
PhTx3-4, a Spider Toxin Calcium Channel Blocker, Reduces NMDA-Induced Injury of the Retina

 and
and
Abstract
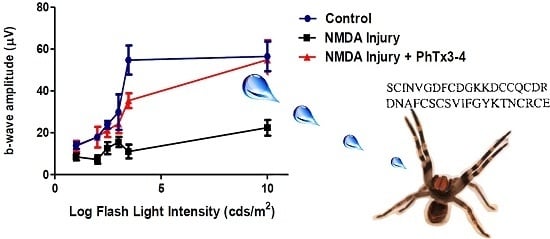
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
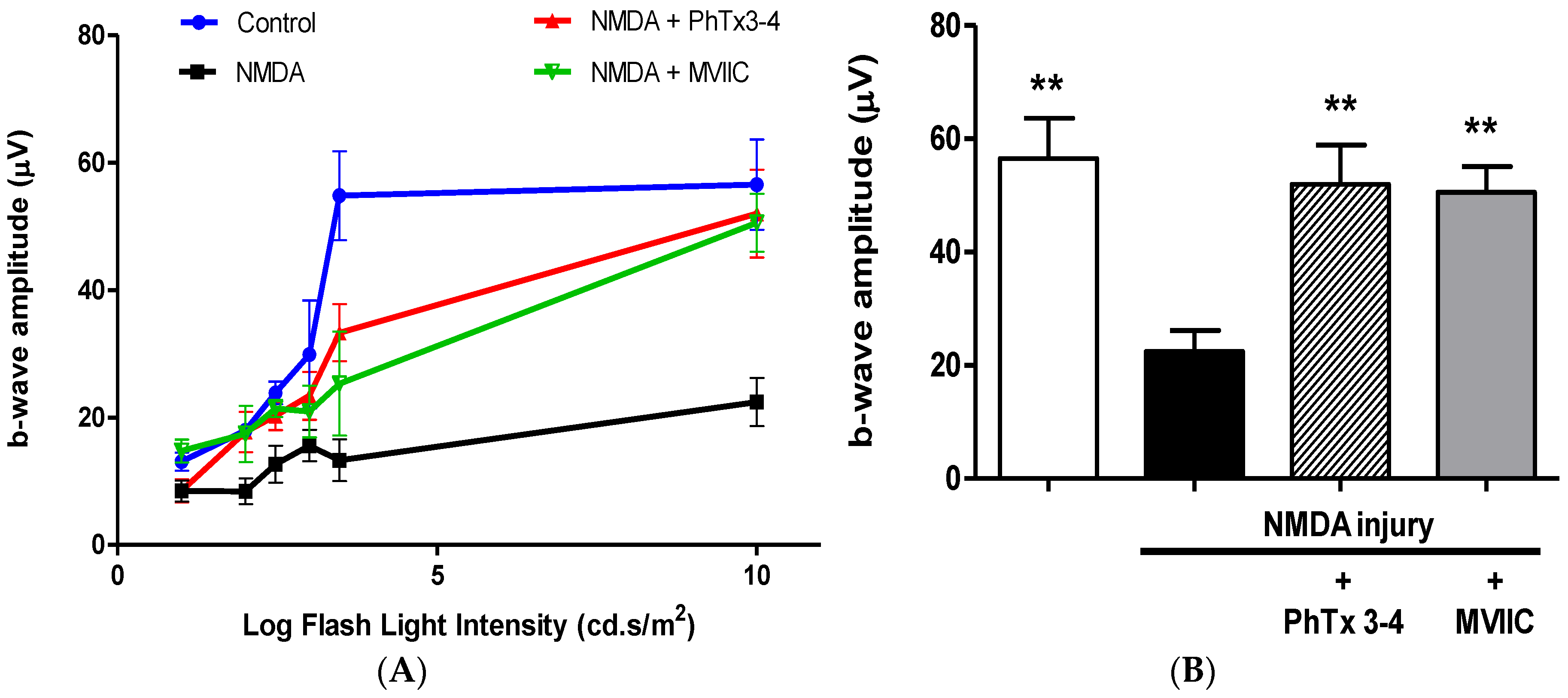
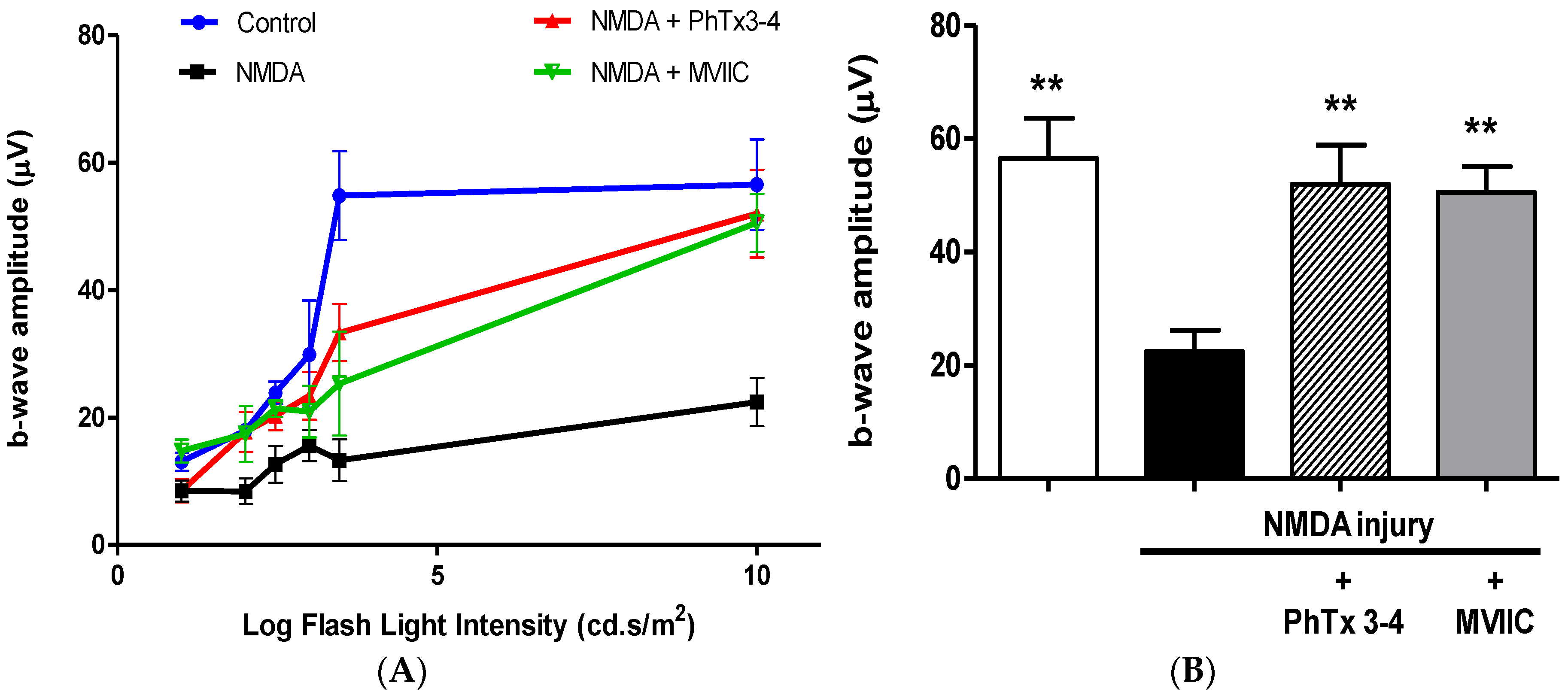
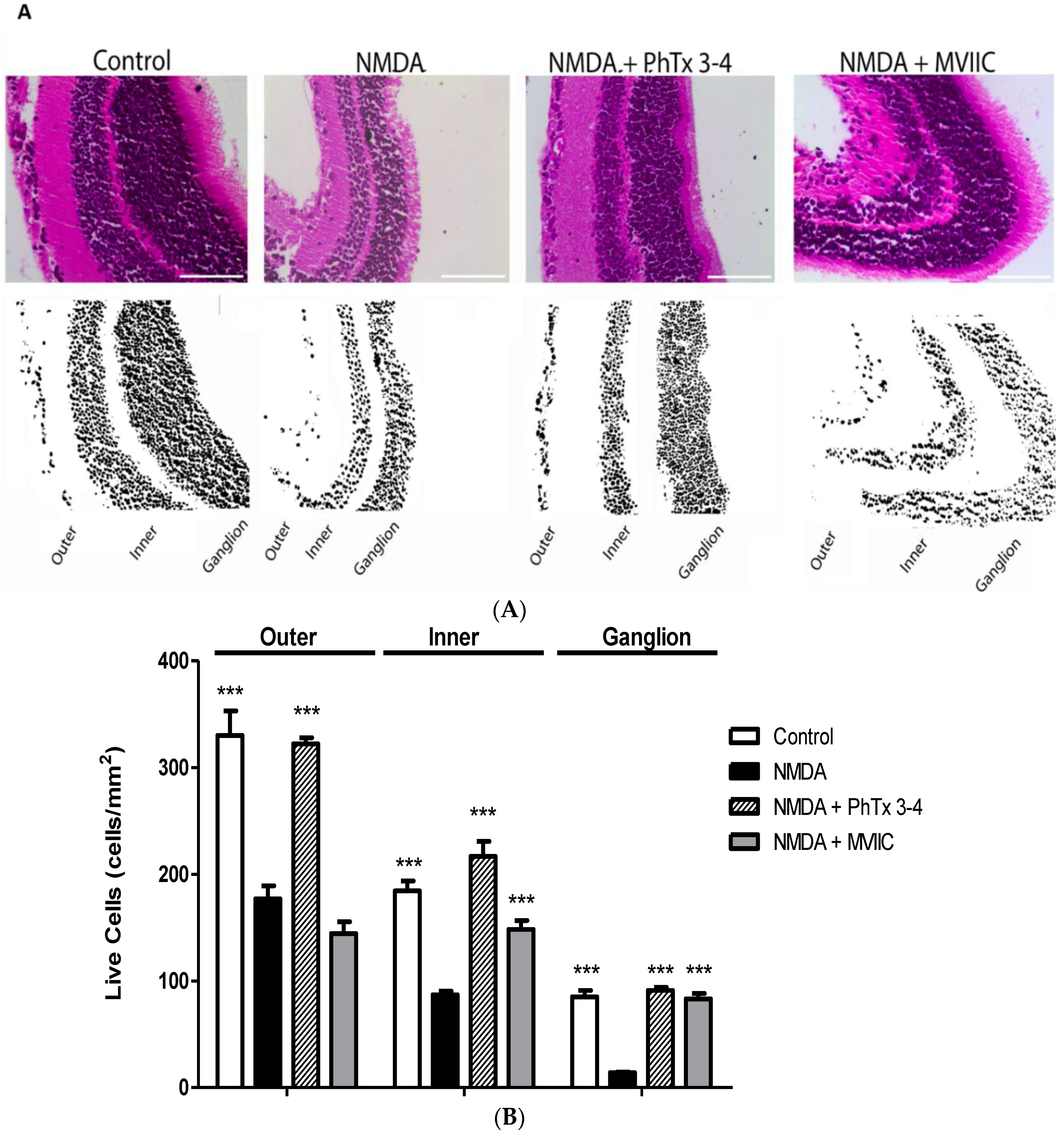
2.1. PhTx3-4 Reverses NMDA-Induced Retinal Injury Disfunctions of the Retina
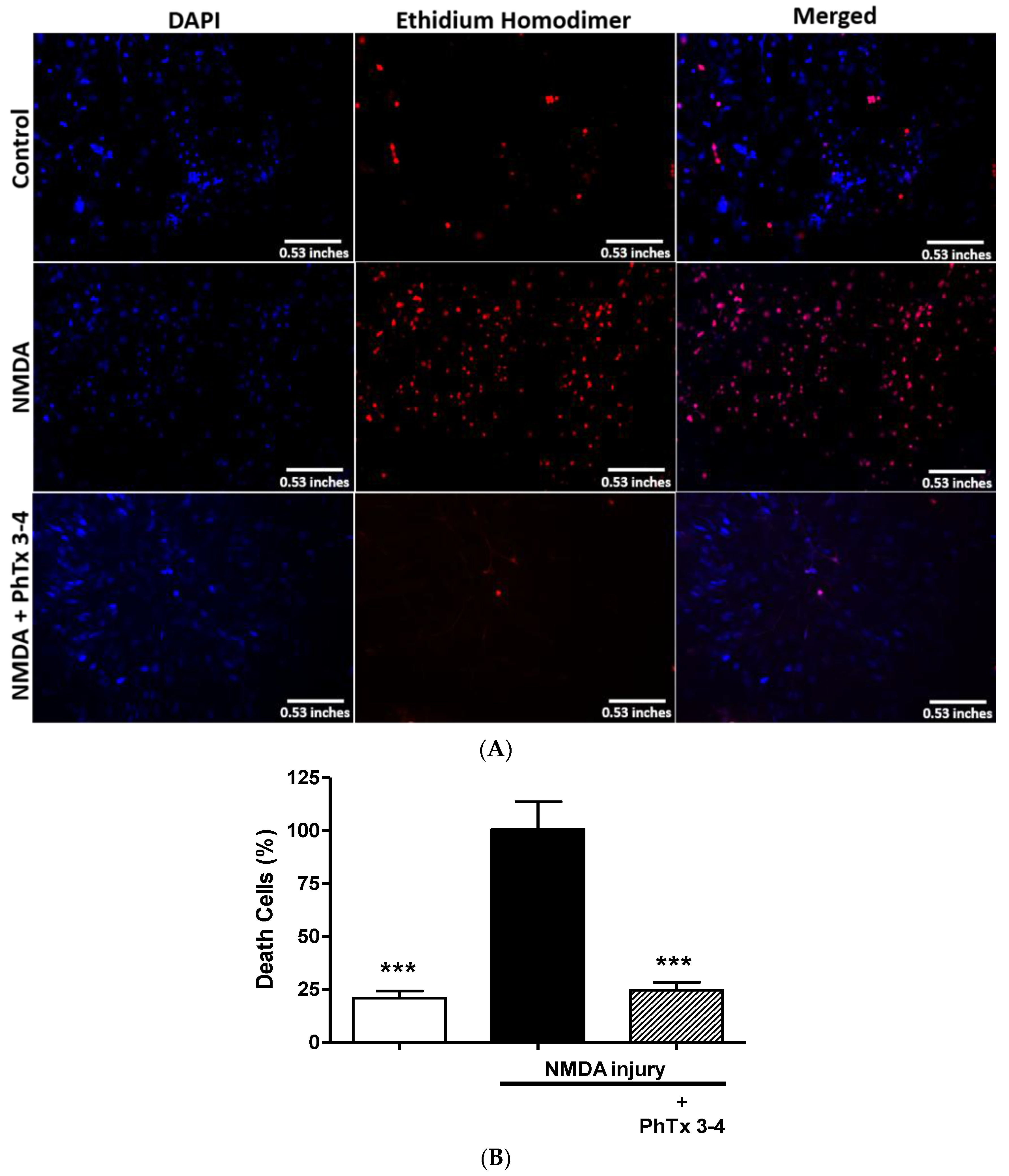
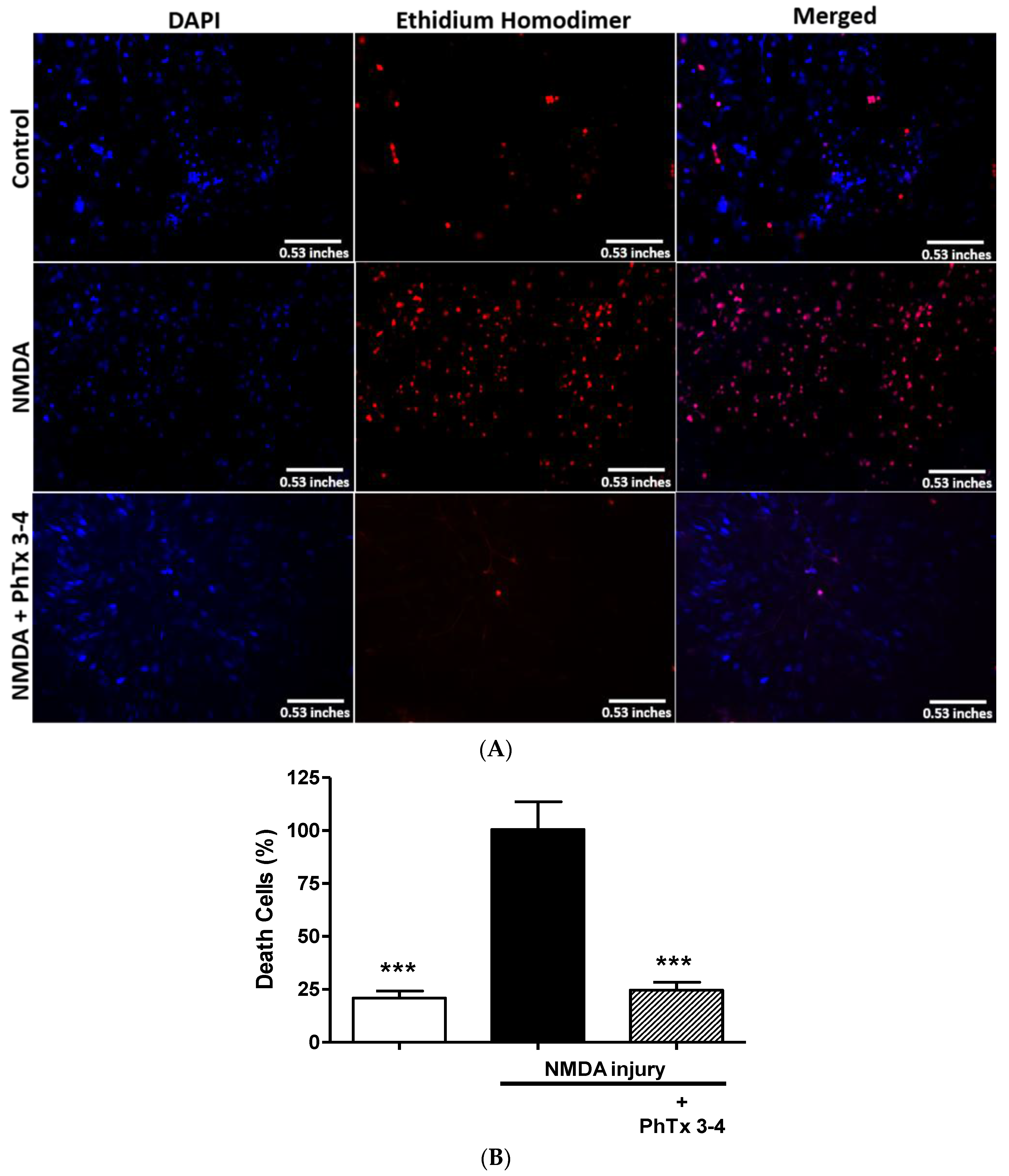
2.2. PhTx3-4 Causes Cell Protection of NMDA-Induced Injury of the Retina
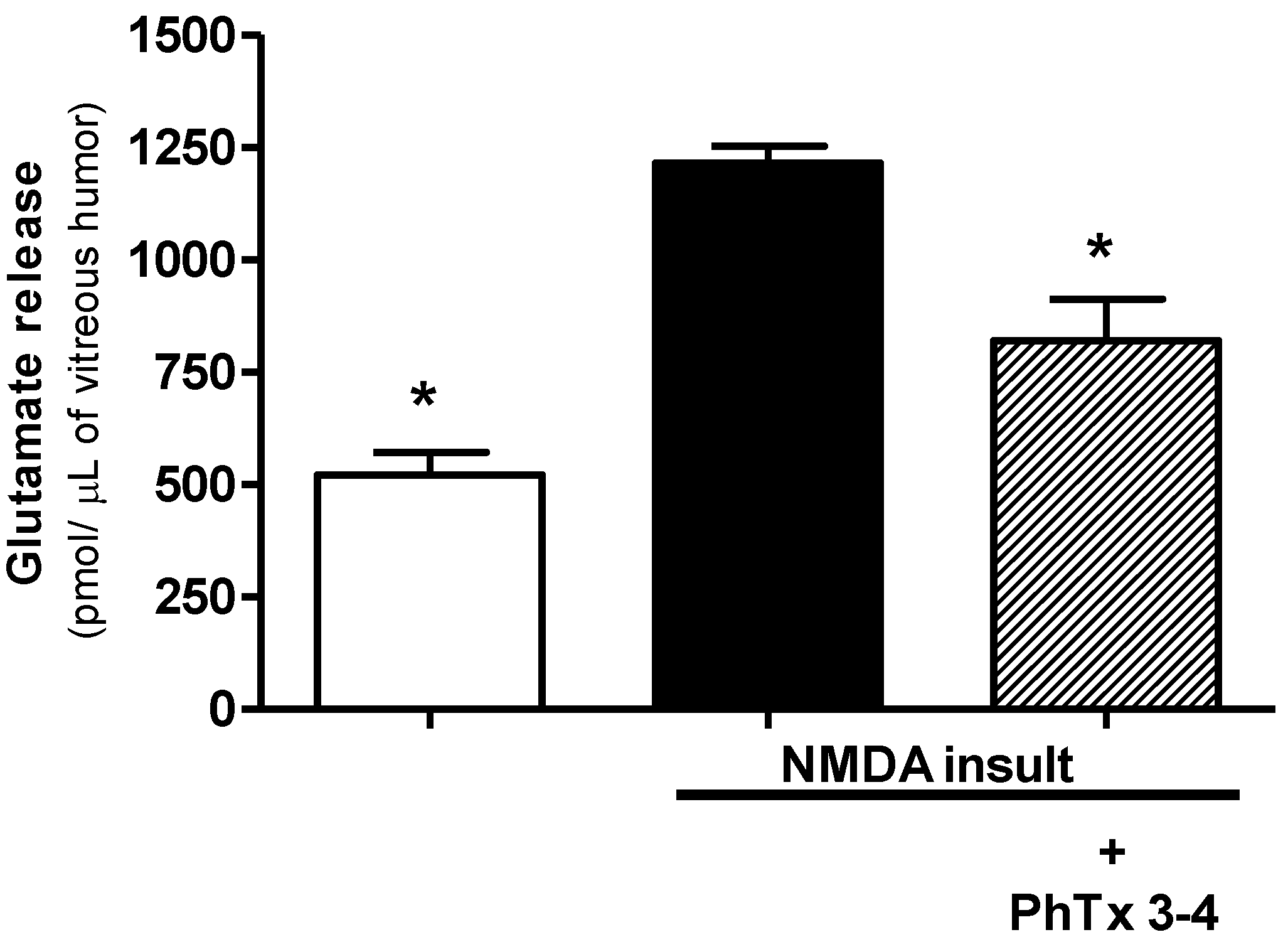
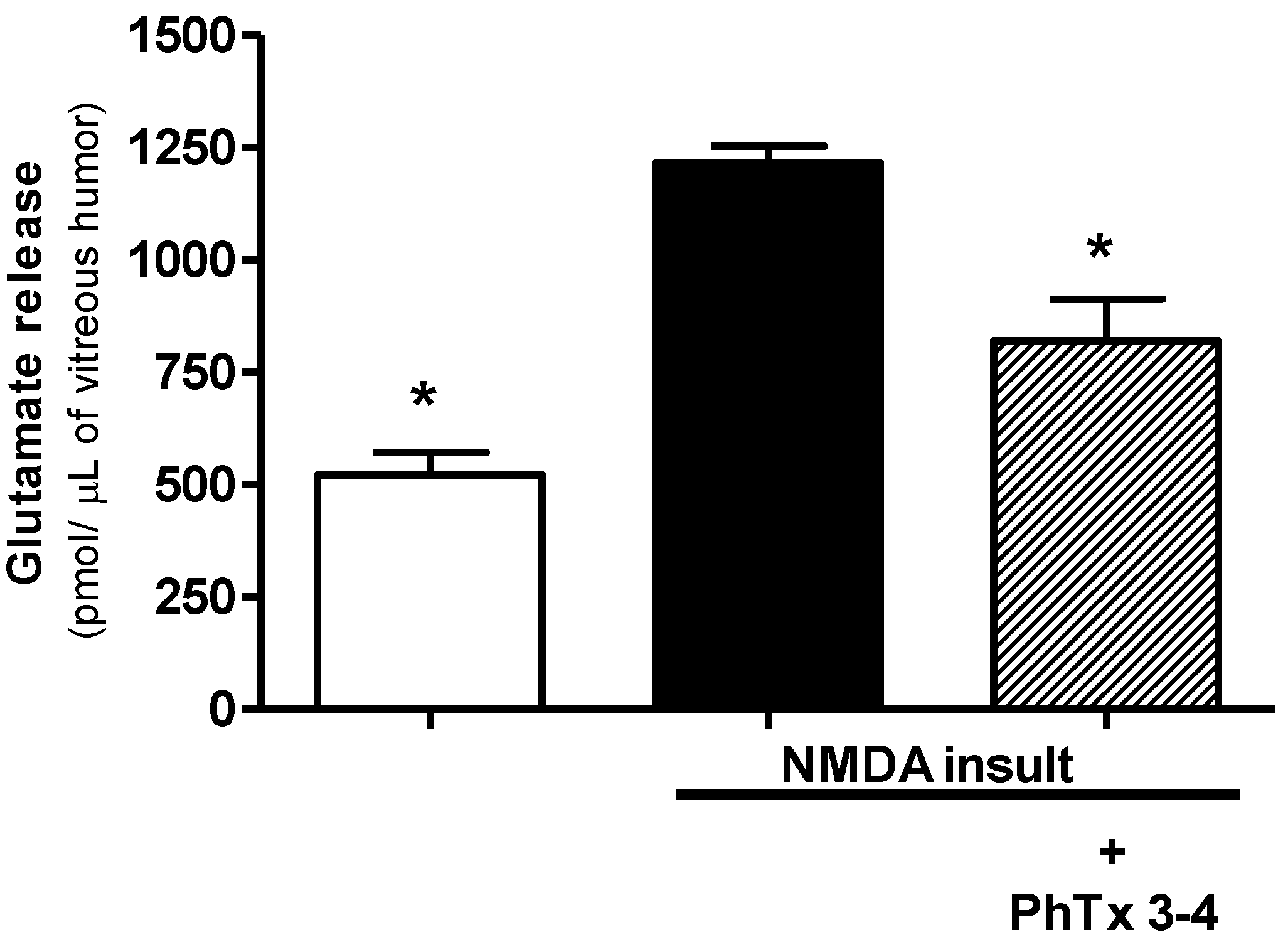
2.3. PhTx3-4 Treatment Reduces Glutamate Release in NMDA-Induced Injury of the Retinas
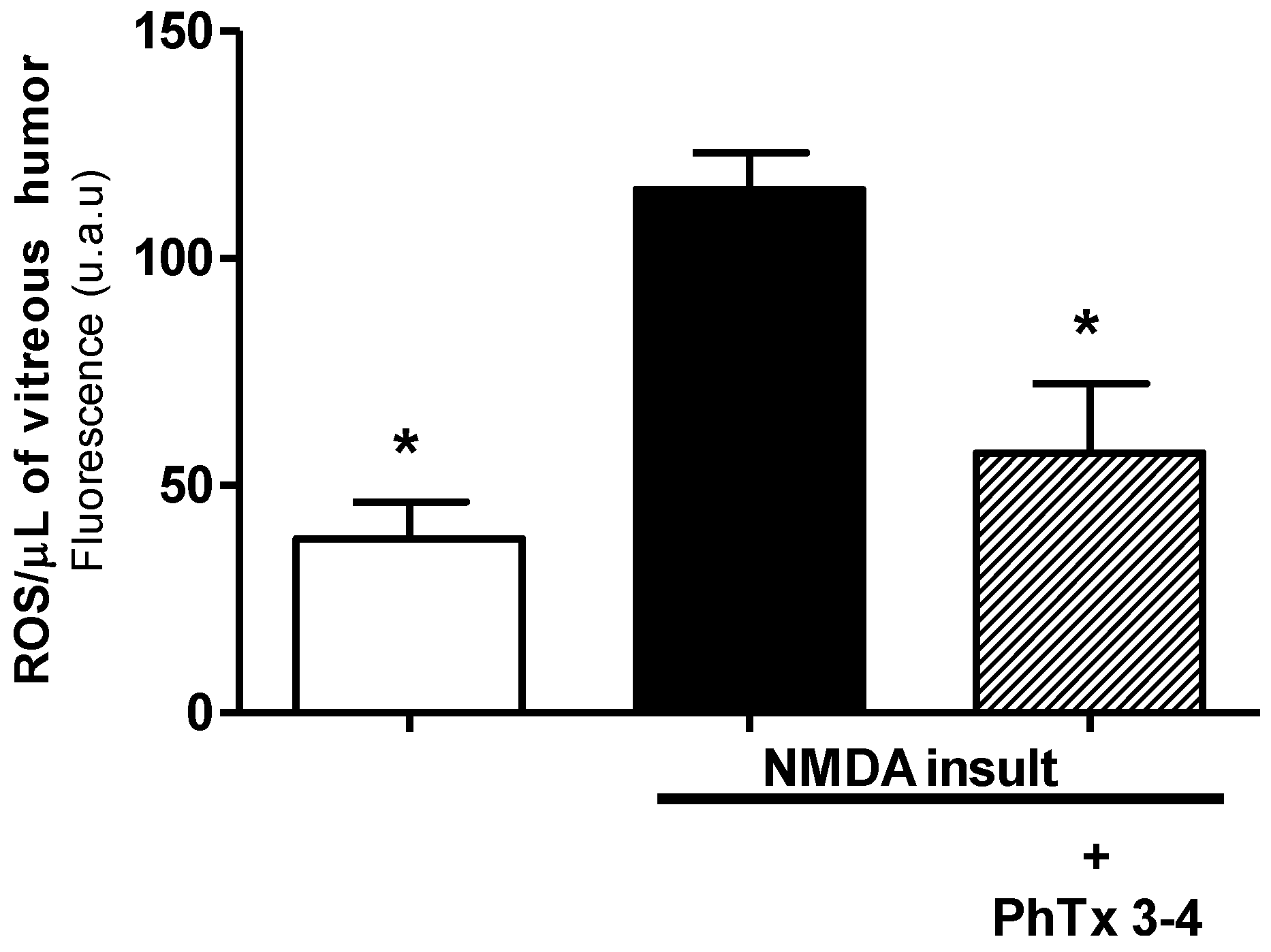
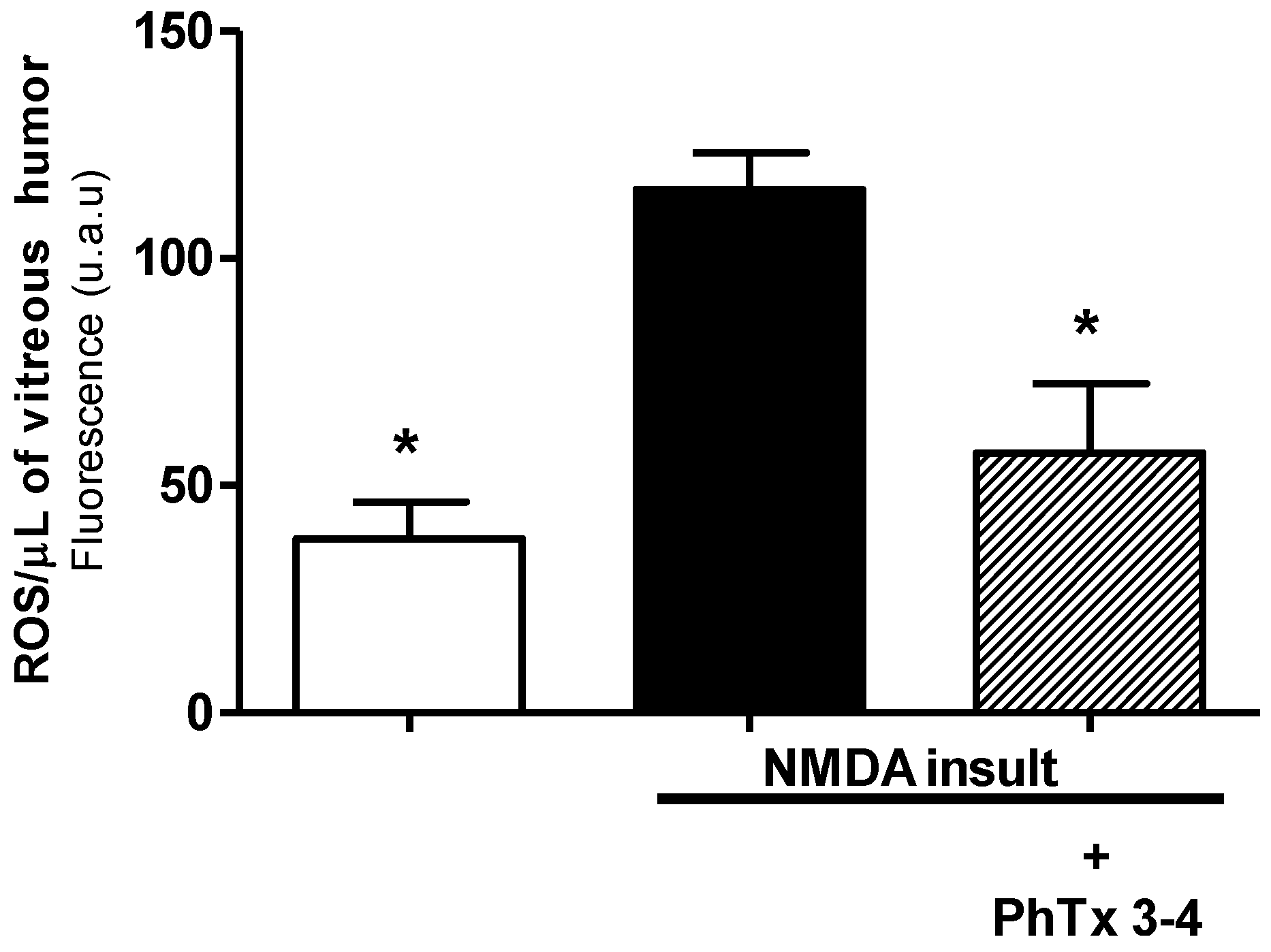
2.4. PhTx3.4 Treatment Reduces ROS in NMDA-Induced Injury of the Retinas
2.5. PhTx3-4 Treatment Reduces Oxidative Stress in NMDA-Induced Injury of the Retinas
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Retinal Injury
4.3. Electroretinography
4.4. Fluorescence Microscopy and Imaging Analysis
4.5. Histopathology Study
4.6. Glutamate Assay
4.7. Free Radicals (ROS) Content of the Vitreous Humor
4.8. Lipid Peroxidation
4.9. Data Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Minhas, G.; Morishita, R.; Anand, A. Preclinical models to investigate retinal ischemia: Advances and drawbacks. Front. Neurol. 2012, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Salido, E.M.; Dorfman, D.; Bordone, M.; Chianelli, M.; Gonzalez Fleitas, M.F.; Rosenstein, R.E. Global and ocular hypothermic preconditioning protect the rat retina from ischemic damage. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Wax, M.B. Hypoxia-inducible factor 1α in the glaucomatous retina and optic nerve head. Arch. Ophthalmol. 