
Influence of Honeybee Sting on Peptidome Profile in Human Serum


Abstract
:1. Introduction
2. Results and Discussion
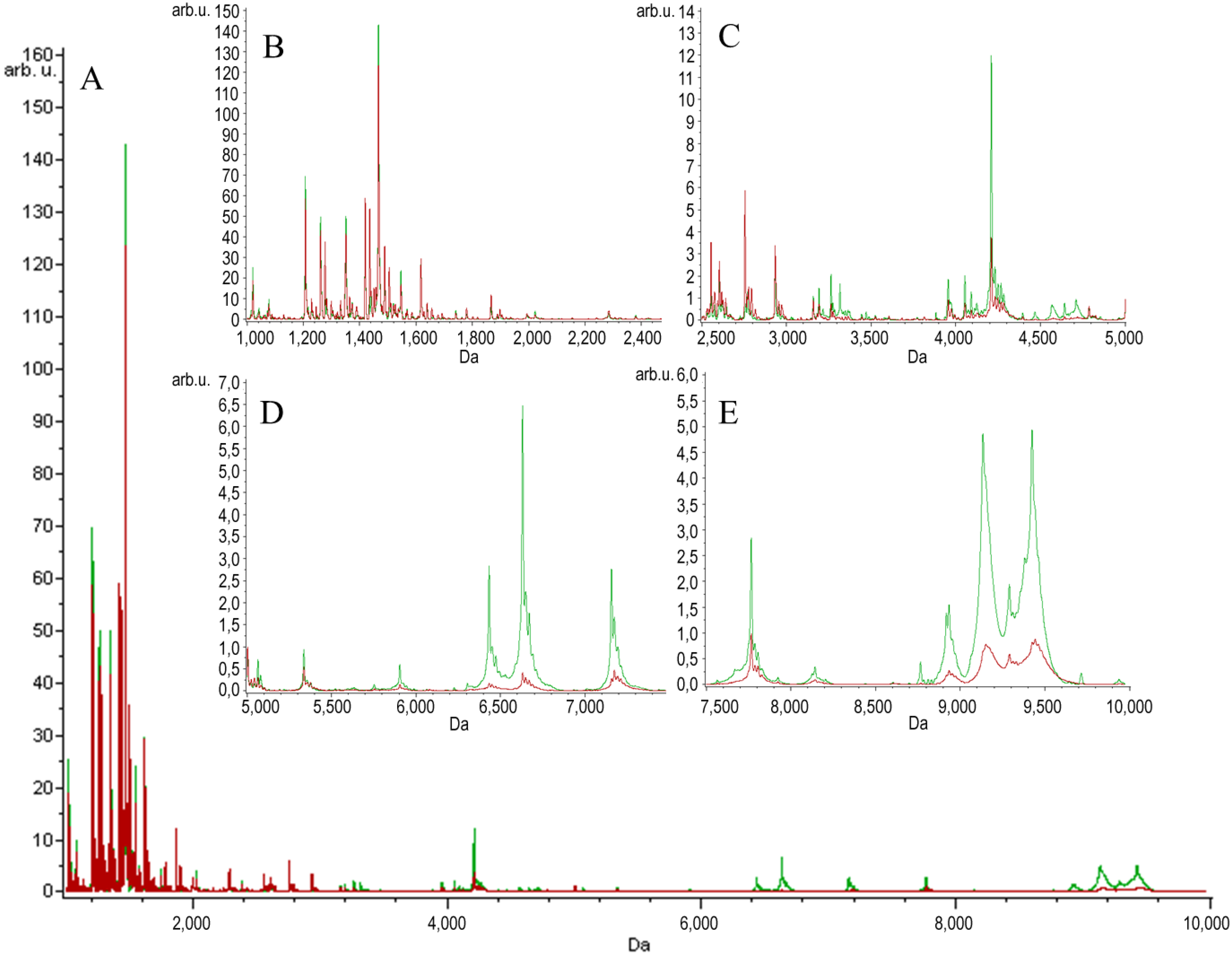
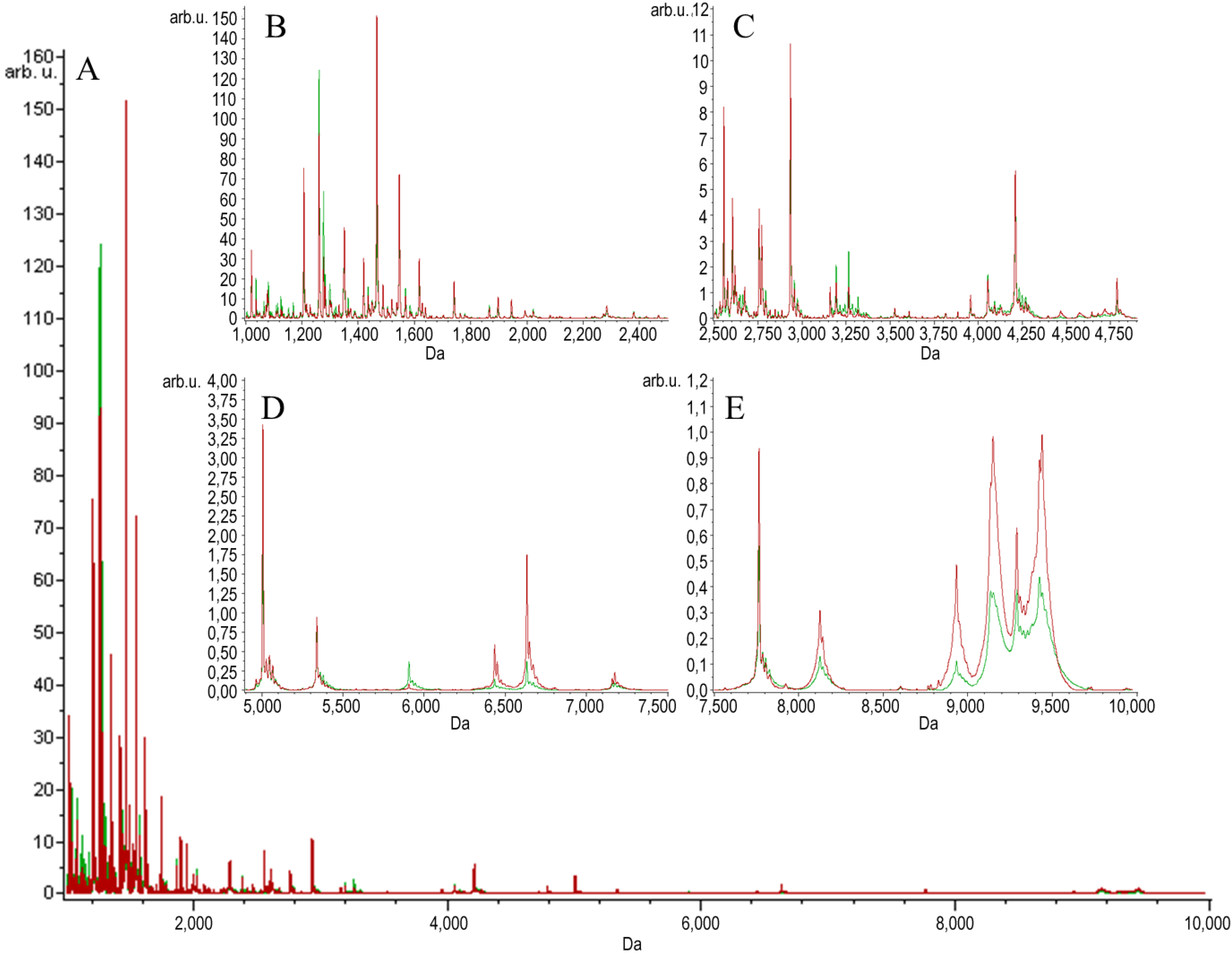
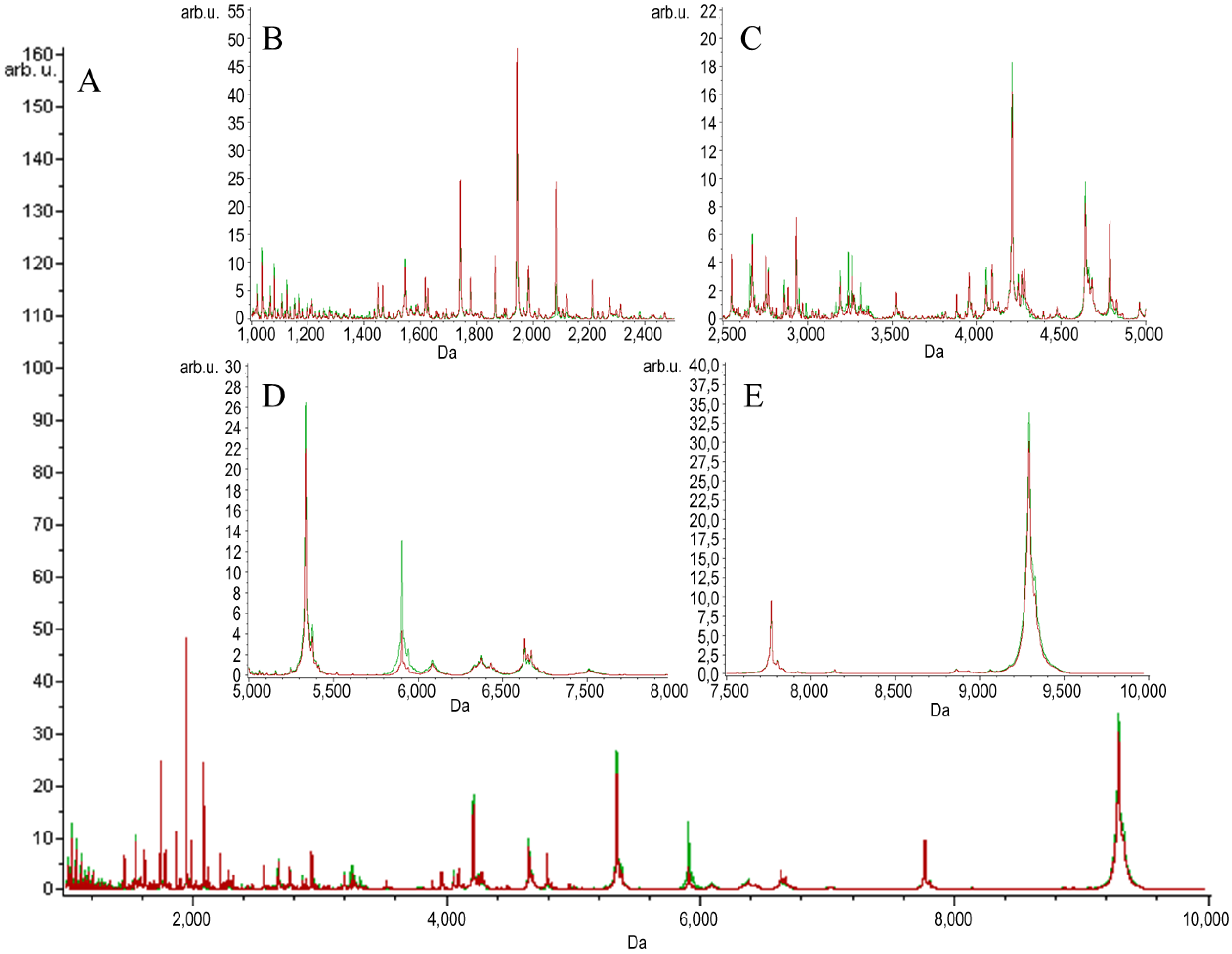

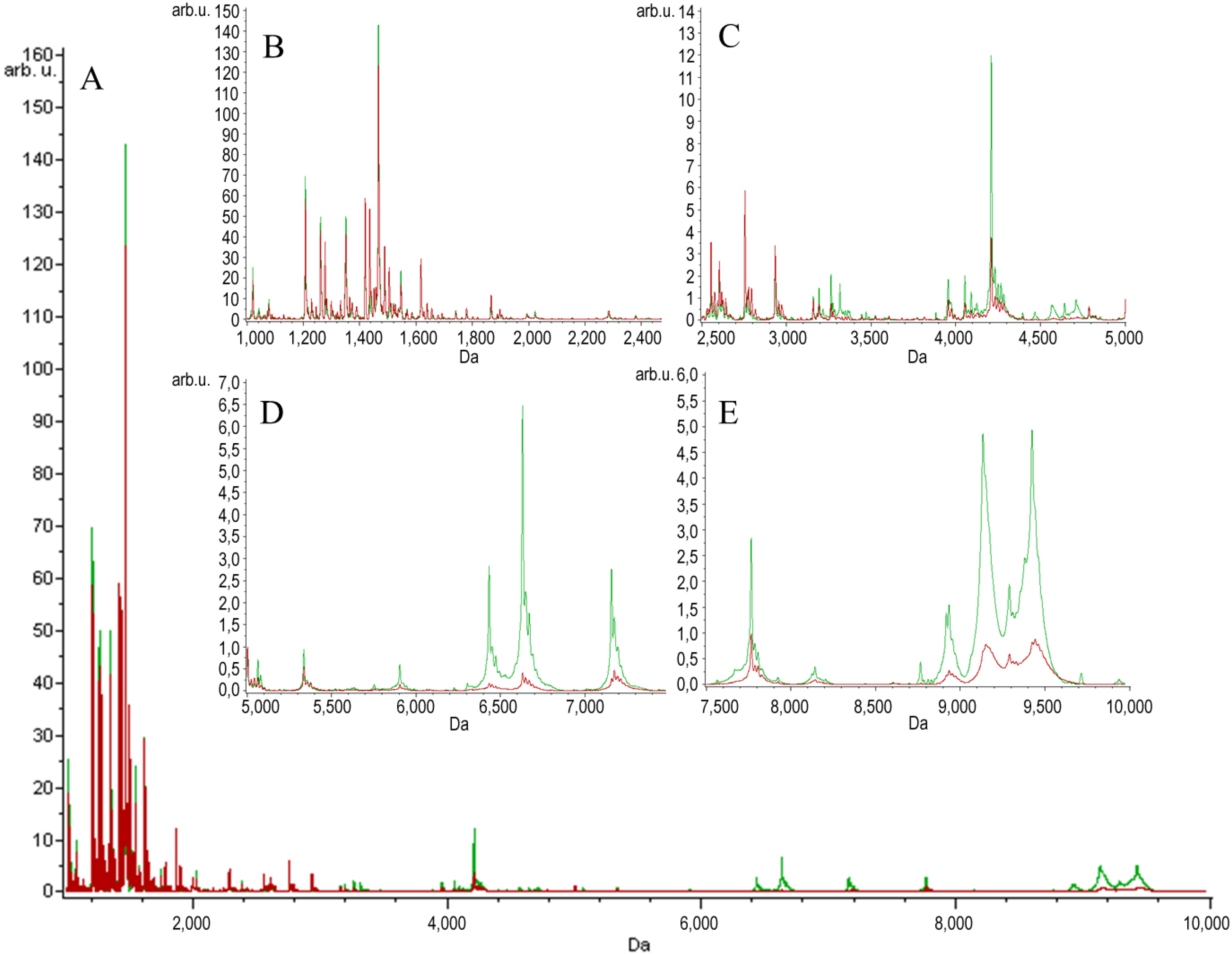{kind=link}
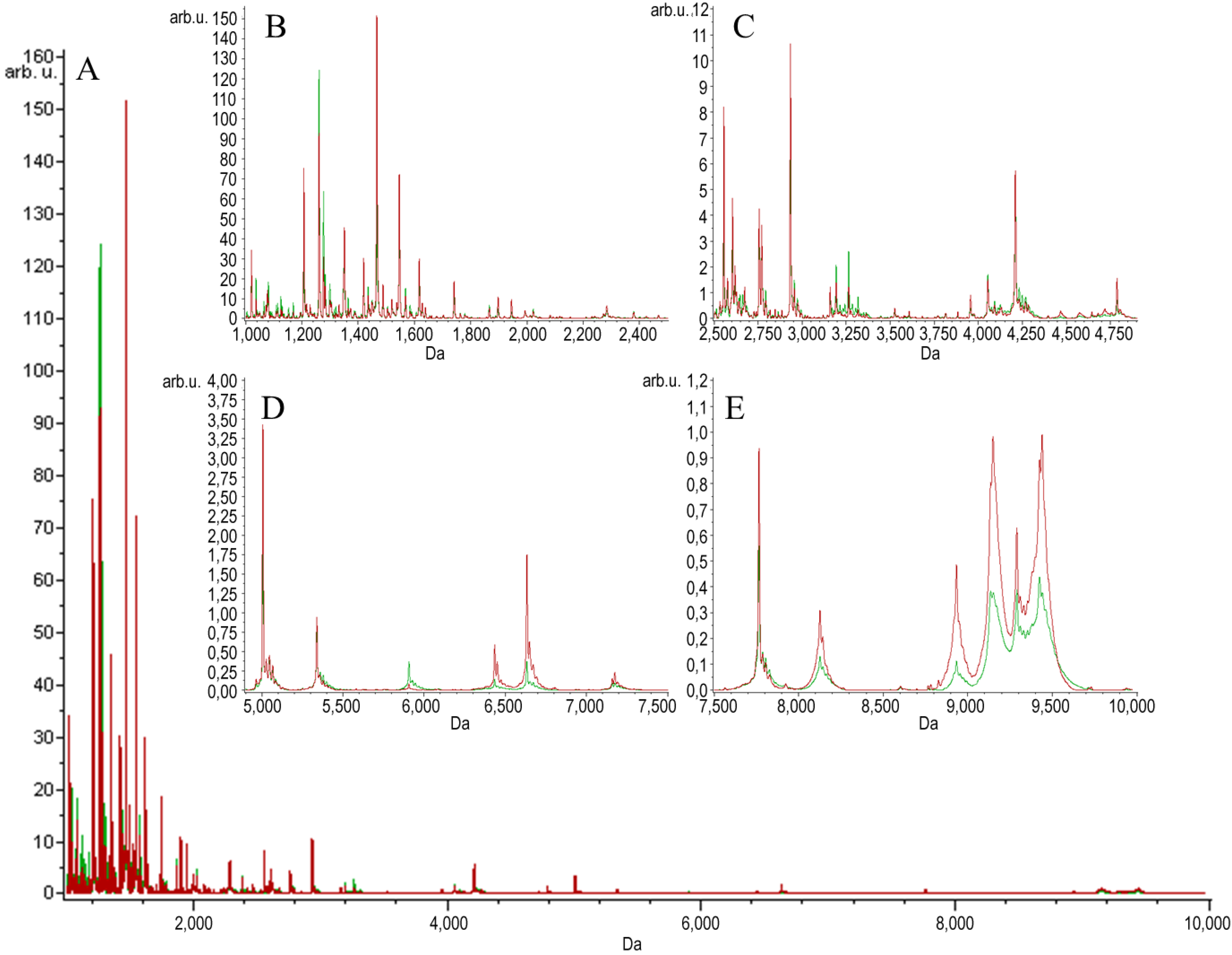{kind=link}
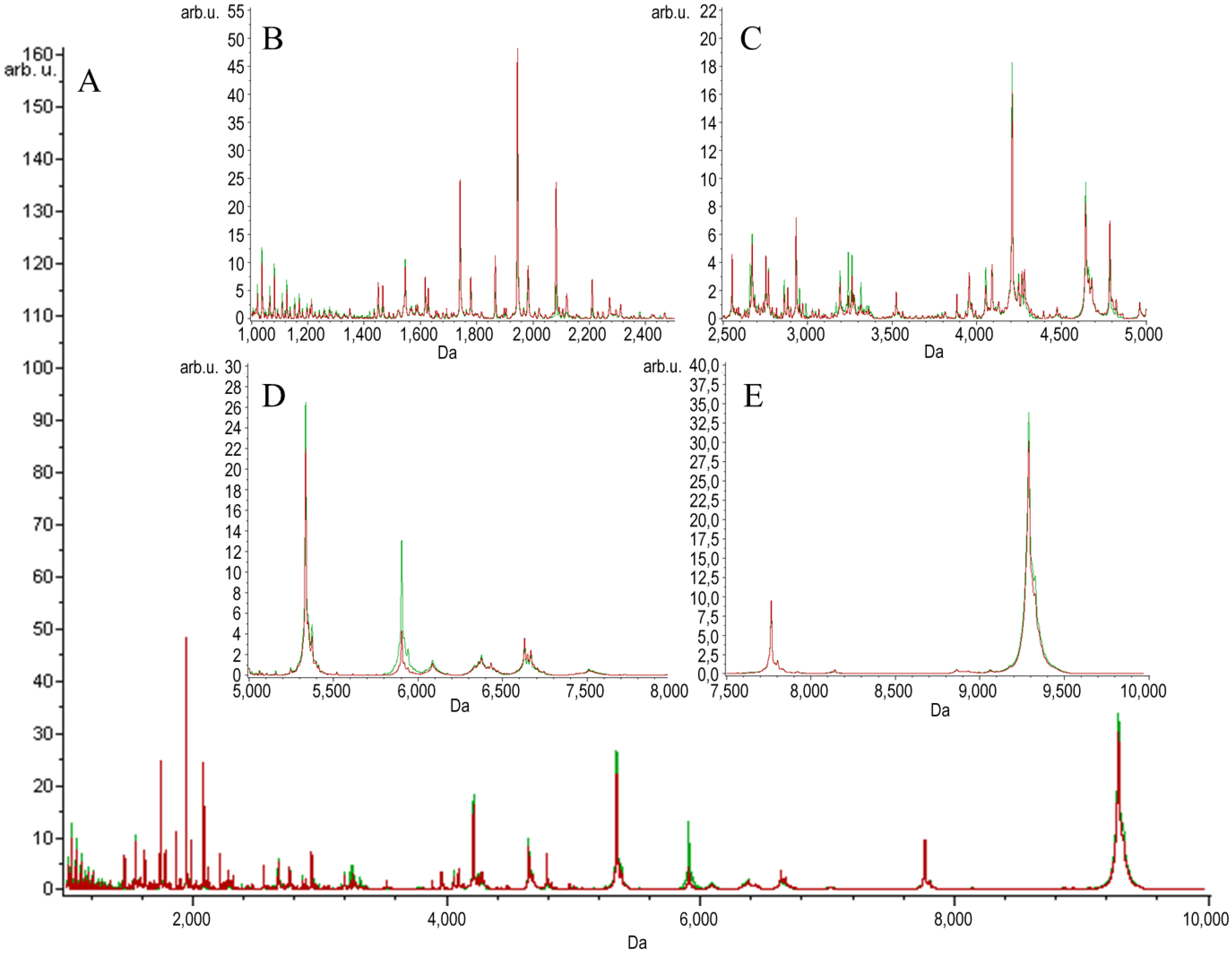{kind=link}
| Sample enrichment strategy | Algorithm | Cross validation (%) | Recognition capability (%) | Number of rejected spectra (%) |
|---|---|---|---|---|
| Omix | QC | 94.81 | 94.65 | 12.96 |
| GA | 97.01 | 100.00 | ||
| SNN | 97.31 | 100.00 | ||
| ZipTips | QC | 84.97 | 87.20 | 0.00 |
| GA | 87.08 | 97.52 | ||
| SNN | 53.57 | 50.57 | ||
| MB-WCX | QC | 80.41 | 85.64 | 0.00 |
| GA | 90.54 | 99.38 | ||
| SNN | 85.16 | 98.13 |
| Enrichment Strategy | Omix | Ziptips | MB-WCX | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Algorithm | QC | GA | SNN | QC | GA | SNN | QC | GA | SNN |
| m/z value of peaks used for classification (Da) | 1277.63 | 1299.62 | 1656.15 | 1037.36 | 1993.78 | 8933.73 | 1450.64 | 4661.58 | 3240.99 |
| 1299.62 | 1458.18 | 1261.58 | 1078.03 | 1037.36 | 1628.00 | 1779.41 | 1866.30 | ||
