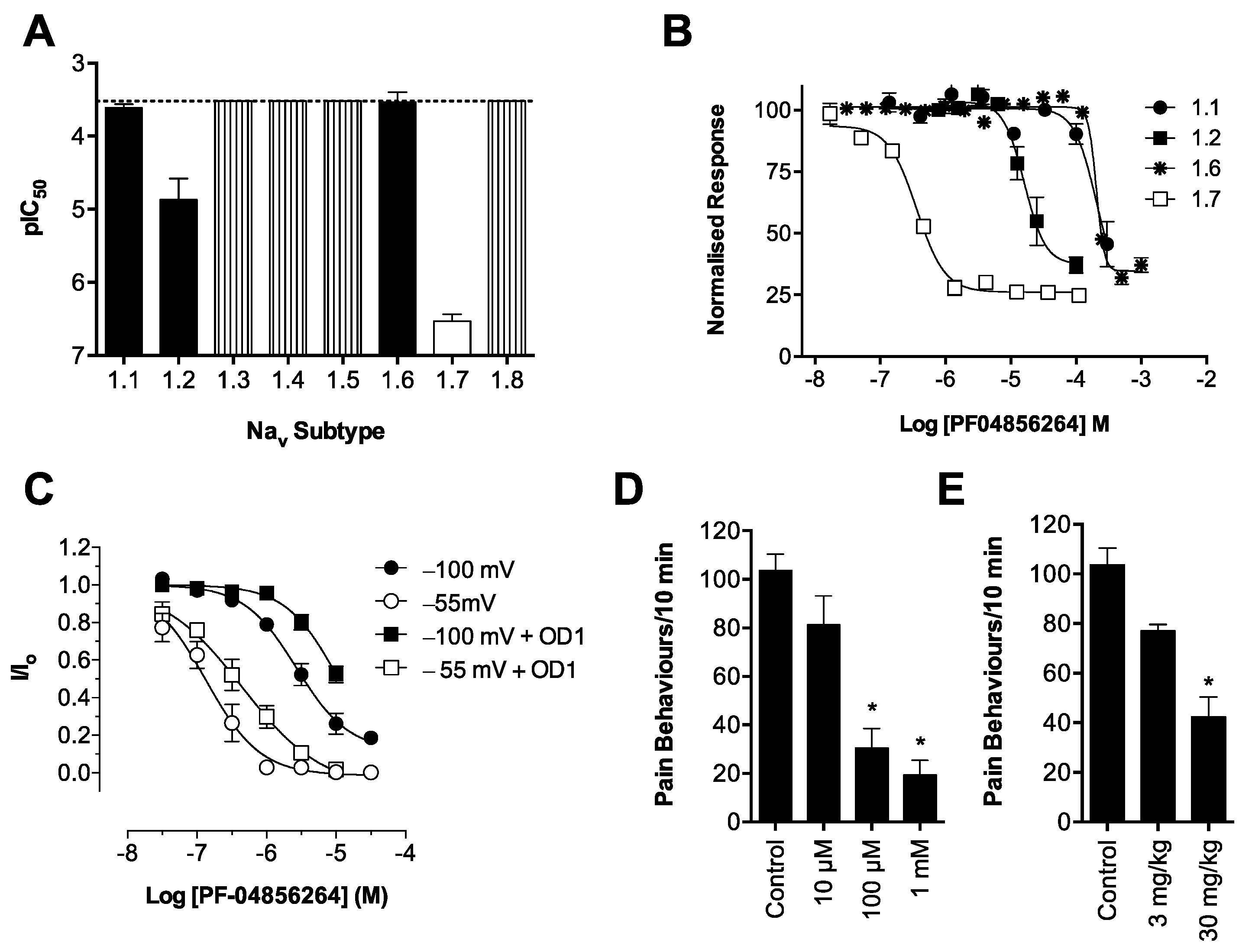
Consistent with its reported activity at Na
V1.7 [
20], GpTx-1 was found to significantly reduce OD1-induced spontaneous pain behaviors when administered locally. The reasons for the potent
in vivo effects of GpTx-1 are not entirely clear. Peptide Na
V inhibitors can have particularly slow off-rates, which could contribute to high apparent
in vivo potency. In addition, it is currently unclear what level of Na
V1.7 inhibition is required to cause analgesia, although it is reasonable to assume that complete inhibition of Na
V1.7 current may not be required to prevent Na
V1.7-mediated action potential firing. Thus, for compounds that are able to achieve relevant free drug concentrations at peripheral nerve endings and that can achieve full inhibition of current at relevant states, the
in vivo IC
50 may be lower than the observed
in vitro IC
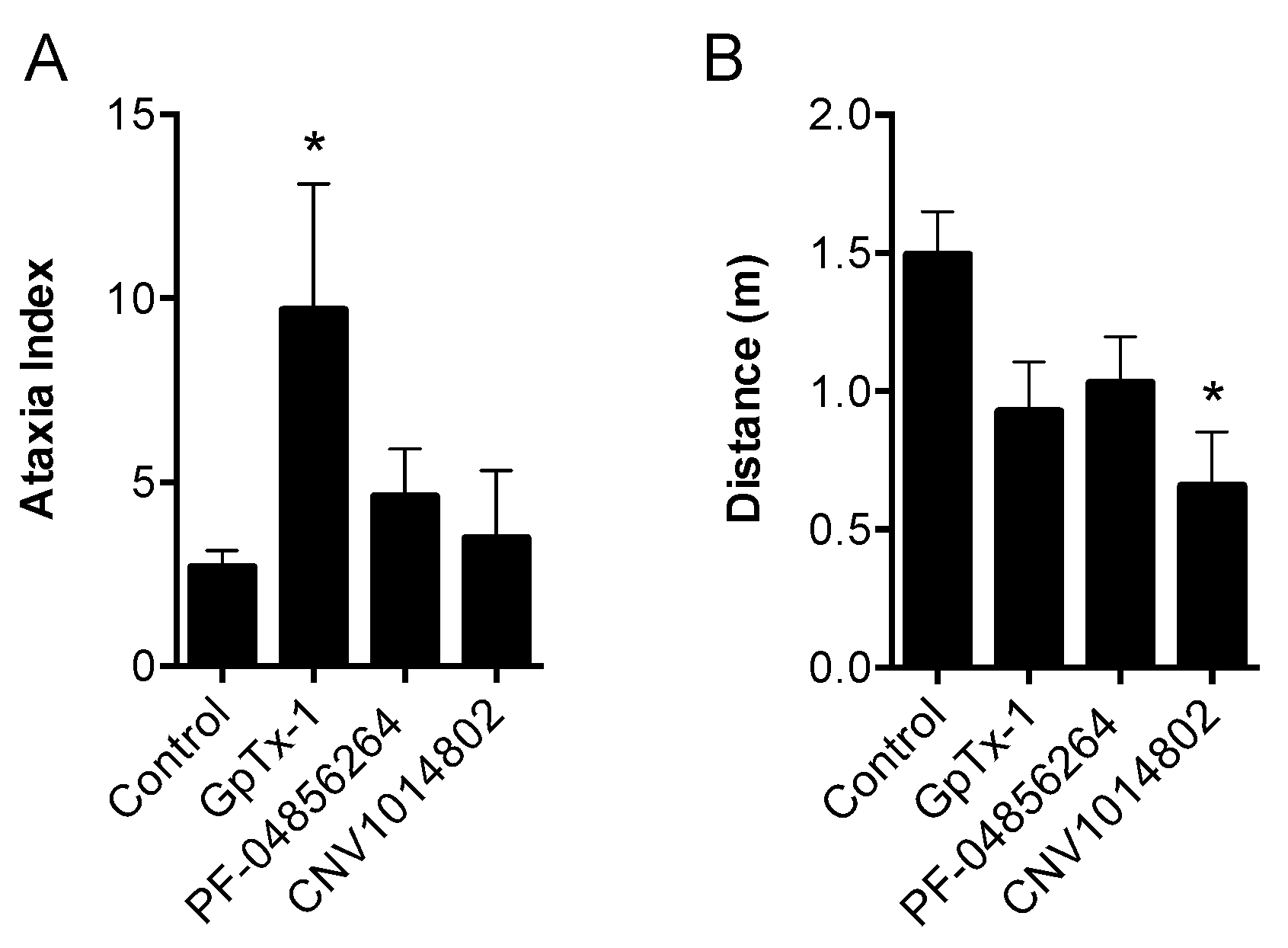
50. Although effective locally, we were not able to reproduce this effect with systemic delivery of GpTx-1. This is likely due to dose-limiting side effects, as the maximum tolerated systemic dose (0.1 mg/kg) may not have achieved sufficiently high concentration at peripheral nerve endings in the hind paw to inhibit Na
V1.7. Motor adverse effects were observed at doses above 0.1 mg/kg, limiting administration of higher doses, and were most likely attributable to activity at Na
V1.6, which is expressed in the periphery on sensory and motor neurons, where loss of function leads to movement disorders and hind limb paralysis [
34]. This suggests that greater than the 30-fold selectivity for Na
V1.7 over Na
V1.6 seen with GpTx-1 is required to have an acceptable therapeutic window for systemic use; however, off-target activity at other Na
V isoforms may also contribute to the adverse effects.