2004, 122, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Casson, R.J.; Wood, J.P.; Chidlow, G.; Graham, M.; Melena, J. Retinal ischemia: Mechanisms of damage and potential therapeutic strategies. Prog. Retin. Eye Res. 2004, 23, 91–147. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.C. Cell types using glutamate as a neurotransmitter in the vertebrate retina. Prog. Retin. Res. 1990, 9, 399–425. [Google Scholar] [CrossRef]
- Fujita, T.; Hirooka, K.; Nakamura, T.; Itano, T.; Nishiyama, A.; Nagai, Y.; Shiraga, F. Neuroprotective effects of angiotensin II type 1 receptor (AT1-R) blocker via modulating AT1-R signaling and decreased extracellular glutamate levels. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4099–4110. [Google Scholar] [CrossRef] [PubMed]
- Vorwerk, C.K.; Lipton, S.A.; Zurakowski, D.; Hyman, B.T.; Sabel, B.A.; Dreyer, E.B. Chronic low-dose glutamate is toxic to retinal ganglion cells. Toxicity blocked by memantine. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1618–1624. [Google Scholar] [PubMed]
- Sucher, N.J.; Lei, S.Z.; Lipton, S.A. Calcium channel antagonists attenuate NMDA receptor-mediated neurotoxicity of retinal ganglion cells in culture. Brain Res. 1991, 551, 297–302. [Google Scholar] [CrossRef]
- Siesjo, B.K. Pathophysiology and treatment of focal cerebral ischemia. Part II: Mechanisms of damage and treatment. J. Neurosurg. 1992, 77, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Siesjo, B.K. Pathophysiology and treatment of focal cerebral ischemia. Part I: Pathophysiology. J. Neurosurg. 1992, 77, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.G.; van Renterghem, C.; Martin-Moutot, N.; Mansuelle, P.; Cordeiro, M.N.; Diniz, C.R.; Mori, Y.; de Lima, M.E.; Seagar, M. Phoneutria nigriventer ω-phonetoxin IIA blocks the Cav2 family of calcium channels and interacts with ω-conotoxin-binding sites. J. Biol. Chem. 2002, 277, 13856–13862. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.V.; Kalapothakis, E.; Guatimosim, C.; Prado, M.A. Phoneutria nigriventer venom: A cocktail of toxins that affect ion channels. Cell. Mol. Neurobiol. 