
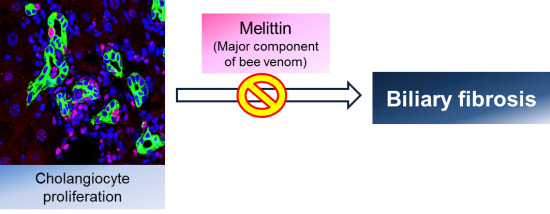
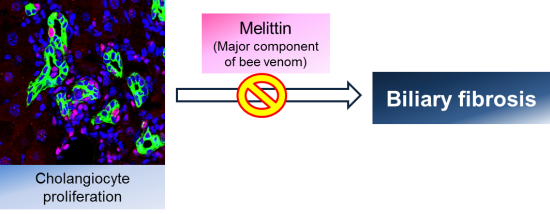
Effects of Melittin Treatment in Cholangitis and Biliary Fibrosis in a Model of Xenobiotic-Induced Cholestasis in Mice

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
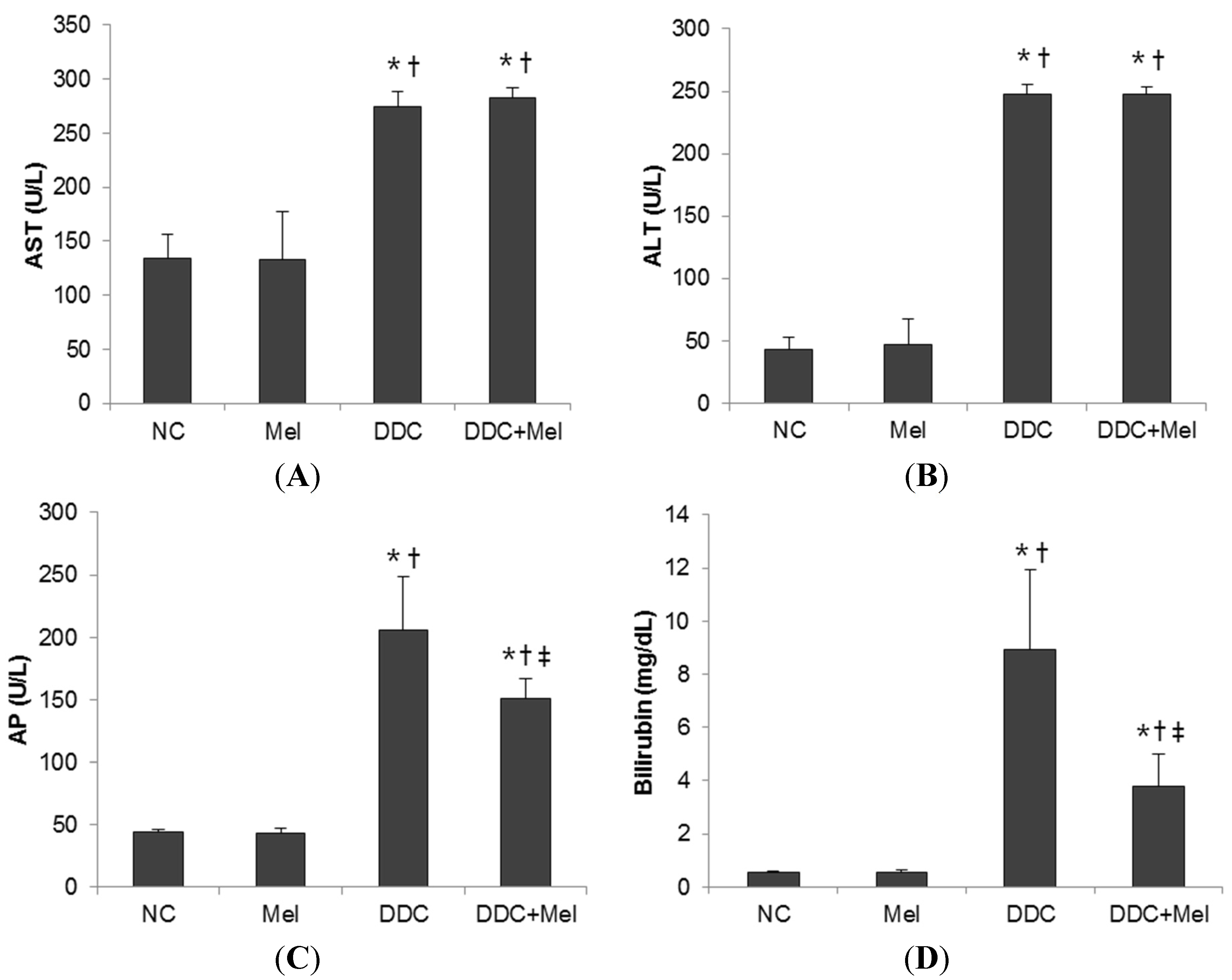
2.1. Effects of Melittin on DDC-Fed Mice
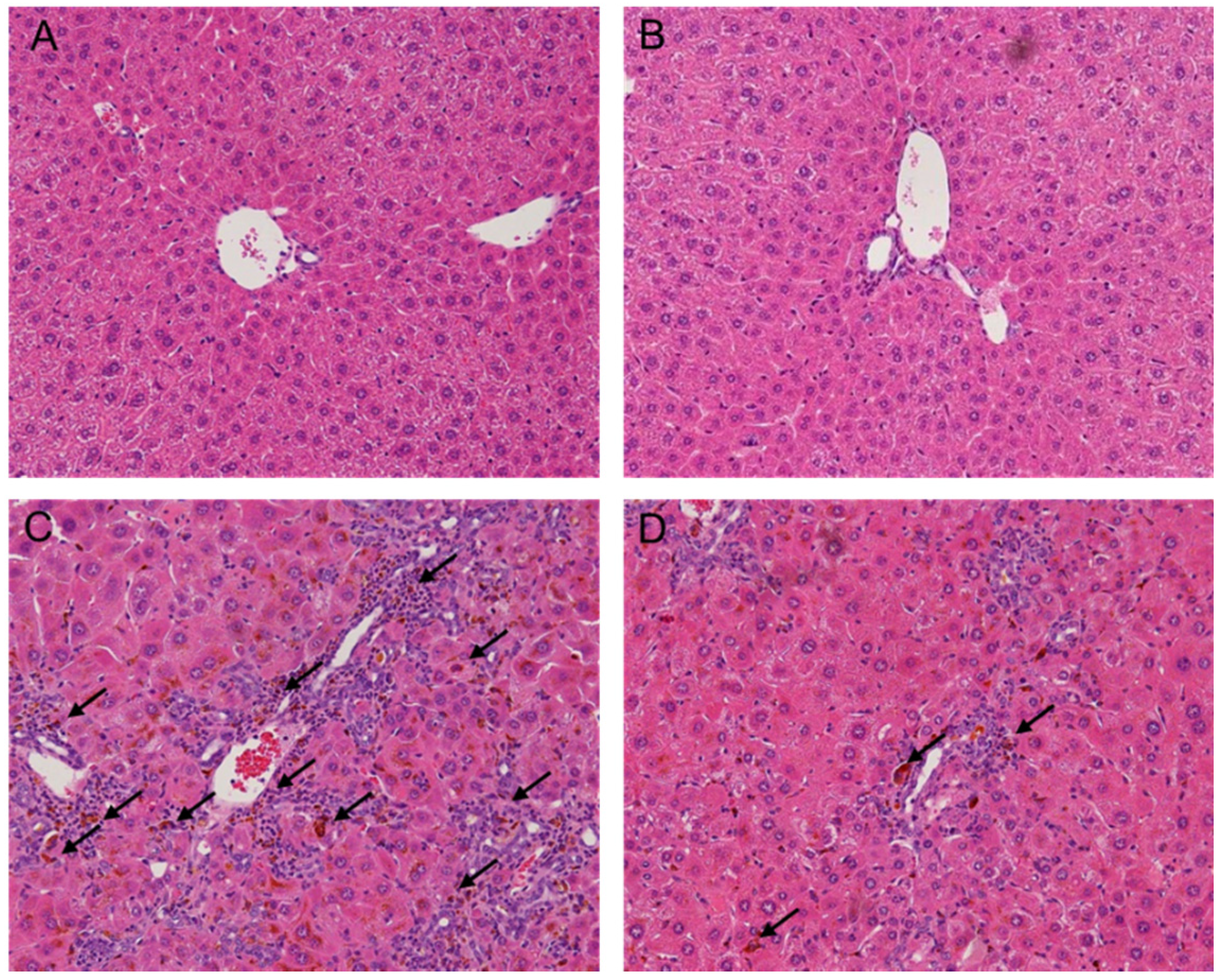
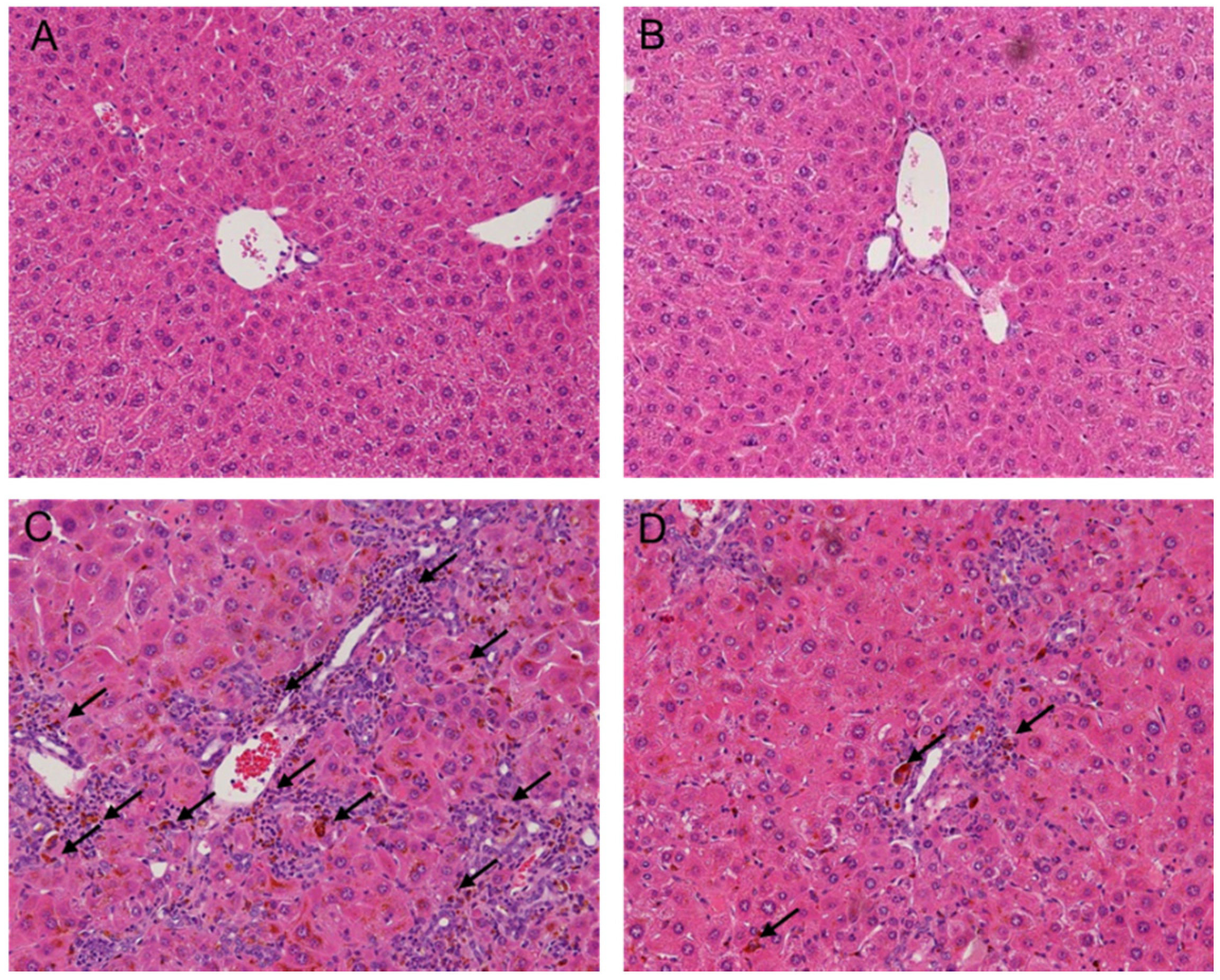
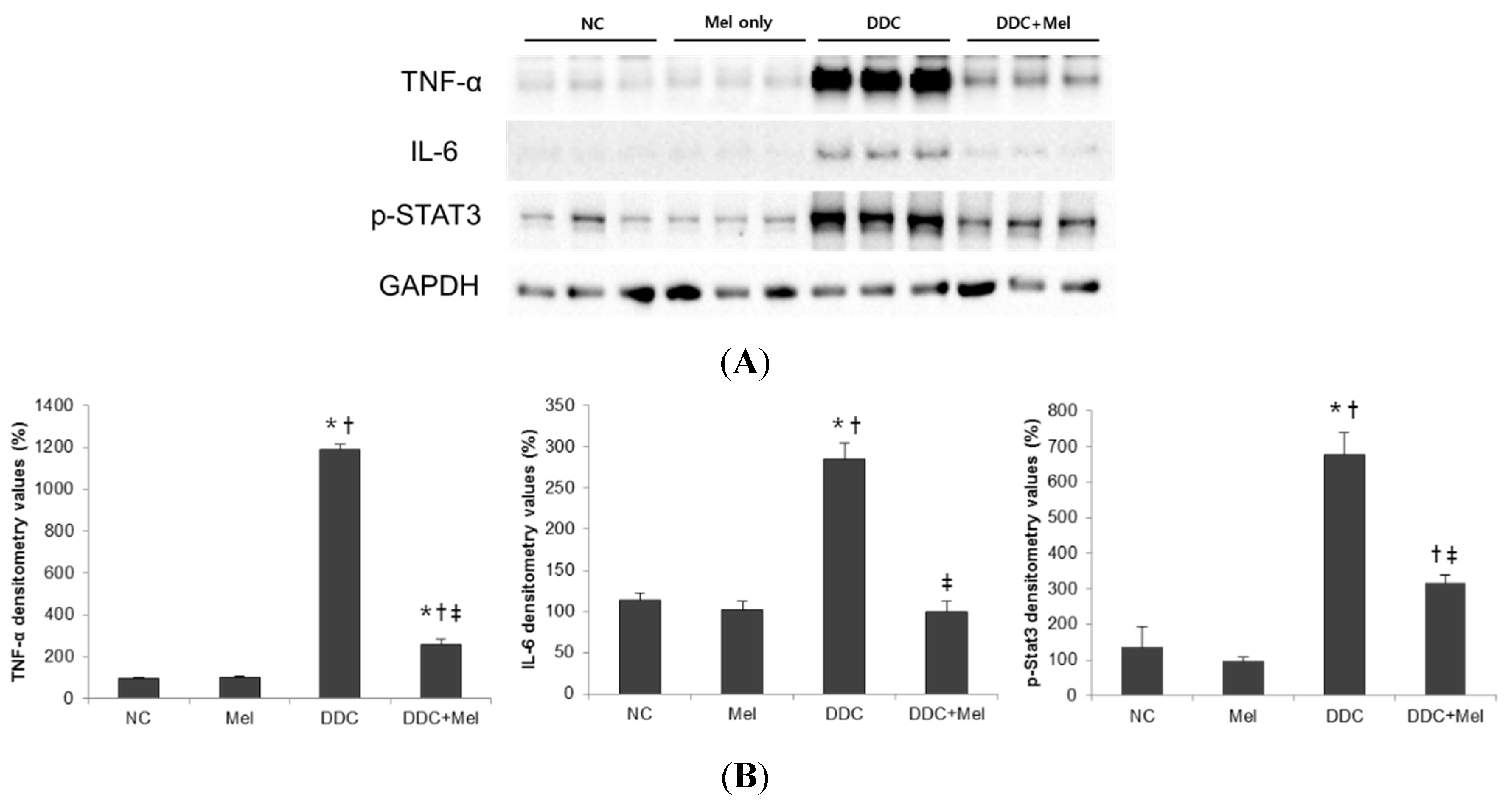
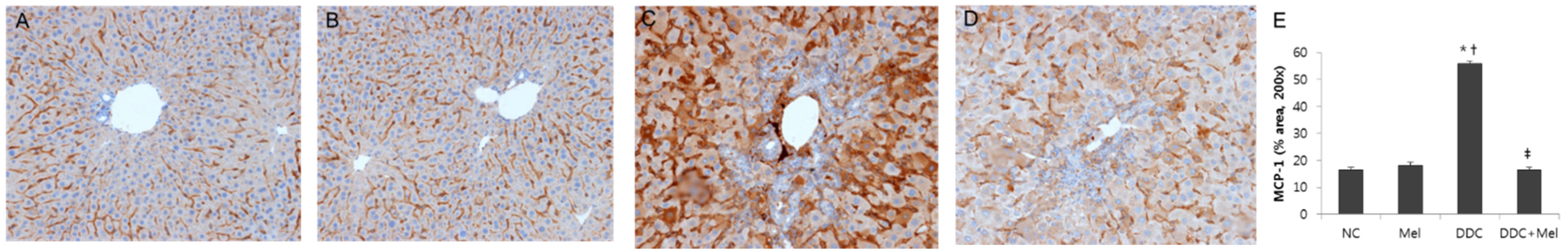
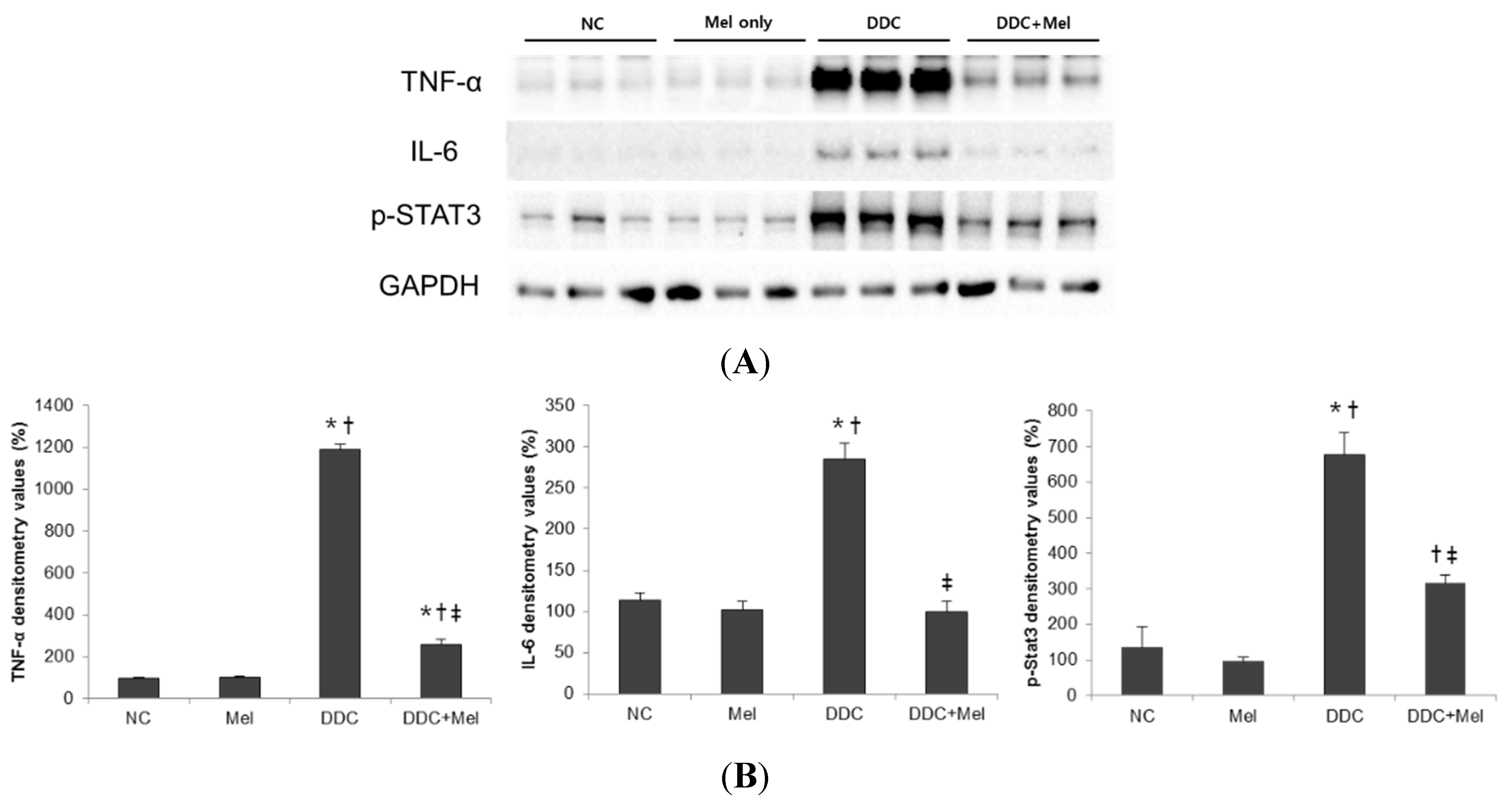
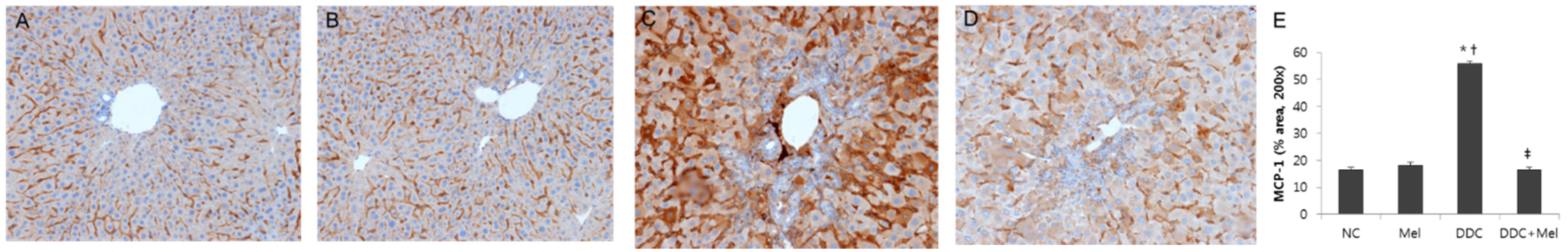
2.2. Melittin Treatment Attenuates DDC-Induced Inflammatory Changes

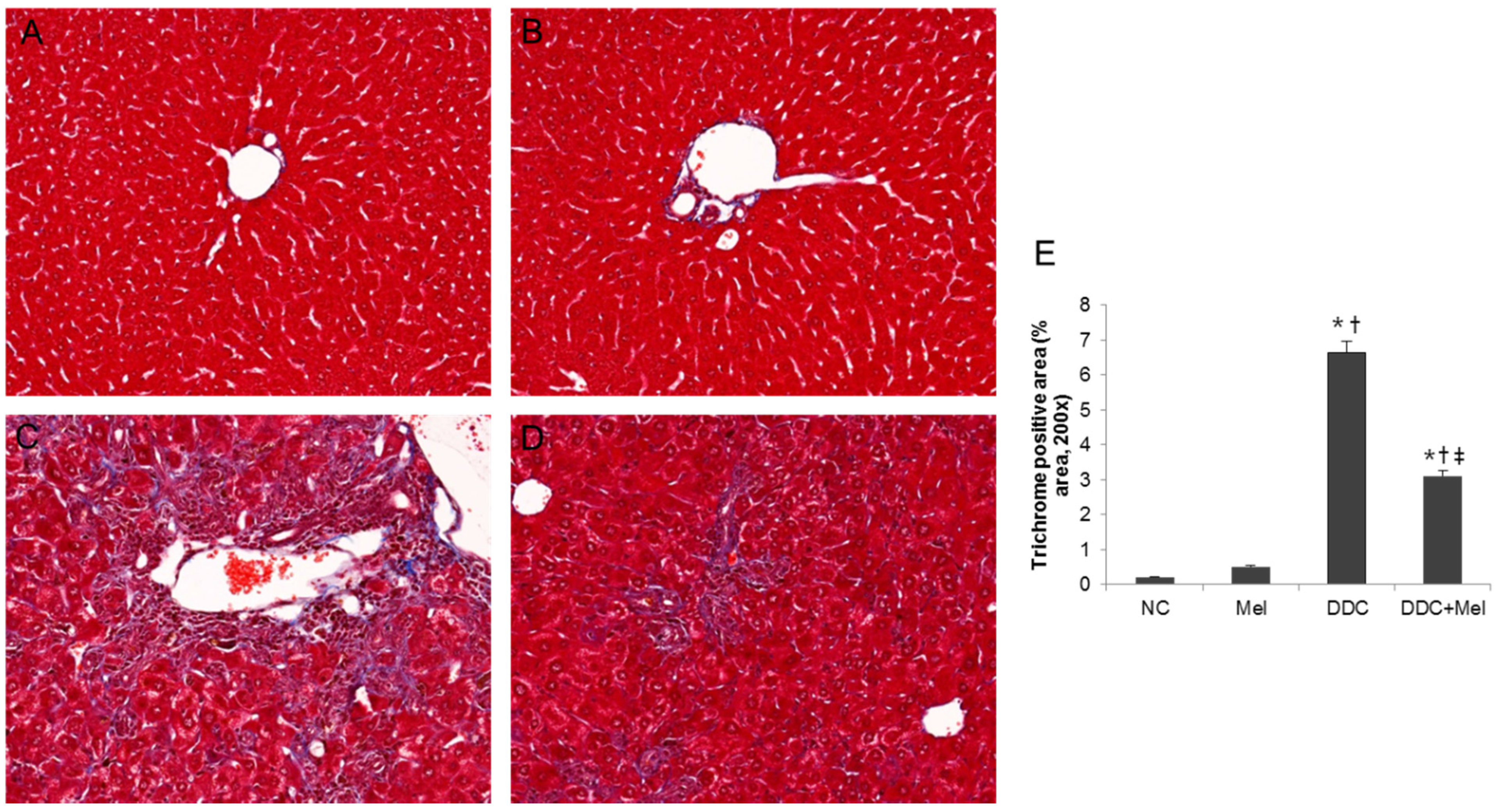
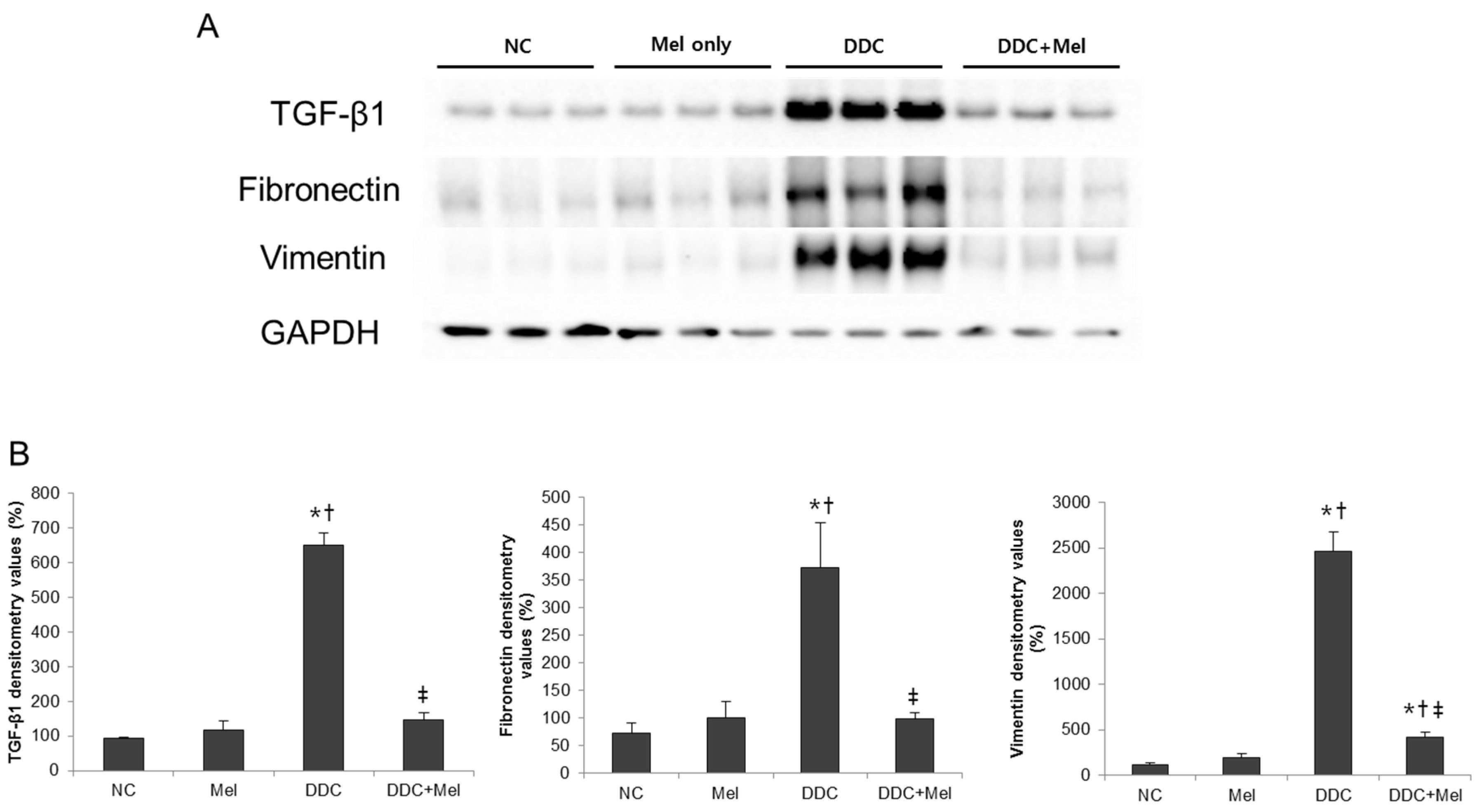
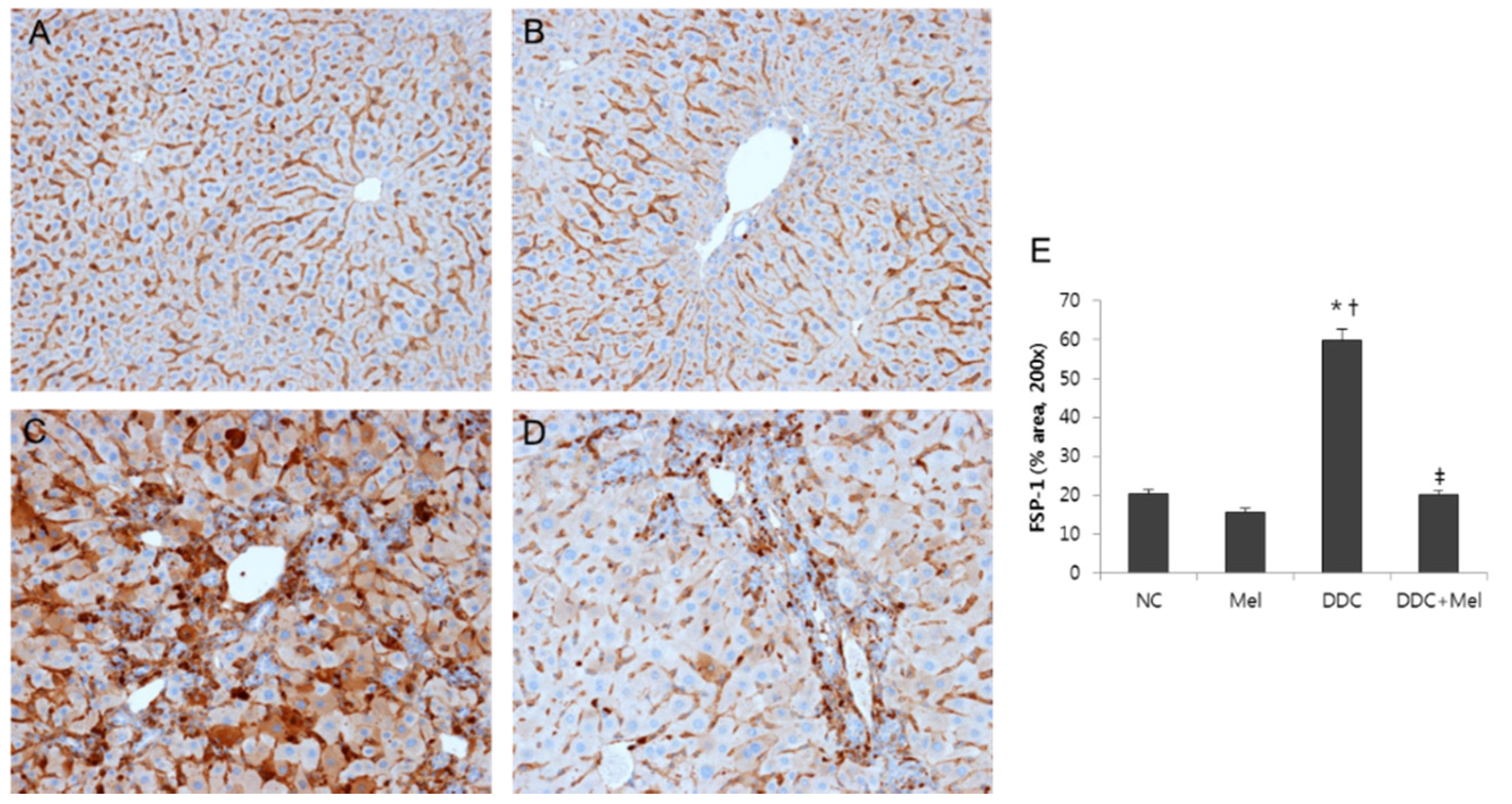
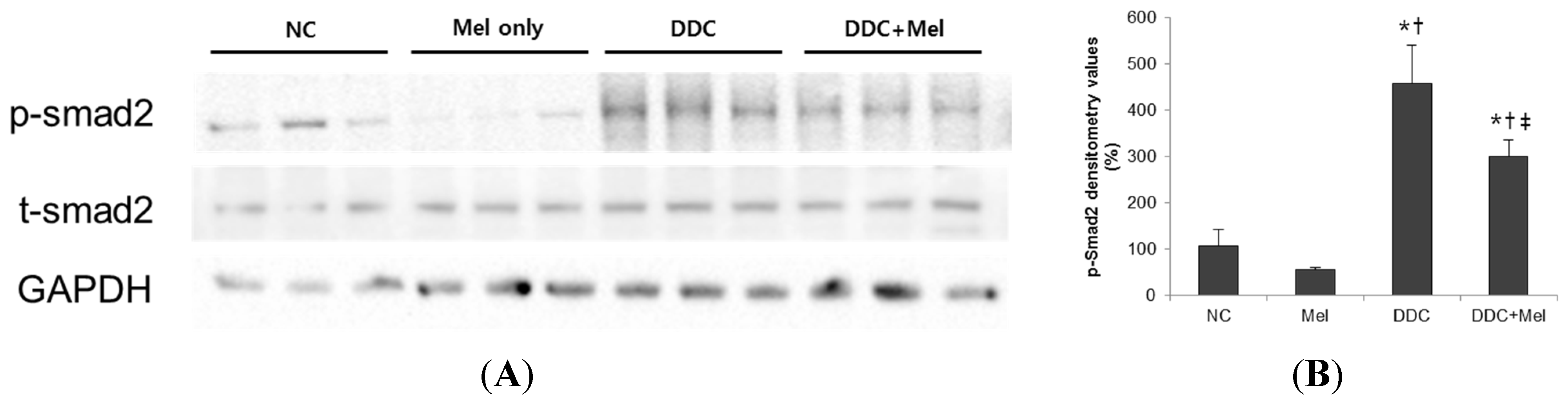
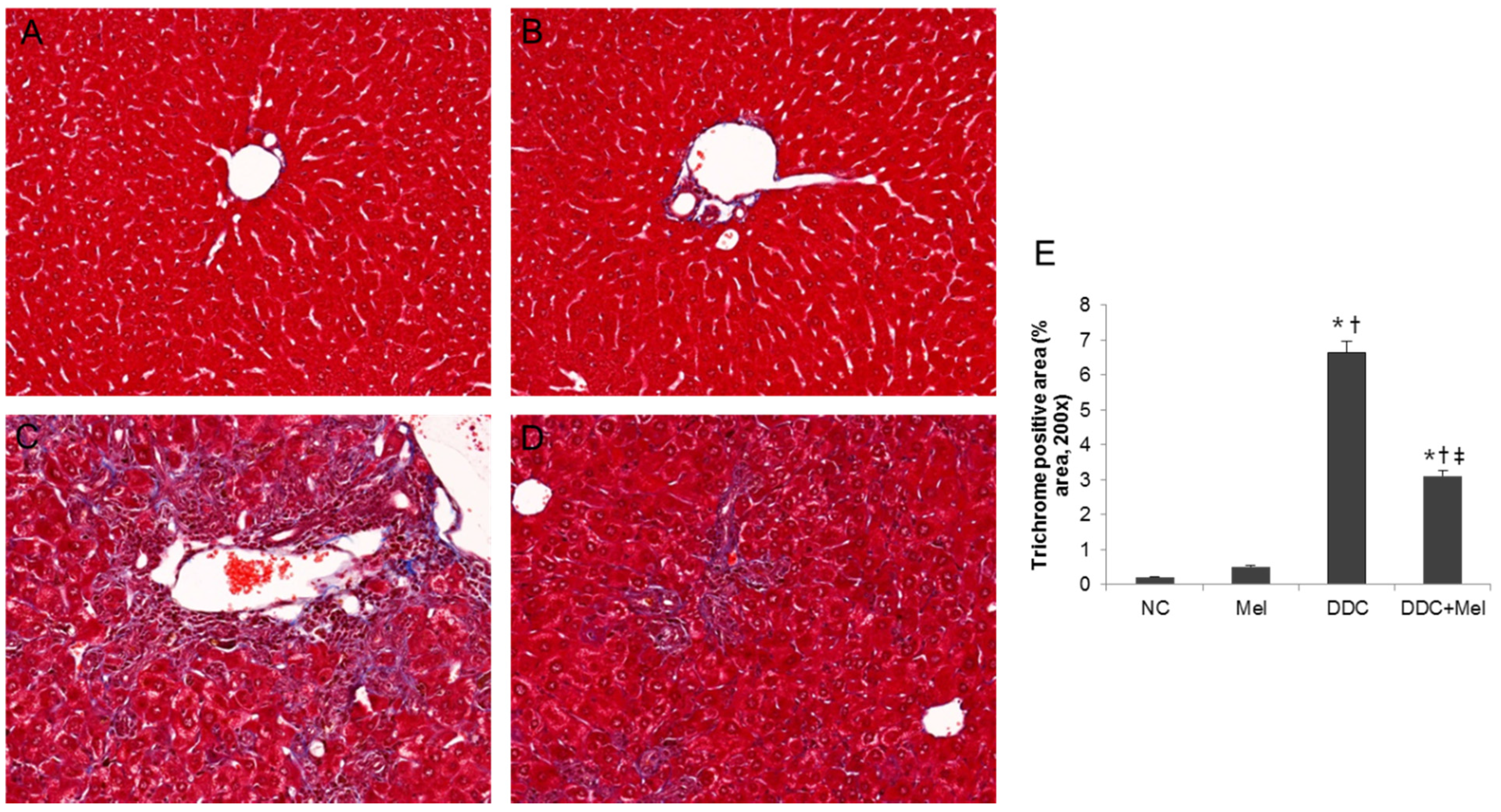
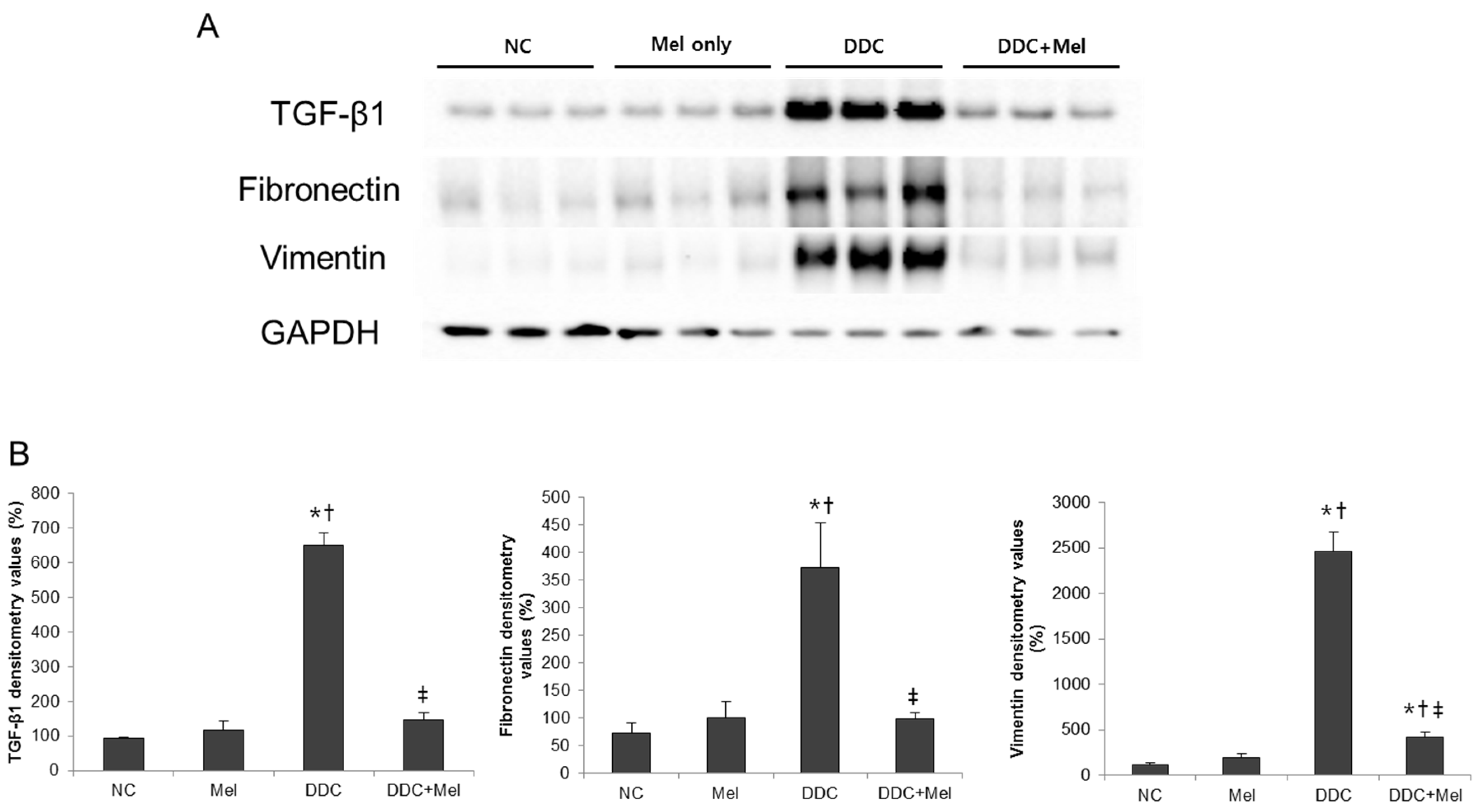
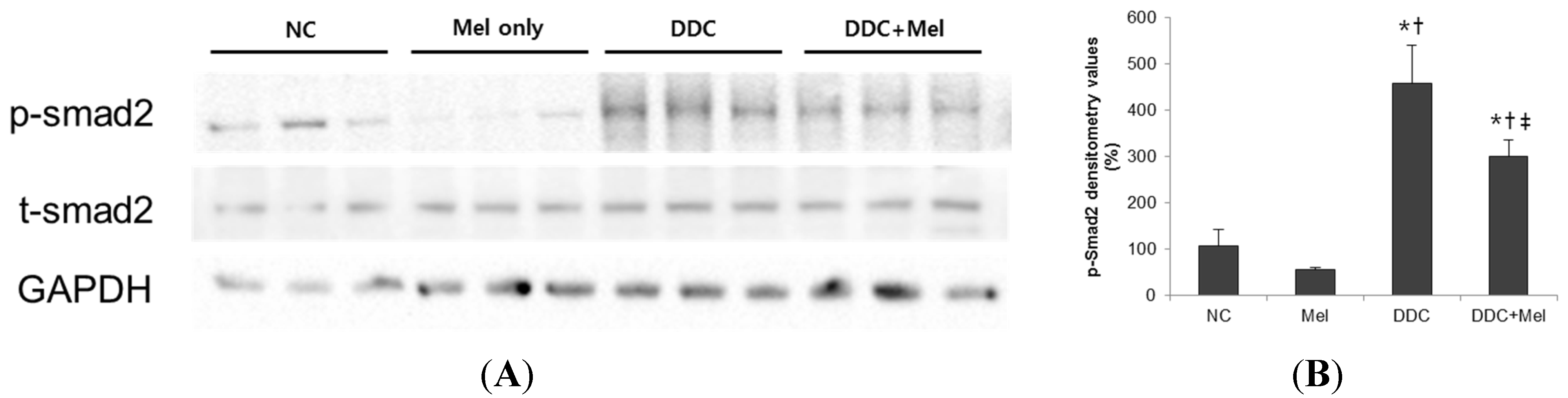
2.3. Melittin Suppresses Liver Fibrosis in the Livers of DDC-Fed Mice
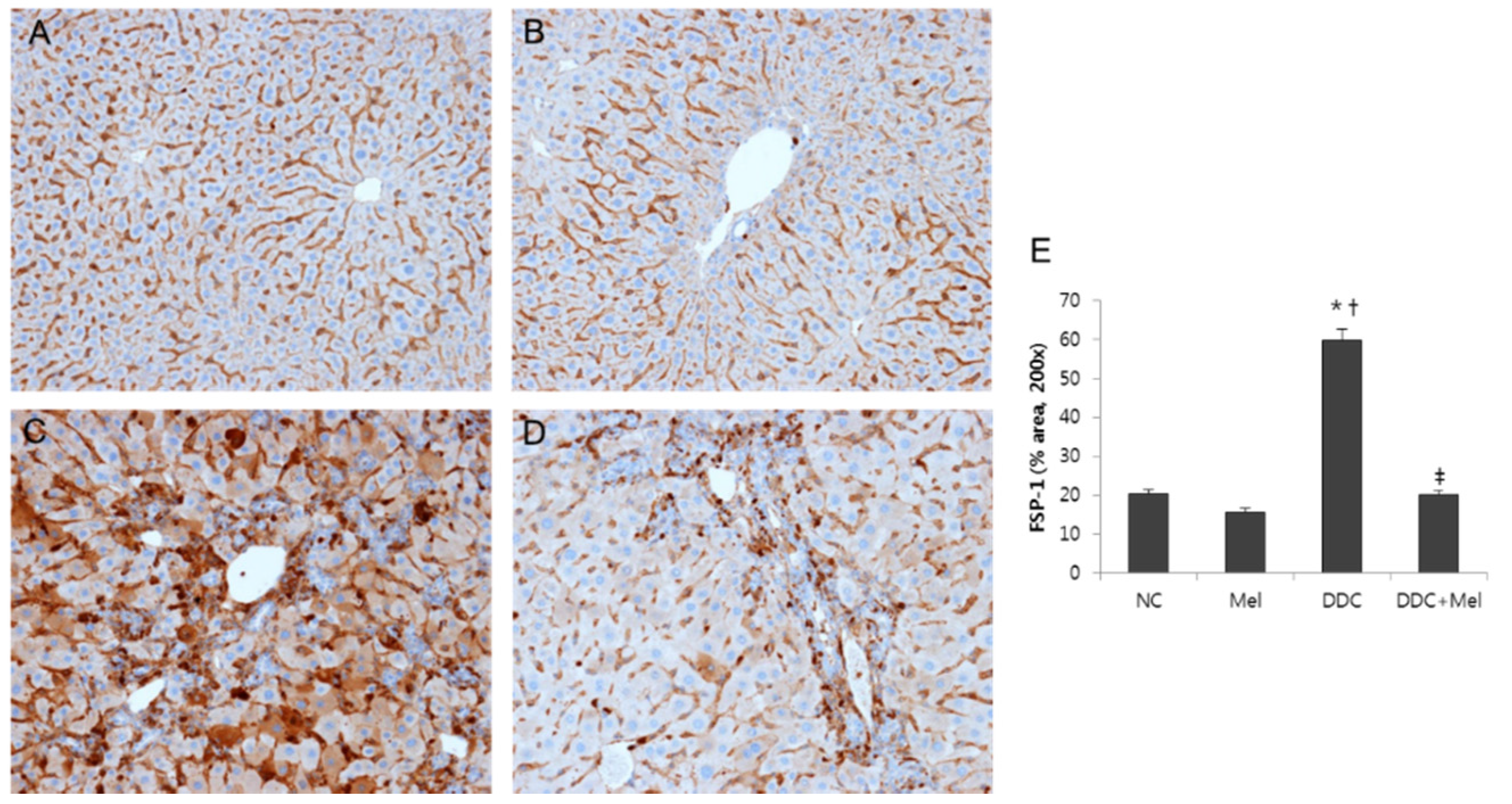
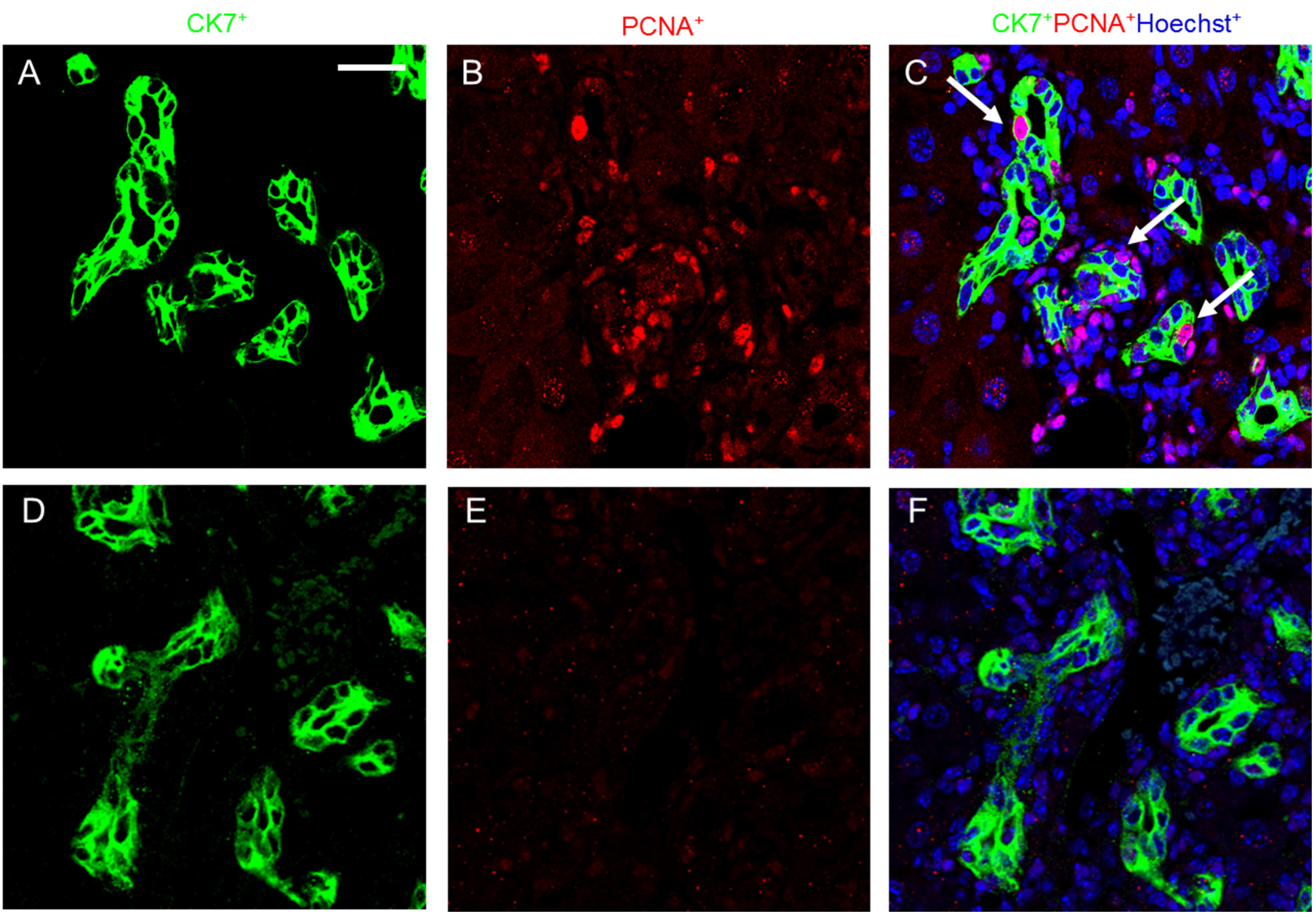
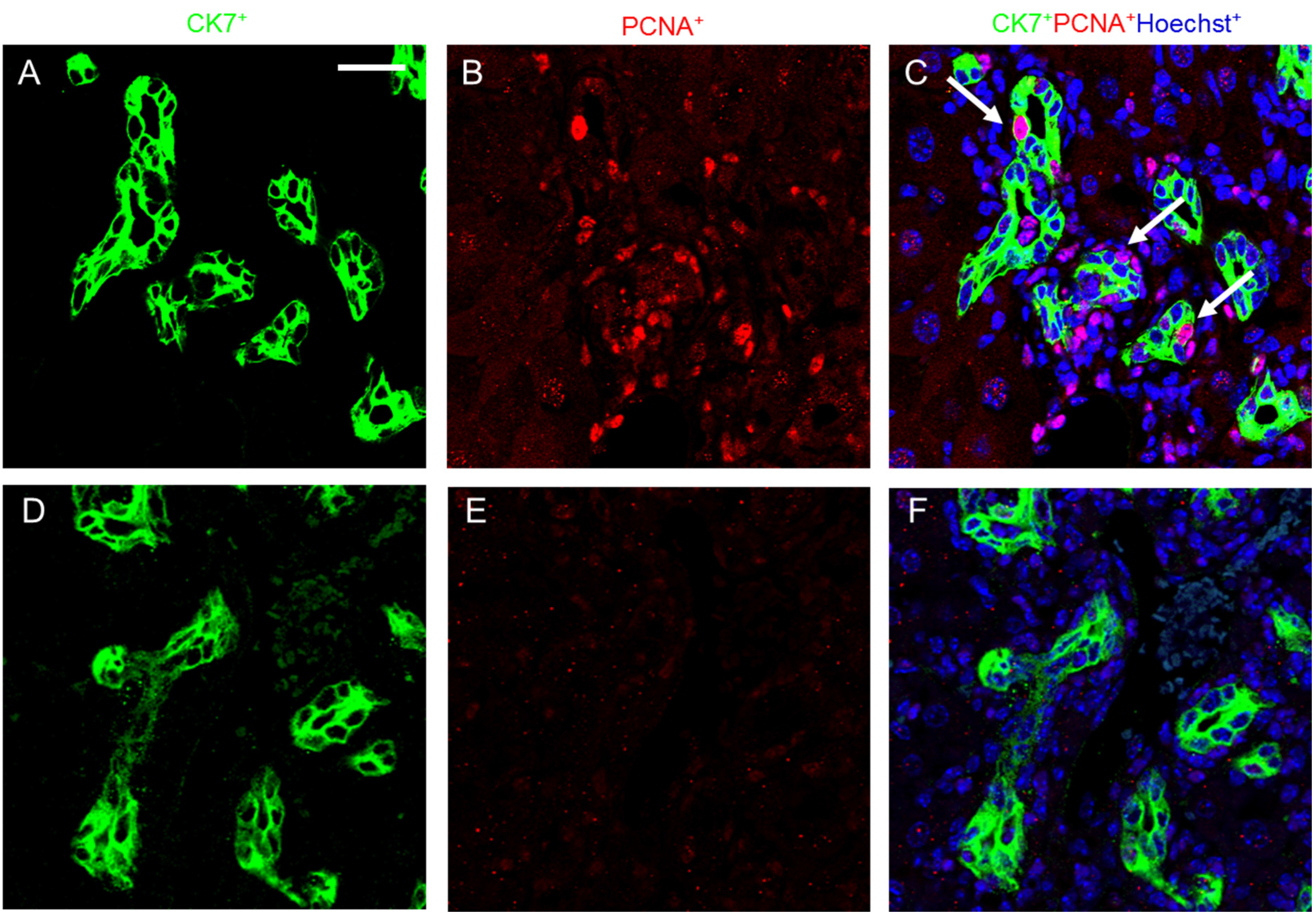
2.4. Melittin Effectively Suppresses Proliferating Cholangiocytes in DDC-Fed Mice
3. Discussion
4. Experimental Section
4.1. Animal Model
4.2. Histopathology and Immunohistochemistry