In contrast, the aryl sulfonamide PF-04856264 displayed excellent selectivity (>1000 fold) for Na
V1.7 over Na
V1.4–Na
V1.6. This selectivity profile was reflected
in vivo, with relatively high systemic doses (30 mg/kg) causing no significant motor adverse effects. PF-04856264 was effective at reversing OD1-induced spontaneous pain behaviors when delivered both locally and systemically, although higher than expected doses were required to elicit analgesia by either routes relative to its potency in the FLIPR membrane potential (IC
50 300 nM) and electrophysiology (IC
50 130 nM) assays. However, given that PF-04856264 preferentially binds to Na
V1.7 in the inactivated state, it is possible that the delayed inactivation produced by OD1 promoted an unfavorable channel state for PF-04856264 binding. Indeed, the presence of OD1 led to a three-fold loss of potency in the electrophysiological assays. State-dependent inhibition of Na
V1.7 may also explain the inability of PF-04856264 to fully block membrane potential changes using the fluorescence assay, where residual responses could be mediated by veratridine-modified channels resistant to inactivation that in turn are unaffected by PF-04856264 [
35]. The physiological relevance of a preference for the inactivated state of Na
V1.7 in other pathological pain conditions remains to be established, although such a mechanism might imply lack of efficacy in paroxysmal extreme pain disorder, where Na
V1.7 channels are relatively resistant to fast inactivation.
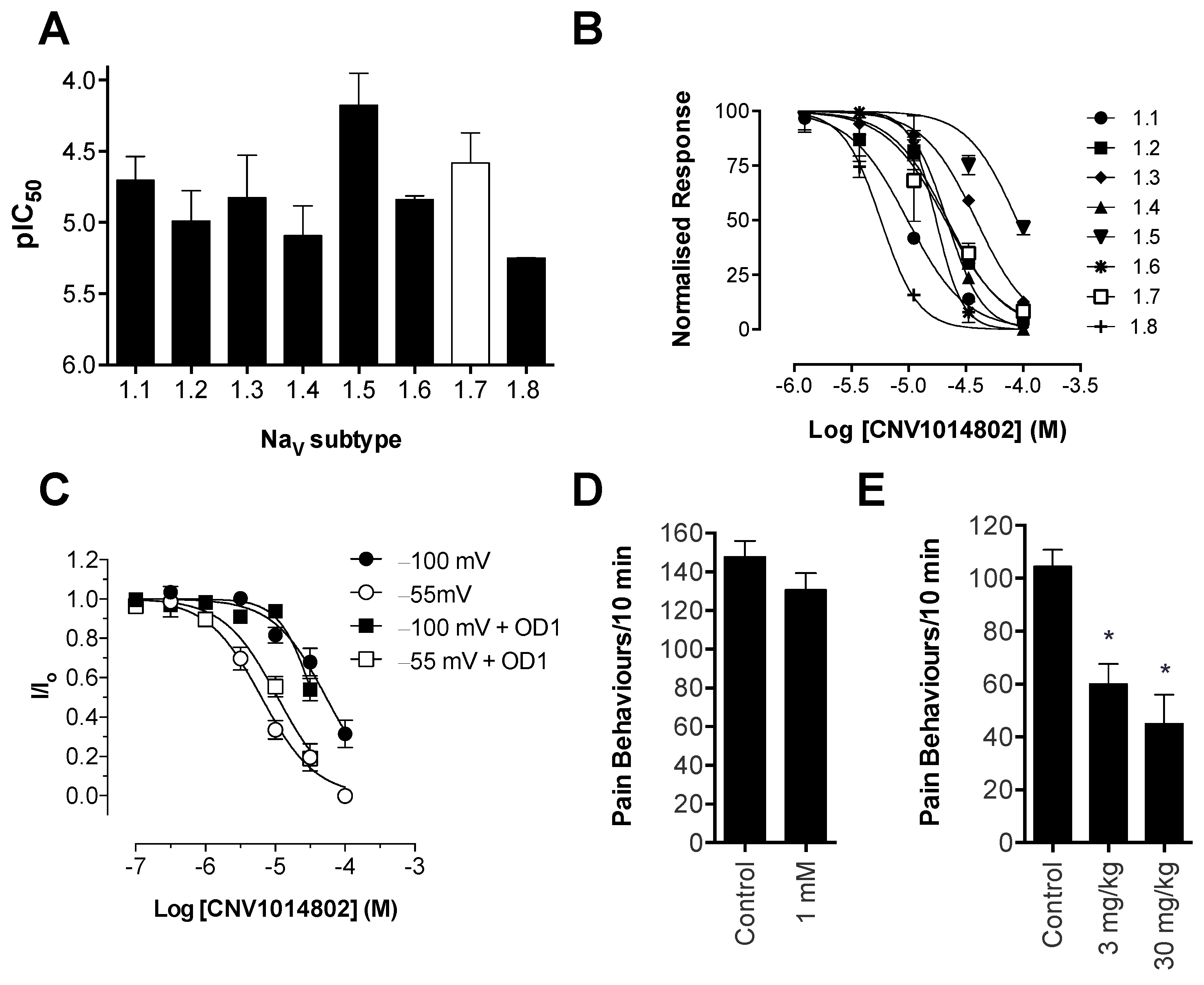
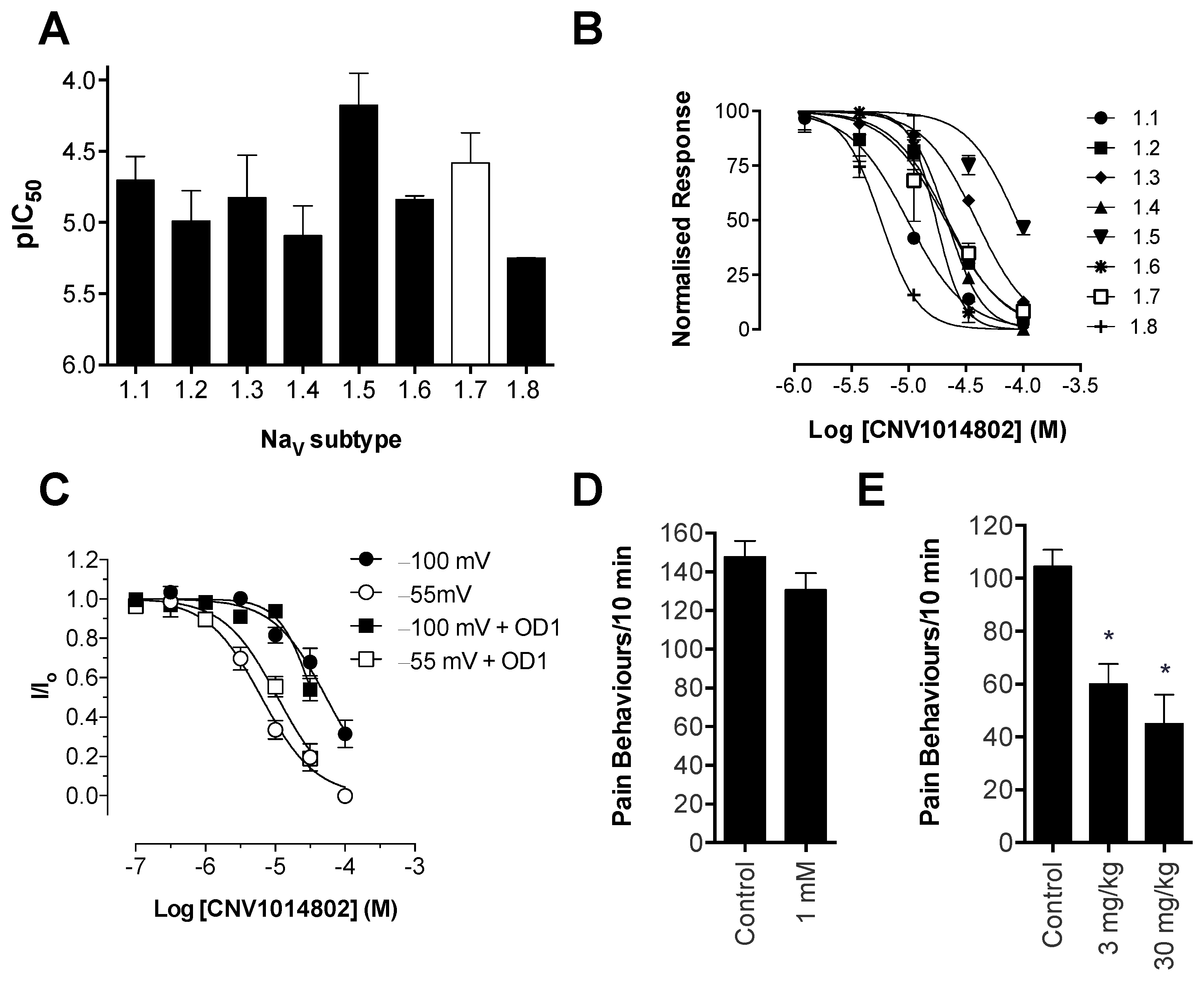
Consistent with previous reports, CNV1014802 also was a state-dependent inhibitor of Na
V1.7 in our hands, although it lacked the Na
V subtype selectivity of PF-04856264. Surprisingly, CNV1014802 lacked
in vivo analgesic efficacy when delivered locally at a concentration of 1 mM, perhaps reflecting its relatively poor potency at Na
V1.7 in membrane potential (32 μM) and electrophysiology (6.3 μM) assays. In addition, CNV1014802 (formally GSK1014802 and now raxatrigine) is reported to be highly (94%) protein bound, which could also lead to a decrease in free effective drug at the site of action after local administration [
36]. In contrast to local administration, CNV1014802 was effective when administered systemically, although some sedative adverse effects were observed at the highest dose (30 mg/kg). These effects are consistent with the reported ability of CNV1014802 to penetrate the central nervous system, which likely is crucial for its efficacy in animal models of seizures and schizophrenia [
36,
37], suggesting that analgesic activity after systemic dosing may also be due to central effects.