2002, 22, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Miranda, D.M.; Romano-Silva, M.A.; Kalapothakis, E.; Diniz, C.R.; Cordeiro, M.N.; Moraes-Santos, T.; de Marco, L.; Prado, M.A.; Gomez, M.V. Spider neurotoxins block the beta scorpion toxin-induced calcium uptake in rat brain cortical synaptosomes. Brain Res. Bull. 2001, 54, 533–536. [Google Scholar] [CrossRef]
- Miranda, D.M.; Romano-Silva, M.A.; Kalapothakis, E.; Diniz, C.R.; Cordeiro, M.N.; Santos, T.M.; Prado, M.A.; Gomez, M.V. Phoneutria nigriventer toxins block tityustoxin-induced calcium influx in synaptosomes. Neuroreport 1998, 9, 1371–1373. [Google Scholar] [CrossRef] [PubMed]
- Guatimosim, C.; Romano-Silva, M.A.; Cruz, J.S.; Beirao, P.S.; Kalapothakis, E.; Moraes-Santos, T.; Cordeiro, M.N.; Diniz, C.R.; Gomez, M.V.; Prado, M.A. A toxin from the spider Phoneutria nigriventer that blocks calcium channels coupled to exocytosis. Br. J. Pharmacol. 1997, 122, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Agostini, R.M.; do Nascimento Pinheiro, A.C.; Binda, N.S.; Romano Silva, M.A.; do Nascimento Cordeiro, M.; Richardson, M.; Sena Guimaraes, A.L.; Gomez, M.V. Phoneutria spider toxins block ischemia-induced glutamate release and neuronal death of cell layers of the retina. Retina 2011, 31, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Januschowski, K.; Mueller, S.; Dollinger, R.; Schnichels, S.; Hofmann, J.; Spitzer, M.S.; Bartz-Schmidt, K.U.; Szurman, P.; Thaler, S. Investigating retinal toxicity of tempol in a model of isolated and perfused bovine retina. Graefes Arch. Clin. Exp. Ophthalmol. 2014, 252, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Coleman, K.; Fitzgerald, D.; Eustace, P.; Bouchier-Hayes, D. Electroretinography, retinal ischaemia and carotid artery disease. Eur. J. Vasc. Surg. 1990, 4, 569–573. [Google Scholar] [CrossRef]