| 1436.04 | 1332.67 | 6432.85 | 1125.57 | 2379.82 | 1779.41 | 3240.99 | 1628.00 | ||
| 1505.18 | 1217.39 | 2755.95 | 1207.37 | 1299.90 | 1866.30 | 3934.87 | 2883.66 | ||
| 1656.15 | 8766.23 | 1299.62 | 1277.47 | 1217.26 | 2082.49 | 1153.78 | 2120.27 | ||
| 4210.32 | 1277.63 | 1299.90 | 2120.27 | 2082.49 | |||||
| 4467.44 | 1436.04 | 1364.67 | 2210.55 | 1450.64 | |||||
| 4568.85 | 1505.18 | 1466.78 | 2554.59 | 1617.58 | |||||
| 4711.11 | 7157.01 | 1584.84 | 2660.80 | 3955.05 | |||||
| 6432.85 | 1217.39 | 1897.73 | 2754.17 | 4053.49 | |||||
| 6450.18 | 2973.00 | 4466.21 | 3240.99 | 2092.96 | |||||
| 6472.33 | 1904.97 | 8933.73 | 4053.49 | 5001.79 | |||||
| 6528.33 | 7766.34 | 5940.75 | 9062.91 | ||||||
| 6631.12 | 6631.12 | 2688.19 | |||||||
| 6648.76 | 6509.33 | ||||||||
| 6670.30 | 6527.47 | ||||||||
| 7672.67 | 8863.22 | ||||||||
| 8919.94 | 4963.28 | ||||||||
| 9136.06 | |||||||||
| 9290.45 | |||||||||
| 9310.43 | |||||||||
| 9333.33 | |||||||||
| 9384.58 | |||||||||
| 9425.00 | |||||||||
3. Experimental Section
3.1. Study Participants and Serum Samples
3.2. Chemicals and Reagents
3.3. MALDI-TOF-MS Analysis