Based on our pharmacological validation, the OD1-induced spontaneous pain model can be used to assess the
in vivo activity of compounds when delivered by both the intraplantar and a systemic route. An advantage of the intraplantar route is that it uses considerably less material and thus may be the preferable route of administration for screening compounds for
in vivo activity in the early stages of lead identification. However, as was the case for PF-04856264 and CNV1014802,
in vivo activity in the OD1 model did not correspond well with the
in vitro potency found in the electrophysiology and FLIPR membrane potential assays. At least in the case of PF-04856264, species-specific effects are unlikely to contribute to this discrepancy, as equipotency at human and mouse Na
V1.7 has been previously reported [
17]. In addition, although PF-04856264 lost potency in the presence of OD1, consistent with preferential binding to the inactivated state, this change was relatively small and would not explain the relative lack of potency
in vivo. Interestingly, we observed incomplete inhibition of Na
V1.7-mediated responses in the FLIPR membrane potential assay by PF-04856264 and CNV1014802 (
Figure S1), although both compounds completely inhibited currents from Na
V1.7 patch-clamp recordings. While it is unclear whether this effect correlates to the ability of these compounds to prevent activity-induced membrane depolarization at peripheral sensory nerve endings
in vivo, such effects may contribute to the poor
in vivo activity observed. In addition, it is plausible that other factors, such as the pharmacokinetic properties of these compounds, affect
in vivo efficacy. Nonetheless, these results highlight the need for a simple animal model, such as the OD1 model described here, which permits rapid evaluation of
in vivo on-target effects, as the
in vitro properties do not always correspond well to
in vivo activity.
Scorpion α-toxins, such as OD1, are known to bind to Domain IV [
38]; however, we have demonstrated that efficacy in the OD1 model is not dependent on competitive binding at this site. For example, TTX, which binds to the pore of the channel, and GpTx-1, which binds to Domain II [
39], were both able to completely abolish OD1-induced spontaneous pain behaviors. Although not as potent
in vivo, the state-dependent inhibitors PF-04856264, predicted to share an overlapping binding site with scorpion α-toxins on Domain IV [
17], and CNV1014802 (binding site remains to be determined) were also effective. Thus, the OD1 model is suitable to evaluate
in vivo activity of Na
V1.7 inhibitors irrespective of their mechanism of action or binding site.
In conclusion, we have established a Na
V1.7-mediated mouse model of pain based on intraplantar injection of the scorpion toxin OD1 and pharmacologically characterized the activity of the spider peptide GpTx-1, the small molecule PF-04856264 and the clinical candidate CNV1014802. The model is suitable to evaluate
in vivo on-target activity of Na
V1.7 inhibitors; however, it is unclear if activity in OD1-induced pain translates to anti-nociceptive or anti-allodynic effects in other rodent models or analgesia in humans. While efficacy in the OD1 model is useful to validate
in vivo on-target activity at Na
V1.7, species differences need to be considered carefully when using animal models of human disease. Given the relatively high homology between mouse and human Na
V isoforms, significant pharmacological differences have rarely been reported. Indeed, PF-04856264 is equipotent at mouse and human Na
V1.7 [
17], and the effects of OD1 on rodent Na
V1.7 closely match the effects on human Na
V1.7 [
15]. While Na
V1.7 is arguably a well-validated pain target in humans based on genetic evidence from loss-of-function and gain-of-function conditions, including congenital insensitivity to pain, erythromelalgia and paroxysmal extreme pain disorder, it remains to be established if pharmacological inhibition of Na
V1.7 can achieve global graded analgesia.