- Louzada-Junior, P.; Dias, J.J.; Santos, W.F.; Lachat, J.J.; Bradford, H.F.; Coutinho-Netto, J. Glutamate release in experimental ischaemia of the retina: An approach using microdialysis. J. Neurochem. 1992, 59, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Vorwerk, C.K.; Zurakowski, D.; McDermott, L.M.; Mawrin, C.; Dreyer, E.B. Effects of axonal injury on ganglion cell survival and glutamate homeostasis. Brain Res. Bull. 2004, 62, 485–490. [Google Scholar] [CrossRef]
- Van Buskirk, E.M.; Cioffi, G.A. Glaucomatous optic neuropathy. Am. J. Ophthalmol. 1992, 113, 447–452. [Google Scholar] [CrossRef]
- Leske, M.C. The epidemiology of open-angle glaucoma: A review. Am. J. Epidemiol. 1983, 118, 166–191. [Google Scholar] [PubMed]
- Chen, E.; Looman, M.; Laouri, M.; Gallagher, M.; van Nuys, K.; Lakdawalla, D.; Fortuny, J. Burden of illness of diabetic macular edema: Literature review. Curr. Med. Res. Opin. 2010, 26, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Giuliari, G.P. Diabetic retinopathy: Current and new treatment options. Curr. Diabetes Rev. 2012, 8, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, E.B.; Zurakowski, D.; Schumer, R.A.; Podos, S.M.; Lipton, S.A. Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch Ophthalmol 1996, 114, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Sugawara, T.; Hosobe, Y.; Gotoh, T.; Kato, C.; Tazawa, Y. The effect of calcium antagonist on ischemia-reperfusion injury of the rabbit retina]. Nippon Ganka Gakkai Zasshi 1996, 100, 773–776. [Google Scholar] [PubMed]
- McKinnon, S.J. Glaucoma, apoptosis, and neuroprotection. Curr. Opin. Ophthalmol. 1997, 8, 28–37. [Google Scholar] [PubMed]
- Block, F.; Schwarz, M. The b-wave of the electroretinogram as an index of retinal ischemia. Gen. Pharmacol. 1998, 30, 281–287. [Google Scholar] [CrossRef]
- Fishman, G.A. Eletrophysiologic Testing in Disorders of the Retina, Optic Nerve and Visual Pathway; Amercian Academy of Ophthalmology: San Francisco, CA, USA, 1990. [Google Scholar]