3.4. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rueff, F.; Jappe, U.; Przybilla, B. Standards and pitfalls of in vitro diagnostics of hymenoptera venom allergy. Hautarzt 2010, 61, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Rieger-Ziegler, V.; Rieger, E.; Kranke, B.; Aberer, W. Hymenoptera venom allergy: Time course of specific IgE concentrations during the first weeks after a sting. Int. Arch. Allergy Immunol. 1999, 120, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Kemeny, D.M.; Harries, M.G.; Youlten, L.J.; Mackenzie-Mills, M.; Lessof, M.H. Antibodies to purified bee venom proteins and peptides. I. Development of a highly specific RAST for bee venom antigens and its application to bee sting allergy. J. Allergy Clin. Immunol. 1983, 71, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Matysiak, J.; Matysiak, J.; Bręborowicz, A.; Kokot, Z.J. Diagnosis of hymenoptera venom allergy—With special emphasis on honeybee (Apis mellifera) venom allergy. Ann. Agric. Environ. Med. 2013, 20, 875–879. [Google Scholar] [PubMed]
- Müller, U.R. New developments in the diagnosis and treatment of hymenoptera venom allergy. Int. Arch. Allergy Immunol. 2001, 124, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Simons, F.E.; Frew, A.J.; Ansotegui, I.J.; Bochner, B.S.; Golden, D.B.; Finkelman, F.D. Risk assessment in anaphylaxis: Current and future approaches. J. Allergy Clin. Immunol. 2007, 120, 2–24. [Google Scholar] [CrossRef]
- Matysiak, J.; Dereziński, P.; Klupczyńska, A.; Matysiak, J.; Kaczmarek, E.; Kokot, Z.J. Effects of a honeybee sting on the serum free amino acid profile in humans. PLoS One 2014, 9, 1–12. [Google Scholar] [CrossRef]
- Szefler, S.J.; Wenzel, S.; Brown, R.; Erzurum, S.C.; Fahy, J.V.; Hamilton, R.G.; Hunt, J.F.; Kita, H.; Liu, A.H.; Panettieri, R.A.; et al. Asthma outcomes: Biomarkers. J. Allergy Clin. Immunol. 2012, 129, 9–23. [Google Scholar] [CrossRef]
- Taylor, R.D. Using biomarkers in the assessment of airways disease. J. Allergy Clin. Immunol. 2011, 128, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Cristea, I.M.; Gaskell, S.J.; Whetton, A.D. Proteomics techniques and their application to hematology. Blood 2004, 103, 3624–3634. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tang, H.; Wu, Z.; Gong, J.; Gruidl, M.; Zou, J.; Tockman, M.; Clark, R.A. Data mining techniques for cancer detection using serum proteomic