4. Experimental Section
4.1. Chemicals
OD1 was synthesized as previously described [
15]. PF-04856264 was from SYNthesis Med Chem (Parkville, VIC, Australia) and CNV1014802 was from Axon MedChem (Groningon, The Netherlands). GIIIA was synthesized by Boc chemistry using methods previously described [
40]. TTX was from Tocris Bioscience (Bristol, UK), and veratridine was from Alomone Labs (Jerusalem, Israel). All other reagents were from Sigma Aldrich (Castle Hill, NSW, Australia), unless otherwise stated. Peptides were routinely diluted in 0.1% bovine serum albumin (BSA).
4.2. Synthesis of GpTx-1
Solvents for reversed-phase high-performance liquid chromatography (HPLC) consisted of 0.05% trifluoroacetic acid (TFA)/H2O (A) and 90% acetonitrile/0.043% TFA/H2O (B). Analytical HPLC was performed on a Shimadzu LC20AT system (Shimadzu, Kyoto, Japan) using a Thermo Hypersil GOLD 2.1 × 100 mm C18 column (ThermoScientific, Brendale, QLD, Australia) heated at 40 °C with flow rate of 0.3 mL/min. A gradient of 10–55% B over 30 min was used, with detection at 214 nm. Preparative HPLC was performed on a Vydac 218TP1022 column (Grace, Columbia, MD, USA) running at a flow rate of 16 mL/min using a gradient of 5%–45% B over 40 min. Mass spectrometry was performed on an API2000 (ABI Sciex, Mt Waverley, VIC, Australia) mass spectrometer in positive ion mode.
Protected 9-fluorenylmethyloxycarbonyl (Fmoc) amino acid derivatives and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from Iris Biotech (Marktredwitz, Germany); Rink-amide resin, N,N-dimethylformamide (DMF), N,N-diisopropylethylamine (DIEA) and TFA from Auspep (Tullamarine, VIC, Australia). All other reagents were obtained from Sigma-Aldrich (Castle Hill, NSW, Australia).
GpTx-1 was assembled on a Symphony (Protein Technologies Inc., Tucson, AZ, USA) automated peptide synthesizer on a Rink-amide (loading 0.67 mmol/g) polystyrene resin on a 0.1 mmol scale. Fmoc deprotections were achieved using 30% piperidine/DMF (1 × 1.5 min, then 1 × 4 min). Couplings were performed in DMF using 5 equivalents of Fmoc-amino acid/HBTU/DIEA (1:1:1) relative to resin loading for 2 × 20 min. Amino acid side-chains were protected as Asn(Trt), Asp(OtBu), Arg(Pbf), Cys(Trt), His(Trt), Lys(Boc), Ser(tBu), Thr(tBu), Trp(Boc), Tyr(tBu). Cleavage from the resin and removal of side-chain protecting groups was achieved by treatment with 95% TFA/2.5% TIPS/2.5% H2O at room temperature for 2 h. After most of the cleavage solution was evaporated under a stream of N2, the product was precipitated and washed with cold Et2O and lyophilized from 50% acetonitrile/0.1% TFA/H2O 223 mg; ESI-MS (m/z): calc. (average) 1021.0 [M + 4H]4+, found 1020.8.
Purified reduced peptide (41 mg), reduced glutathione (100 equiv) and oxidized glutathione (10 equiv.) were dissolved in 6 M GnHCl (21 mL) then added to a solution of 0.36 M NH4OAc (pH 8.0, 230 mL) and stirred at room temperature with exposure to air for 48 h. The single major product was isolated by preparative HPLC: 13.5 mg; ESI-MS (m/z): calc. (average) 1019.5 [M + 4H]4+, found 1019.4. The crude product was purified by preparative HPLC to give 42 mg of hexathiol GpTx-1 (99% purity).
4.3. Animals
Ethical approval for in vivo experiments in animals was obtained from The University of Queensland animal ethics committee (PHARM/261/13/ARC and IMB/PACE/326/15). Experiments involving animals were conducted in accordance with the Animal Care and Protection Regulation Qld (2012), the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, 8th edition (2013, and the International Association for the Study of Pain Guidelines for the Use of Animals in Research. All efforts were made to minimize animal suffering and to reduce the number of animals used.
For behavioral assessment, we used adult male C57BL/6J mice aged 6–8 weeks. Animals were housed in groups of 3 or 4 per cage, under 12-h light-dark cycles and had standard rodent chow and water
ad libitum. Advillin-Cre constitutive Na
V1.7 knockout mice were kindly provided by Professor John N. Wood (University College London) [
41].
