
Does Bacillus anthracis Lethal Toxin Directly Depress Myocardial Function? A Review of Clinical Cases and Preclinical Studies

Abstract
:1. Introduction
2. Lethal Toxin and Its Intracellular Actions
3. Effects of B. Anthracis Infection on the Heart in Clinical Studies

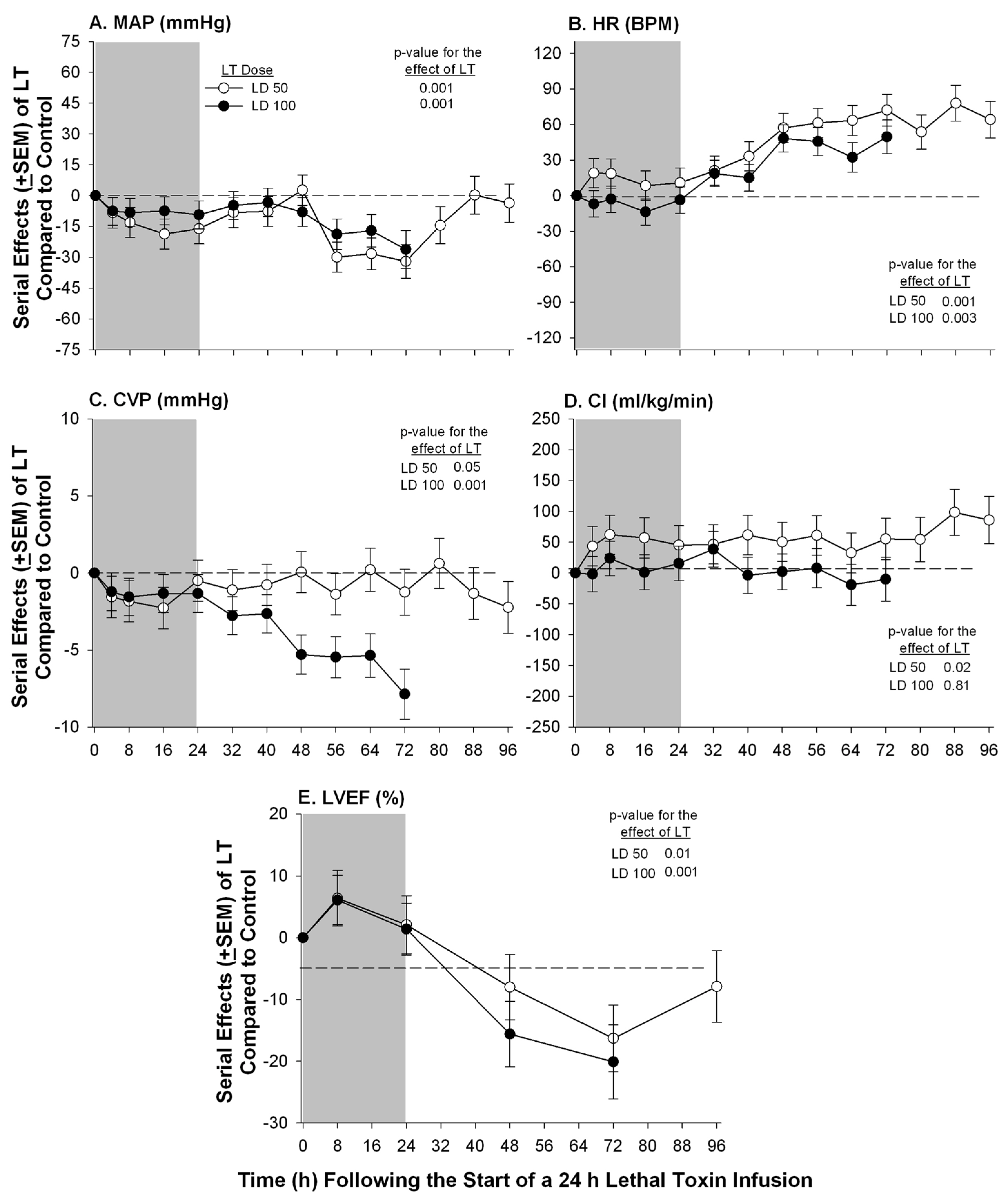{kind=link}
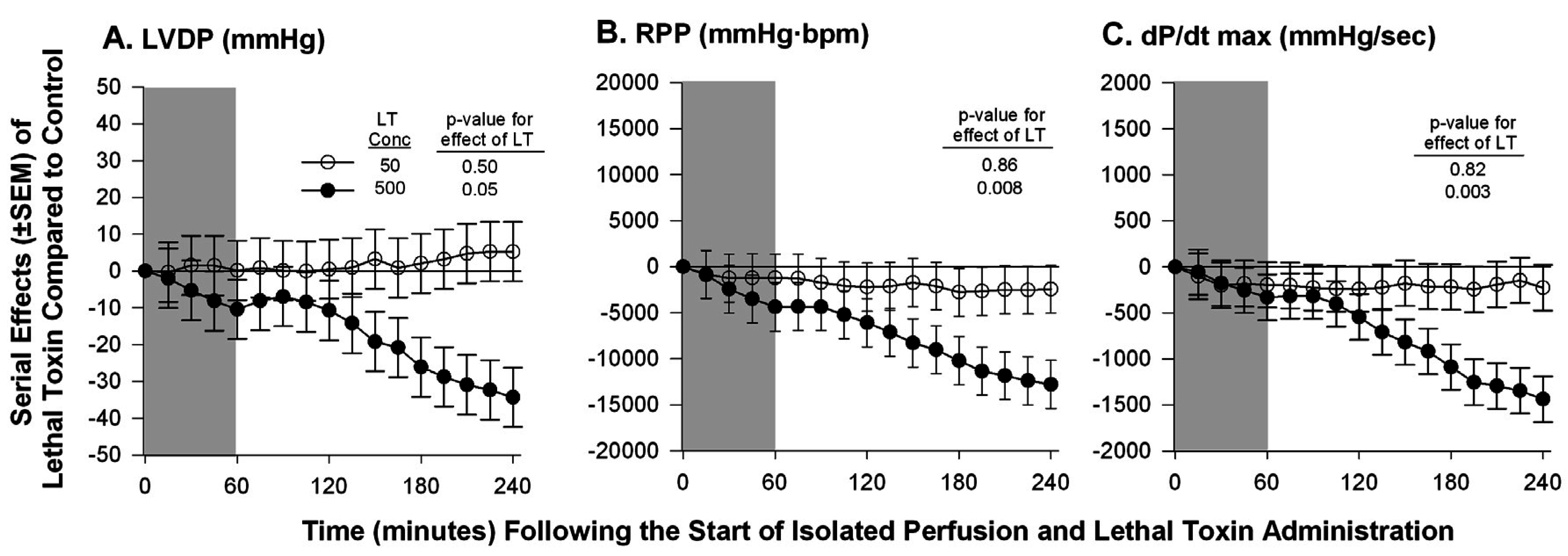
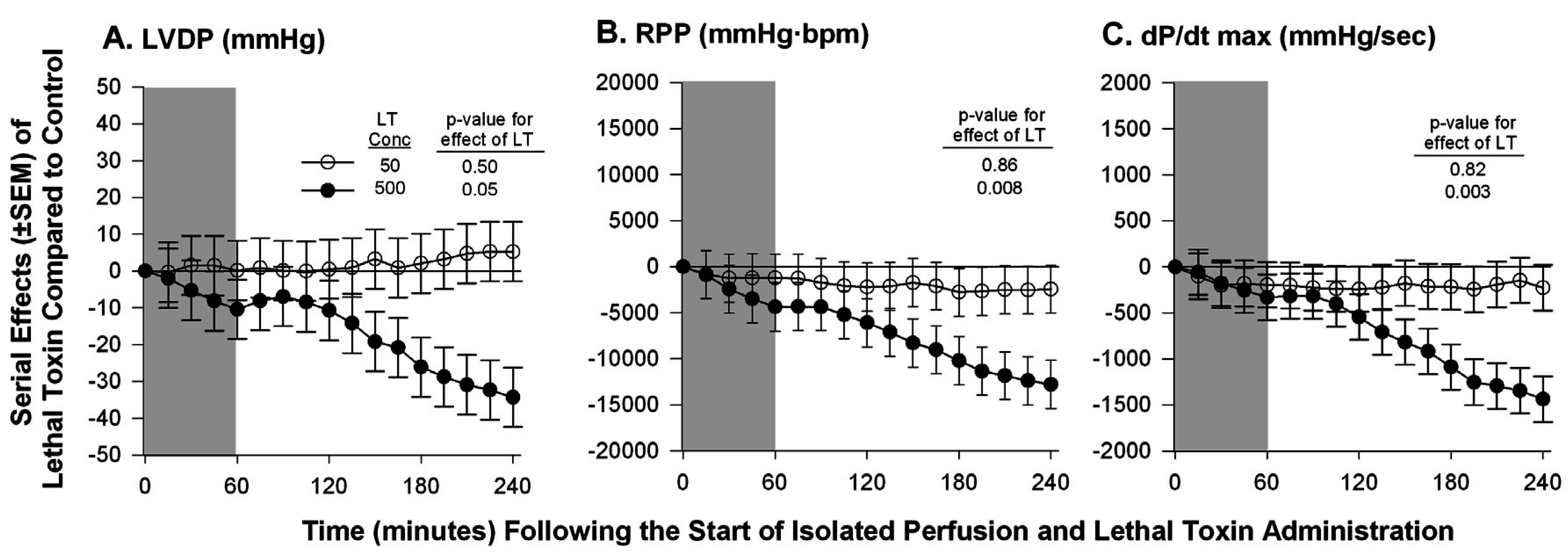{kind=link}
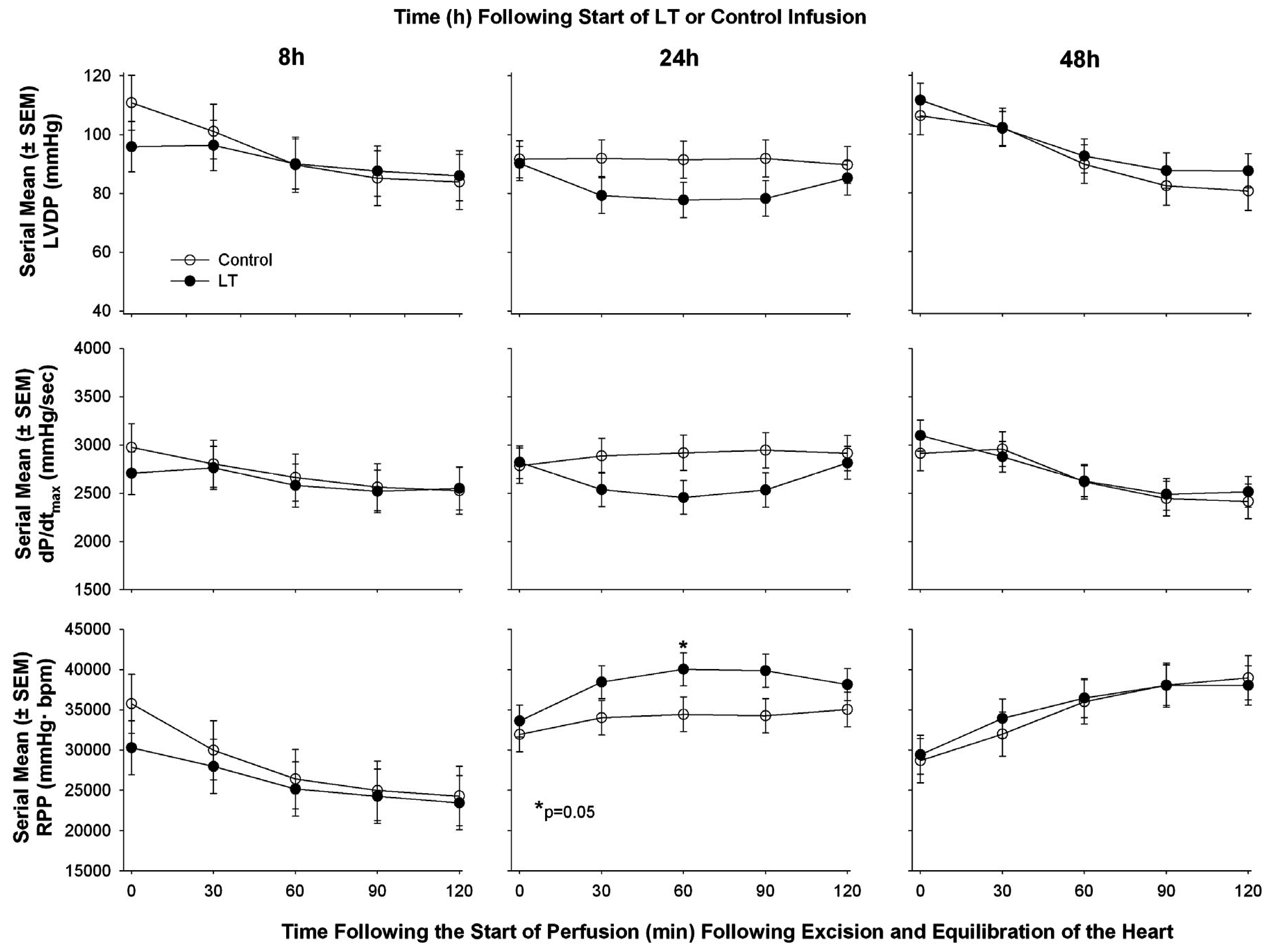{kind=link}
| Publication | Number of Patients | Type of Anthrax Infection | Clinical or Histopathological Cardiac Findings |
|---|---|---|---|
| Albrink et al. 1960 [32] | 3 | Inhalational | Moderate subendocardial hemorrhage into myocardium of left ventricle noted at autopsy |
| Abramova et al. 1993 [11] Grinberg et al. 2001 [12] | 42 | Inhalational | No specific cardiac histopathological findings were noted. However, occasional cases showed myocyte hypereosinophilia and rare focal contraction band necrosis attributed to agonal hypotension and hypoxemia |
| Mayer et al. 2001 [44] | 2 | Inhalational | No functional cardiac assessment completed |
| Borio et al. 2001 [29] | 2 | Inhalational | ECG showing atrial fibrillation, no other specific findings noted |