profiling. Artif. Intell. Med. 2004, 32, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Pietrowska, M.; Polańska, J.; Suwiński, R.; Wideł, M.; Rutkowski, T.; Marczyk, M.; Domińczyk, I.; Ponge, L.; Marczak, Ł.; Polański, A.; et al. Comparison of peptide cancer signatures identified by mass spectrometry in serum of patients with head and neck, lung and colorectal cancers: Association with tumor progression. Int. J. Oncol. 2012, 40, 148–156. [Google Scholar] [PubMed]
- Pietrowska, M.; Polanska, J.; Marczak, L.; Behrendt, K.; Nowicka, E.; Stobiecki, M.; Polanski, A.; Tarnawski, R.; Widlak, P. Mass spectrometry-based analysis of therapy-related changes in serum proteome patterns of patients with early-stage breast cancer. J. Trans. Med. 2010, 8. [Google Scholar] [CrossRef]
- He, J.; Zeng, Z.; Xiang, Z.; Yang, P. Mass spectrometry-based serum peptide profiling in hepatocellular carcinoma with bone metastasis. World J. Gastroenterol. 2014, 20, 3025–3032. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Jiang, P.; Zhou, L.; Zhang, W. Evaluation of novel serum biomarkers and the proteomic differences of endometriosis and adenomyosis using MALDI-TOF–MS. Arch. Gynecol. Obstet. 