4.3. Serum Biochemical Analysis
4.4. Western Blot Analysis
4.5. Immunofluorescence Staining and Confocal Microscopy
4.6. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lazaridis, K.N.; Strazzabosco, M.; Larusso, N.F. The cholangiopathies: Disorders of biliary epithelia. Gastroenterology 2004, 127, 1565–1577. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.; Gores, G.J. Apoptosis and hepatobiliary disease. Hepatology 1995, 21, 1725–1741. [Google Scholar] [PubMed]
- Peters, M.G. Pathogenesis of primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune cholangiopathy. Clin. Liver Dis. 1998, 2, 235–247, vii–viii. [Google Scholar] [CrossRef]
- Glaser, S.S.; Gaudio, E.; Miller, T.; Alvaro, D.; Alpini, G. Cholangiocyte proliferation and liver fibrosis. Expert Rev. Mol. Med. 2009, 11, e7. [Google Scholar] [CrossRef] [PubMed]
- LeSage, G.; Glaser, S.; Alpini, G. Regulation of cholangiocyte proliferation. Liver 2001, 21, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Mancino, M.G.; Glaser, S.; Gaudio, E.; Marzioni, M.; Francis, H.; Alpini, G. Proliferating cholangiocytes: A neuroendocrine compartment in the diseased liver. Gastroenterology 2007, 132, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Sell, S. Comparison of liver progenitor cells in human atypical ductular reactions with those