4.4. Behavioral Assessment Following Injection of OD1
OD1 was diluted in phosphate-buffered saline/0.1% BSA (10–300 nM) and administered by shallow subcutaneous (intraplantar) injection to the left hind paw of mice in a volume of 40 μL under light isoflurane (3%) anesthesia. Mice were then placed individually into polyvinyl boxes (10 × 10 × 10 cm), and spontaneous pain behavior (licks and flinches) was counted by an investigator unaware of the treatments received from video recordings for 40 min post-injection at 5-min intervals. Once spontaneous pain behavior had ceased, mechanical and thermal allodynia was assessed using an electronic von Frey apparatus (MouseMet Electronic von Frey, TopCat Metrology, Ely, UK) and the Hargreaves method (Plantar Analgesia Meter, IITC, Woodland Hills, CA, USA), as previously described [
42].
4.5. Effect of Pharmacological Inhibitors and Clinical Compounds
OD1 (300 nM) was administered as described above, and cumulative spontaneous pain behaviors were quantified for 10 min immediately post-injection of OD1 by a blinded observer. Compounds delivered by the intraplantar route were co-injected with OD1 at the concentrations stated. Compounds delivered by the intraperitoneal route were administered 15 min prior to injection of OD1 at the doses stated in a volume of 10 μL/g. All compounds were diluted in phosphate-buffered saline, except PF-04856264 and CNV1014802, which were diluted in phosphate-buffered saline/10% DMSO due to poor solubility.
4.6. Motor Performance Assessment
Motor performance was assessed using the Parallel Rod Floor Test and analyzed using ANY-Maze software (Stoelting Co., version 4.70, Wood Dale, IL, USA). GpTx-1 (0.3 mg/kg), PF-04856264 (30 mg/kg) and CNV1014802 (30 mg/kg) were administered by the intraperitoneal route, as described above, 15 min prior to assessment of motor performance. Mice were then placed in the Parallel Rod Floor Test apparatus, and the distance travelled (m) and number of foot slips were recorded over 1 min using the ANY-Maze software. The ataxia index was calculated by dividing the number of foot slips by the distance travelled (m).
4.7. Skin-Nerve Preparation
The effect of OD1 on A- and C-fibers was assessed using single fiber recordings from isolated mouse skin-saphenous nerve preparations, as previously described [
43]. Briefly, the saphenous nerve along with the skin of the dorsal hind paw and lower leg of adult C57BL/6J mice was removed and placed in an organ chamber, perfused with carbogenated synthetic interstitial fluid containing the following (in mM): NaCl (107.8), KCl (3.5), MgSO
4 (0.69), NaHCO
3 (26.2), NaH
2PO
4 (1.67), Na-gluconate (9.64), glucose (5.55), sucrose (7.6) and CaCl
2 (1.53); pH 7.3. The saphenous nerve was placed in a separate recording chamber immersed in paraffin oil and was desheathed and teased apart until a single fiber recording was identified by mechanically probing the corresponding receptive field. Fibers were then classified based on conduction velocity assessed by electrical stimulation of the receptive field with a microelectrode (C-fiber < 1 m/s, A-fiber 1.6–12 m/s). The receptive field was isolated using a plastic ring, and OD1 (30 nM) was continuously perfused at a rate of 8 mL/min at 32 °C. Data were recorded and analyzed using DAPSYS, Version 8.
4.8. Cell Culture
Human embryonic kidney (HEK 293) cells stably expressing hNaV1.1, hNaV1.2, hNaV1.3, hNaV1.4, hNaV1.5, hNaV1.7 and hNaV1.8 (SB Drug Discovery, Glasgow, UK) were cultured in MEM containing 10% v/v fetal bovine serum supplemented with L-glutamine (2 mM) and selection antibiotics as recommended by the manufacturer. HEK 293 cells stably expressing hNaV1.6 (kind gift from Dr. Frank Lehmann-Horn, University of Ulm, Germany) were cultured in DMEM containing 10% v/v fetal bovine serum and geneticin 0.5 mg/mL. CHO cells stably expressing hNav1.7 (Chantest, OH, USA) were cultured in Ham’s F12 containing 10% v/v fetal bovine serum and selection antibiotics, as recommended by the manufacturer. Cells were grown in a humidified 5% CO2 incubator at 37 °C, grown to 70%–80% confluence and passaged every 3–4 days using TrypLE Express (Invitrogen, Scoresby, VIC, Australia).
4.9. FLIPR Membrane Potential Assays
HEK 293 cells stably expressing hNaV1.1–1.8 were plated 48 h before the assay on 384-well black-walled imaging plates at a density of 10,000–15,000 cells per well and were loaded with red membrane potential dye (Molecular Devices, San Francisco, CA, USA) according to the manufacturer’s instructions for 30 min at 37 °C. After the addition of GpTx-1, PF-04856264 and CNV1014802 using the FLIPRTETRA, cells were incubated a further 5 min before stimulating NaV using veratridine (60 μM, NaV1.1–NaV1.7) or deltamethrin (150 μM, NaV1.8). Changes in membrane potential were assessed using the FLIPRTETRA (excitation 515–545 nm, emission 565–625 nm) every second for 300 s after adding agonists. To quantitate the effect of test compounds on NaV responses, the area under the curve (AUC) corresponding to 300 s after the addition of veratridine was computed using ScreenWorks (Molecular Devices, Version 3.2.0.14) and normalized to buffer responses (0%) and veratridine control responses (100%).