- Tse, D.Y.; Chung, I.; Wu, S.M. Pharmacological inhibitions of glutamate transporters EAAT1 and EAAT2 compromise glutamate transport in photoreceptor to on-bipolar cell synapses. Vision Res. 2014, 103, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.R.; Newhouse, J.P. The toxic effect of sodium l-glutamate on the inner layers of the retina. AMA Arch. Ophthalmol. 1957, 58, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Lindsey, J.D.; Angert, M.; Patel, A.; Weinreb, R.N. Glutamate receptor activation triggers OPA1 release and induces apoptotic cell death in ischemic rat retina. Mol. Vis. 2008, 14, 2629–2638. [Google Scholar] [PubMed]
- Sattler, R.; Tymianski, M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol. Neurobiol. 2001, 24, 107–129. [Google Scholar] [CrossRef]
- Choi, D.W. Calcium-mediated neurotoxicity: Relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988, 11, 465–469. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef]
- Goto, W.; Ota, T.; Morikawa, N.; Otori, Y.; Hara, H.; Kawazu, K.; Miyawaki, N.; Tano, Y. Protective effects of timolol against the neuronal damage induced by glutamate and ischemia in the rat retina. Brain Res. 2002, 958, 10–19. [Google Scholar] [CrossRef]
- Xu, H.P.; Zhao, J.W.; Yang, X.L. Expression of voltage-dependent calcium channel subunits in the rat retina. Neurosci. Lett. 2002, 329, 297–300. [Google Scholar] [CrossRef]
- Schmid, S.; Guenther, E. Voltage-activated calcium currents in rat retinal ganglion cells in situ: Changes during prenatal and postnatal development. J. Neurosci. 1999, 19, 3486–3494. [Google Scholar] [PubMed]
- Thoreson, W.B.; Witkovsky, P. Glutamate receptors and circuits in the vertebrate retina. Prog. Retin. Eye Res. 1999, 18, 765–810. [Google Scholar] [CrossRef]
- Taschenberger, H.; Grantyn, R. Several types of Ca2+ channels mediate glutamatergic synaptic responses to activation of single thy-1-immunolabeled rat retinal ganglion neurons. J. Neurosci. 1995, 15, 2240–2254. [Google Scholar] [PubMed]