seen in experimental models of liver injury. Hepatology 1998, 27, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Fickert, P.; Stoger, U.; Fuchsbichler, A.; Moustafa, T.; Marschall, H.U.; Weiglein, A.H.; Tsybrovskyy, O.; Jaeschke, H.; Zatloukal, K.; Denk, H.; et al. A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am. J. Pathol. 2007, 171, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Zatloukal, K.; Stumptner, C.; Fuchsbichler, A.; Fickert, P.; Lackner, C.; Trauner, M.; Denk, H. The keratin cytoskeleton in liver diseases. J. Pathol. 2004, 204, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Habermann, E. Bee and wasp venoms. Science 1972, 177, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, C.E. The actions of melittin on membranes. Biochim. Biophys. Acta 1990, 1031, 143–161. [Google Scholar] [CrossRef]
- Jang, H.S.; Kim, S.K.; Han, J.B.; Ahn, H.J.; Bae, H.; Min, B.I. Effects of bee venom on the pro-inflammatory responses in raw264.7 macrophage cell line. J. Ethnopharmacol. 2005, 99, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Wheeler, M.D.; Kono, H.; Bradford, B.U.; Gallucci, R.M.; Luster, M.I.; Thurman, R.G. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology 1999, 117, 942–952. [Google Scholar] [CrossRef]