| Jernigan et al. 2001 [1] | 10 * | Inhalational | ECG showing atrial fibrillation noted in one of the four patients not reported as isolated cases |
| Bush et al. 2001 [28] | 1 | Inhalational | No gross cardiac abnormalities on autopsy found |
| Barakat et al. 2002 [31] | 1 | Inhalational | No functional cardiac assessment completed |
| Mina et al. 2002 [18] | 1 | Inhalational | Echocardiogram showed normal LV function at presentation with small pericardial effusion that enlarged and progressed to tamponade. Pulmonary artery catheterization also completed (see Section 3) |
| Guarner et al. 2003 [27] | 11 ** | Inhalational | No specific cardiac histopathological abnormalities noted |
| Tabei et al. 2004 [33] | 33 | Cutaneous, Inhalational, and Gastrointestinal | No specific cardiac histopathological abnormalities noted |
| Babamahmoodi et al. 2005 [39] | 3 | Gastrointestinal | No functional cardiac abnormalities noted |
| Walsh et al. 2007 [35] | 1 | Inhalational | Echocardiogram showing minimal pericardial effusion |
| Klempner et al. 2010 [37] | 1 | Gastrointestinal | Transthoracic echocardiogram showed a normal ejection fraction, no valvular vegetations and findings consistent with right atrial volume overload, and right ventricular systolic hypertension |
| Doganay et al. 2010 [40] | 22 | Cutaneous | No functional cardiac abnormalities noted |
| Popescu et al. 2011 [41] | 2 | Cutaneous | No functional cardiac abnormalities noted |
| Powell et al. 2011 [42] | 1 | Injectional | No functional cardiac abnormalities noted |
| Gruno et al. 2012 [43] | 3 | Injectional | No functional cardiac abnormalities noted |
| Russel et al. 2013 [19] | 2 | Injectional | Transesophageal echocardiogram reported normal in setting of fulminant septic shock |
| Booth et al. 2014 [2] | 27 | Injectional | Of nine patients reported, three had dysfunction based on echocardiography, lithium dilution or pulse contour cardiac outputs, and one had an elevated troponin. Other patients had no evidence of abnormal cardiac function (see section 3) |
| Sprenkle et al. 2014 [38] | 1 | Inhalational | Echocardiogram showed left ventricular hypertrophy with EF of 40%, normal estimated pulmonary artery pressure, probable decreased right ventricular function and no pericardial effusion |