- Tamura, N.; Yokotani, K.; Okuma, Y.; Okada, M.; Ueno, H.; Osumi, Y. Properties of the voltage-gated calcium channels mediating dopamine and acetylcholine release from the isolated rat retina. Brain Res. 1995, 676, 363–370. [Google Scholar] [CrossRef]
- Reis, H.J.; Gomez, M.V.; Kalapothakis, E.; Diniz, C.R.; Cordeiro, M.N.; Prado, M.A.; Romano-Silva, M.A. Inhibition of glutamate uptake by Tx3-4 is dependent on the redox state of cysteine residues. Neuroreport 2000, 11, 2191–2194. [Google Scholar] [CrossRef] [PubMed]
- Reis, H.J.; Prado, M.A.; Kalapothakis, E.; Cordeiro, M.N.; Diniz, C.R.; de Marco, L.A.; Gomez, M.V.; Romano-Silva, M.A. Inhibition of glutamate uptake by a polypeptide toxin (phoneutriatoxin 3-4) from the spider Phoneutria nigriventer. Biochem. J. 1999, 343, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Vallazza-Deschamps, G.; Fuchs, C.; Cia, D.; Tessier, L.H.; Sahel, J.A.; Dreyfus, H.; Picaud, S. Diltiazem-induced neuroprotection in glutamate excitotoxicity and ischemic insult of retinal neurons. Doc. Ophthalmol. 2005, 110, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G. The metabolism of ω-3 polyunsaturated fatty acids in the eye: The possible role of docosahexaenoic acid and docosanoids in retinal physiology and ocular pathology. Prog. Clin. Biol. Res. 1989, 312, 95–112. [Google Scholar] [PubMed]
- Pellegrini-Giampietro, D.E.; Cherici, G.; Alesiani, M.; Carla, V.; Moroni, F. Excitatory amino acid release and free radical formation may cooperate in the genesis of ischemia-induced neuronal damage. J. Neurosci. 1990, 10, 1035–1041. [Google Scholar] [PubMed]