- Burra, P.; Hubscher, S.G.; Shaw, J.; Elias, E.; Adams, D.H. Is the intercellular adhesion molecule-1/leukocyte function associated antigen 1 pathway of leukocyte adhesion involved in the tissue damage of alcoholic hepatitis? Gut 1992, 33, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Taub, R. Liver regeneration: From myth to mechanism. Nat. Rev. Mol. Cell Biol. 2004, 5, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lafdil, F.; Kong, X.; Gao, B. Signal transducer and activator of transcription 3 in liver diseases: A novel therapeutic target. Int. J. Biol. Sci. 2011, 7, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Meindl-Beinker, N.M.; Dooley, S. Transforming growth factor-beta and hepatocyte transdifferentiation in liver fibrogenesis. J. Gastroenterol. Hepatol. 2008, 23, S122–S127. [Google Scholar] [CrossRef] [PubMed]
- Penz-Osterreicher, M.; Osterreicher, C.H.; Trauner, M. Fibrosis in autoimmune and cholestatic liver disease. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Aron, J.H.; Bowlus, C.L. The immunobiology of primary sclerosing cholangitis. Semin. Immunopathol. 2009, 31, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Aoki, C.A.; Bowlus, C.L.; Gershwin, M.E. The immunobiology of primary sclerosing cholangitis. Autoimmun. Rev. 2005, 4, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, H.; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep. 2007, 27, 189–223. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, K.H.; Kim, S.J.; Lee, W.R.; Lee, K.G.; Park, K.K. Bee venom protects hepatocytes from tumor necrosis factor-alpha and actinomycin d. Arch. Pharm. Res. 2010, 33, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C. Honeybee venom: A rich source of pharmacologically active peptides. Endeavour 1988, 12, 60–65. [Google Scholar] [CrossRef]
- Pratt, J.P.; Ravnic, D.J.; Huss, H.T.; Jiang, X.; Orozco, B.S.; Mentzer, S.J. Melittin-induced membrane permeability: A nonosmotic mechanism of cell death. Vitro Cell. Dev. Biol. Anim. 2005, 41, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Son, D.J.; Ha, S.J.; Song, H.S.; Lim, Y.; Yun, Y.P.; Lee, J.W.; Moon, D.C.; Park, Y.H.; Park, B.S.; Song, M.J.; et al. Melittin inhibits vascular smooth muscle cell proliferation through induction of apoptosis via suppression of nuclear factor-kappab and akt activation and enhancement of apoptotic protein expression. J. Pharmacol. Exp. Ther. 2006, 317, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.R.; Kim, K.H.; An, H.J.; Kim, J.Y.; Chang, Y.C.; Chung, H.; Park, Y.Y.; Lee, M.L.; Park, K.K. The protective effects of melittin on propionibacterium acnes-induced inflammatory responses in vitro and in vivo. J. Investig. Dermatol. 2014, 134, 1922–1930. [Google Scholar] [CrossRef] [PubMed]