4.10. Electrophysiology
CHO cells were used for the electrophysiology, because in our experience, they are more robust to clamp compared to HEK cells. CHO hNaV1.7 were passaged 48 h prior to the assay in a T-175 flask and cultured in selection-free media at 37 °C or CHO hNav1.7 EZCells™ were thawed the day of the experiment according to the manufacture’s specifications. To induce expression in the CHO hNaV1.7 cells, tetracycline (1 μg/mL) was added to culture media 24 h prior to the assay. Cells were harvested at 70%–80% confluence and resuspended to 1 × 106 cells/mL in Ex-Cell ACF CHO Medium with 25 mM HEPES and 1× Glutamax (Gibco, Thermo Fisher Scientic, Scoresby, VIC, Australia) before being transferred to the QPatch QStirrer and allowed to recover for 30 min. Extracellular solution (EC) was used to resuspend all test compounds and contained (in mM): NaCl (140), KCl (4), CaCl2 (2), MgCl2 (1), HEPES (10), CdCl2 (0.1) and glucose (10). The pH was adjusted to 7.4 with NaOH, and osmolarity was adjusted with sucrose to 315 mOsm. The intracellular solution contained (in mM): CsF (140), EGTA/CsOH (1/5), HEPES (10) and NaCl (10). The pH was adjusted to 7.4 with CsOH and osmolarity adjusted with sucrose to 320 mOsm. OD1 and GpTx-1 were diluted in EC with the addition of 0.1% BSA.
Whole-cell patch-clamp experiments were performed at room temperature on a QPatch-16 automated electrophysiology platform (Sophion Bioscience, Ballerup, Denmark) using 16-channel planar patch chip plates (QPlates; Sophion Bioscience) with a patch hole diameter of 1 µm and a resistance of 2 ± 0.02 MΩ. Cell positioning and sealing parameters were set as follows: positioning pressure −60 mbar, minimum seal resistance 0.1 GΩ, holding potential −100 mV, holding pressure −20 mbar. Whole-cell currents were filtered at 5 kHz and acquired at 25 kHz.
Cells expressing hNaV1.7 were maintained with a holding potential of −100 mV between voltage protocols. Current-voltage (IV) relationships were determined using a family of 500-ms conditioning pulses from −120 mV to +70 mV in 5-mV steps, followed by depolarization to 0 mV to assess the voltage dependence of fast inactivation (repetition interval: 20 s). Both peak and late current (10 ms post-peak) of the IV family were measured. State-dependence was assessed after a 10-min compound incubation (±OD1 300 nM) to ensure steady-state inhibition for each concentration. In order to assess compound activity at the partially inactivated/open state, a 50-ms depolarization to 0 mV was measured after a conditioning pulse to −55 mV for 8 s, with a 50-ms recovery period between (repetition interval 12 s). To assess possible interaction with the voltage sensor domain, a triple-pulse protocol was used, comprising two steps to 0 mV for 50 ms separated by a strong depolarization step to +200 mV for 50 ms with 20-ms recovery to the −100-mV holding potential between each step.
4.11. Data Analysis and Statistics
Data were plotted and analyzed using GraphPad Prism (GraphPad Softare, Version 6.0, La Jolla, CA, USA). Statistical significance was defined as p < 0.05 and was determined by the t-test or one-way ANOVA with Dunnett’s post-test, as appropriate. Data are expressed as the mean ± standard error of the mean. For electrophysiology experiments, the voltage dependence of steady-state inactivation was calculated by dividing the amplitude of the test current (I) by the maximal current elicited (Io), as defined by: INa = I/Io. The voltage-dependence of activation was derived from IV curves after normalizing for Na+ conductance (GNa) using: GNa = INa/(Vmemb − Vrev), where Vmemb is the membrane potential and Vrev is the reversal potential. Steady-state kinetic parameters were obtained by fitting the data to Boltzmann equations y = bottom + (top − bottom)/(1 + exp((V50 − x)/slope)). Sophion QPatch Assay Software (Sophion, Version 5.0, Ballerup, Denmark) was used to determine the time of decay (τ) for inactivating current using the equation for first-order exponential decay: y = y0 × exp(- k × x) + yp, where y is current, x is time, y0 is peak current, yp is the plateau and k is the rate constant. τ was calculated separately for each conditioning potential, defined as 1/k.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}