- Li, S.Y.; Fu, Z.J.; Ma, H.; Jang, W.C.; So, K.F.; Wong, D.; Lo, A.C. Effect of lutein on retinal neurons and oxidative stress in a model of acute retinal ischemia/reperfusion. Investig. Ophthalmol. Vis. Sci. 2009, 50, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro Mdo, N.; de Figueiredo, S.G.; Valentim Ado, C.; Diniz, C.R.; von Eickstedt, V.R.; Gilroy, J.; Richardson, M. Purification and amino acid sequences of six Tx3 type neurotoxins from the venom of the brazilian “armed” spider Phoneutria nigriventer (keys). Toxicon 1993, 31, 35–42. [Google Scholar] [CrossRef]
- Richardson, M.; Pimenta, A.M.; Bemquerer, M.P.; Santoro, M.M.; Beirao, P.S.; Lima, M.E.; Figueiredo, S.G.; Bloch, C., Jr.; Vasconcelos, E.A.; Campos, F.A.; et al. Comparison of the partial proteomes of the venoms of brazilian spiders of the genus phoneutria. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2006, 142, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Batista, C.V.; del Pozo, L.; Zamudio, F.Z.; Contreras, S.; Becerril, B.; Wanke, E.; Possani, L.D. Proteomics of the venom from the amazonian scorpion tityus cambridgei and the role of prolines on mass spectrometry analysis of toxins. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 803, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Nakahara, T.; Hoshino, M.; Mori, A.; Sakamoto, K.; Ishii, K. Retinal blood vessels are damaged in a rat model of NMDA-induced retinal degeneration. Neurosci. Lett. 2010, 485, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Bessero, A.C.; Chiodini, F.; Rungger-Brandle, E.; Bonny, C.; Clarke, P.G. Role of the c-jun N-terminal kinase pathway in retinal excitotoxicity, and neuroprotection by its inhibition. J. Neurochem. 2010, 113, 1307–1318. [Google Scholar] [PubMed]
- Pinheiro, A.C.; Gomez, R.S.; Massensini, A.R.; Cordeiro, M.N.; Richardson, M.; Romano-Silva, M.A.; Prado, M.A.; de Marco, L.; Gomez, M.V. Neuroprotective effect on brain injury by neurotoxins from the spider Phoneutria nigriventer. Neurochem. Int. 2006, 49, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Sihra, T.S.; Sanchez-Prieto, J. Calcium-dependent and -independent release of glutamate from synaptosomes monitored by continuous fluorometry. J. Neurochem. 