- Osterreicher, C.H.; Trauner, M. Xenobiotic-induced liver injury and fibrosis. Expert Opin. Drug Metab. Toxicol. 2012, 8, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.E. Special considerations in interpreting liver function tests. Am. Family Phys. 1999, 59, 2223–2230. [Google Scholar]
- European Association for the Study of the Liver. Easl clinical practice guidelines: Management of cholestatic liver diseases. J. Hepatol. 2009, 51, 237–267. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kum, Y.S.; Lee, T.I.; Kim, S.J.; Lee, W.R.; Kim, B.I.; Kim, H.S.; Kim, K.H.; Park, K.K. Melittin attenuates liver injury in thioacetamide-treated mice through modulating inflammation and fibrogenesis. Exp. Biol. Med. (Maywood) 2011, 236, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Afford, S.C. The role of cholangiocytes in the development of chronic inflammatory liver disease. Front. Biosci. 2002, 7, e276–e285. [Google Scholar] [PubMed]
- Mair, M.; Zollner, G.; Schneller, D.; Musteanu, M.; Fickert, P.; Gumhold, J.; Schuster, C.; Fuchsbichler, A.; Bilban, M.; Tauber, S.; et al. Signal transducer and activator of transcription 3 protects from liver injury and fibrosis in a mouse model of sclerosing cholangitis. Gastroenterology 2010, 138, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Theise, N.D.; Badve, S.; Saxena, R.; Henegariu, O.; Sell, S.; Crawford, J.M.; Krause, D.S. Derivation of hepatocytes from bone marrow cells in mice after radiation-induced myeloablation. Hepatology 2000, 31, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Invernizzi, P.; Onori, P.; Franchitto, A.; De Santis, A.; Crosignani, A.; Sferra, R.; Ginanni-Corradini, S.; Mancino, M.G.; Maggioni, M.; et al. Estrogen receptors in cholangiocytes and the progression of primary biliary cirrhosis. J. Hepatol. 2004, 41, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.S.; Huang, C.C.; Wu, J.J.; Chaung, H.C.; Wu, C.L.; Chang, N.K.; Chang, Y.M.; Chou, M.H.; Chuang, J.H. Ascending cholangitis provokes il-8 and mcp-1 expression and promotes inflammatory cell infiltration in the cholestatic rat liver. J. Pediatr. Surg. 2001, 36, 1623–1628. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Sedlaczek, N.; Jia, J.D.; Bauer, M.; Herbst, H.; Ruehl, M.; Hahn, E.G.; Schuppan, D. Proliferating bile duct epithelial cells are a major source of connective tissue growth factor in rat biliary fibrosis. Am. J. Pathol. 2001, 158, 1239–1244. [Google Scholar] [CrossRef]