2013, 288, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Taneja, S.; Ahmad, I.; Sen, S.; Kumar, S.; Arora, R.; Gupta, V.K.; Aggarwal, R.; Narayanasamy, K.; Reddy, V.S.; Jameel, S. Plasma peptidome profiling of acute hepatitis E patients by MALDI-TOF/TOF. Proteome Sci. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Pusch, W.; Kostrzewa, M. Application of MALDI-TOF mass spectrometry in screening and diagnostic research. Curr. Pharm. Design 2005, 11, 2577–2591. [Google Scholar] [CrossRef]
- Šalplachta, J.; Řehulka, P.; Chmelík, J. Identification of proteins by combination of size-exclusion chromatography with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and comparison of some desalting procedures for both intact proteins and their tryptic digests. J. Mass Spectrom. 2004, 39, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Palmblad, M.; Vogel, J.S. Quantitation of binding, recovery and desalting efficiency of peptides and proteins in solid phase extraction micropipette tips. J. Chromatogr. B 2005, 814, 309–313. [Google Scholar] [CrossRef]
- Velstra, B.; van der Burgt, Y.E.M.; Mertens, B.J.; Mesker, W.E.; Deelder, A.M.; Tollenaar, R.A. Improved classification of breast cancer peptide and protein profiles by combining two serum workup procedures. J. Cancer Res. Clin. Oncol. 2012, 138, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Instructions for Use. Available online: https://www.bruker.com/fileadmin/user_upload/8-PDF-Docs/Separations_MassSpectrometry/InstructionForUse/IFU_223983_223987_MB-WCX_Rev1.pdf (accessed on 27 March 2015).
- Agilent Bond Elut OMIX Pipette Tips for Micro Extractions. Available online: http://www.chem.agilent.com/Library/primers/Public/5990–9049EN-Omix-Sept11-lo.pdf (accessed on 27 March 2015).
- User Guide for Reversed-Phase ZipTip Pipette Tips for Sample Preparation. Available online: http://personal.rhul.ac.uk/upba/211/Zip-tip.pdf (accessed on 27 March 2015).
- Matysiak, J. Assessment the Risk Factors of Allergic Reactions after a Bee Sting in the Beekeepers and Their Family Members. Ph.D. Thesis, Poznan University of Medical Sciences, Poznań, Poland, May 2012. [Google Scholar]
- Basics on data preparation, model generation and spectra classification, Clinprotools software for biomarker detection and evaluation. In ClinProTools 3.0 User Manual; Bruker Daltonic GmbH: Bremen, Germany, 2011; pp. 51–96.