1987, 49, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, I.R.; Cimarosti, H.; Fochesatto, C.; Salbego, C.; Netto, C.A. Age-related susceptibility to oxygen and glucose deprivation damage in rat hippocampal slices. Brain Res. 2004, 1025, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Binda, N.S.; Porto Petruceli Carayon, C.; Agostini, R.M.; Do Nascimento Pinheiro, A.C.; Nascimento Cordeiro, M.; Romano Silva, M.A.; Figueira Silva, J.; Rita Pereira, E.M.; Da Silva Junior, C.A.; De Castro Junior, C.J.; et al. PhTx3-4, a Spider Toxin Calcium Channel Blocker, Reduces NMDA-Induced Injury of the Retina. Toxins 2016, 8, 70. https://doi.org/10.3390/toxins8030070
Binda NS, Porto Petruceli Carayon C, Agostini RM, Do Nascimento Pinheiro AC, Nascimento Cordeiro M, Romano Silva MA, Figueira Silva J, Rita Pereira EM, Da Silva Junior CA, De Castro Junior CJ, et al. PhTx3-4, a Spider Toxin Calcium Channel Blocker, Reduces NMDA-Induced Injury of the Retina. Toxins. 2016; 8(3):70. https://doi.org/10.3390/toxins8030070
Chicago/Turabian StyleBinda, Nancy Scardua, Charles Porto Petruceli Carayon, Rafael Mourão Agostini, Ana Cristina Do Nascimento Pinheiro, Marta Nascimento Cordeiro, Marco Aurélio Romano Silva, Juliana Figueira Silva, Elizete Maria Rita Pereira, Claudio Antonio Da Silva Junior, Célio José De Castro Junior, and et al. 2016. "PhTx3-4, a Spider Toxin Calcium Channel Blocker, Reduces NMDA-Induced Injury of the Retina" Toxins 8, no. 3: 70. https://doi.org/10.3390/toxins8030070
APA StyleBinda, N. S., Porto Petruceli Carayon, C., Agostini, R. M., Do Nascimento Pinheiro, A. C., Nascimento Cordeiro, M., Romano Silva, M. A., Figueira Silva, J., Rita Pereira, E. M., Da Silva Junior, C. A., De Castro Junior, C. J., Sena Guimarães, A. L., & Gomez, M. V. (2016). PhTx3-4, a Spider Toxin Calcium Channel Blocker, Reduces NMDA-Induced Injury of the Retina. Toxins, 8(3), 70. https://doi.org/10.3390/toxins8030070