- Milani, S.; Herbst, H.; Schuppan, D.; Kim, K.Y.; Riecken, E.O.; Stein, H. Procollagen expression by nonparenchymal rat liver cells in experimental biliary fibrosis. Gastroenterology 1990, 98, 175–184. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.-H.; Sung, H.-J.; Lee, W.-R.; An, H.-J.; Kim, J.-Y.; Pak, S.C.; Han, S.-M.; Park, K.-K. Effects of Melittin Treatment in Cholangitis and Biliary Fibrosis in a Model of Xenobiotic-Induced Cholestasis in Mice. Toxins 2015, 7, 3372-3387. https://doi.org/10.3390/toxins7093372
Kim K-H, Sung H-J, Lee W-R, An H-J, Kim J-Y, Pak SC, Han S-M, Park K-K. Effects of Melittin Treatment in Cholangitis and Biliary Fibrosis in a Model of Xenobiotic-Induced Cholestasis in Mice. Toxins. 2015; 7(9):3372-3387. https://doi.org/10.3390/toxins7093372
Chicago/Turabian StyleKim, Kyung-Hyun, Hyun-Jung Sung, Woo-Ram Lee, Hyun-Jin An, Jung-Yeon Kim, Sok Cheon Pak, Sang-Mi Han, and Kwan-Kyu Park. 2015. "Effects of Melittin Treatment in Cholangitis and Biliary Fibrosis in a Model of Xenobiotic-Induced Cholestasis in Mice" Toxins 7, no. 9: 3372-3387. https://doi.org/10.3390/toxins7093372
APA StyleKim, K.-H., Sung, H.-J., Lee, W.-R., An, H.-J., Kim, J.-Y., Pak, S. C., Han, S.-M., & Park, K.-K. (2015). Effects of Melittin Treatment in Cholangitis and Biliary Fibrosis in a Model of Xenobiotic-Induced Cholestasis in Mice. Toxins, 7(9), 3372-3387. https://doi.org/10.3390/toxins7093372