- Villanueva, J.; Lawlor, K.; Toled-Crow, R.; Tempst, P. Automated serum peptide profiling. Nat. Protoc. 2006, 1, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Philip, J.; Chaparro, C.A.; Li, Y. Correcting common errors in identifying cancer-specific serum peptide signatures. J. Proteome Res. 2005, 4, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.; Hirschberg, D.; Palmberg, C.; Jornvall, H.; Bergman, T. Integrated sample preparation and MALDI mass spectrometry on a microfluid compact disk. Anal. Chem. 2004, 76, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Tiss, A.; Smith, C.; Camuzeau, S.; Kabir, M.; Gayther, S.; Menon, U.; Waterfield, M.; Timms, J.; Jacobs, I.; Cramer, R. Serum peptide profiling using MALDI mass spectrometry avoiding the pitfalls of coated magnetic beads using well-established ZipTip technology. Pract. Proteomics 2007, 7, 77–89. [Google Scholar] [CrossRef]
- Lin, J.; Bruni, F.M.; Fu, Z.; Maloney, J.; Bardina, L.; Boner, A.L.; Gimenez, G.; Sampson, H.A. A bioinformatics approach to identify patients with symptomatic peanut allergy using peptide microarray immunoassay. J. Allergy Clin. Immunol. 2012, 129, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Sandanayake, N.S.; Camuzeaux, S.; Sinclair, J.; Blyuss, O.; Andreola, F.; Chapman, M.H.; Webster, G.J.; Smith, R.C.; Timms, J.F.; Pereira, S.P. Identification of potential serum peptide biomarkers of biliary tract cancer using MALDI MS profiling. BMC Clin. Pathol. 2014, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A.; Lin, Y.; Kurek, K.; Calicchio, M.; Wang, Y.; Monigatti, F.; Campagne, F.; Lee, R.; Horwitz, B.; Steen, H.; Bachur, R. Discovery and validation of urine markers of acute pediatric appendicitis using high-accuracy mass spectrometry. Ann. Emerg. Med. 2010, 55, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Petricoin, E.F.; Ardekani, A.M.; Hitt, B.A.; Levine, P.J.; Fusaro, V.A.; Steinberg, S.M.; Mills, G.B.; Simone, C.; Fishman, A.; Kohn, E.C.; et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 2002, 359, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Hu, Y.; He, X.; Li, J.; Pan, T.; Liu, H.; Wu, X.; He, H.; Ge, W.; Yu, J.; et al. Serum profiling by mass spectrometry combined with bioinformatics for the biomarkers discovery in diffuse large B-cell lymphoma. Tumour Biol. 2014, 36, 2193–2199. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.P.; Lin, Y.W.; Lai, H.C.; Chu, T.Y.; Kuo, Y.L.; Liu, H.S. SELDI-TOF MS profiling of plasma proteins in ovarian cancer. Taiwan J. Obstet. Gynecol. 2006, 45, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Bruegel, M.; Planert, M.; Baumann, S.; Focke, A.; Bergh, F.T.; Leichtle, A.; Ceglarek, U.; Thiery, J.; Fiedler, G.M. Standardized peptidome profiling of human cerebrospinal fluid by magnetic bead separation and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J. Proteomics 2009, 72, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.J.; Chen, L.C.; Chien, K.Y.; Chen, Y.J.; Chang, J.T.; Wang, H.M.; Liao, C.T.; Chen, I.H. Oral cancer plasma tumor marker identified with bead-based affinity-fractionated proteomic technology. Clin. Chem. 2005, 51, 2236–2244. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matysiak, J.; Światły, A.; Hajduk, J.; Matysiak, J.; Kokot, Z.J. Influence of Honeybee Sting on Peptidome Profile in Human Serum. Toxins 2015, 7, 1808-1820. https://doi.org/10.3390/toxins7051808
Matysiak J, Światły A, Hajduk J, Matysiak J, Kokot ZJ. Influence of Honeybee Sting on Peptidome Profile in Human Serum. Toxins. 2015; 7(5):1808-1820. https://doi.org/10.3390/toxins7051808
Chicago/Turabian StyleMatysiak, Jan, Agata Światły, Joanna Hajduk, Joanna Matysiak, and Zenon J. Kokot. 2015. "Influence of Honeybee Sting on Peptidome Profile in Human Serum" Toxins 7, no. 5: 1808-1820. https://doi.org/10.3390/toxins7051808
APA StyleMatysiak, J., Światły, A., Hajduk, J., Matysiak, J., & Kokot, Z. J. (2015). Influence of Honeybee Sting on Peptidome Profile in Human Serum. Toxins, 7(5), 1808-1820. https://doi.org/10.3390/toxins7051808