4. Effects of LT on the Heart in in vivo, in vitro and ex vivo Studies

| Publication | Subjects | Route of Lethal Toxin Exposure | Functional Measurement | Findings |
|---|---|---|---|---|
| Watson et al. 2007 [13] | Sprague-Dawley rat | Intravenous bolus | Echocardiography | ▪20% increase in LVAs and LVAd within 2 h. Specific LVEF measurements not reported; ▪Increase in Vp |
| Watson et al. 2007 [52] | Sprague-Dawley rat | Intravenous bolus | Echocardiography | ▪30% reduction in LVEF in 11/14 rats surviving after 48 h related to acute increase in LVAs. No increase in LVAd noted; ▪Decreased VCFC, Decrease in Vp |
| Cheng et al. 2007 [57] | Canine | Intravenous bolus | Pressure-Volume catheter | ▪Significant LV dysfunction starting at 6 h with development of heart failure at 96 h; ▪Decreases in LVEF, stroke volume, LVESP, contractility, prolonged relaxation time constant, increases in LVEDP |
| Moayeri et al. 2009 [14] | C57BL/6J mouse | Intravenous bolus | Echocardiography | ▪Decreases in ejection fraction and fractional shortening at 24 h after LT challenge without change in stroke volume or CO |
| Sweeney et al. 2010 [17] | Purpose–bred Beagle | Continuous infusion | PA Catheter Echocardiography | ▪Low and high dose (see section 4) of LT caused progressive declines (15%–20%) in LVEF at 72 h; ▪No significant change in PAOP or SVI with either dose but CVP decreased with high dose |
| Lawrence et al. 2011 [20] | Dutch-belted rabbit | Intravenous bolus | Echocardiography | ▪Serial echo measurements at 0 to 48 h showed no significant change in LVAs or LVAd despite elevated markers of myocardial injury |
| Hicks et al. 2011 [59] | Isolated Sprague-Dawley rat heart | Continuous non-recirculating perfusion | Ex-vivo Langendorff Model | ▪No change in LVDP, RPP, or dP/dt max at a known lethal dose of LT; ▪A 10-fold increase in the lethal dose caused decreases in all measured parameters |
| Liu et al. 2013 [10] | Mouse | Intraperitoneal | Echocardiography | ▪Significant decrease in EF at 48 h after LT challenge |
| Golden et al. 2013 [16] | Sprague-Dawley rat | Intravenous bolus | Echocardiography | ▪Abnormal indices of diastolic dysfunction within 2–8 h including prolonged LV deceleration time, elevated E/E’ ratio, left atrial chamber enlargement and pulmonary regurgitation; ▪No change in EF noted |
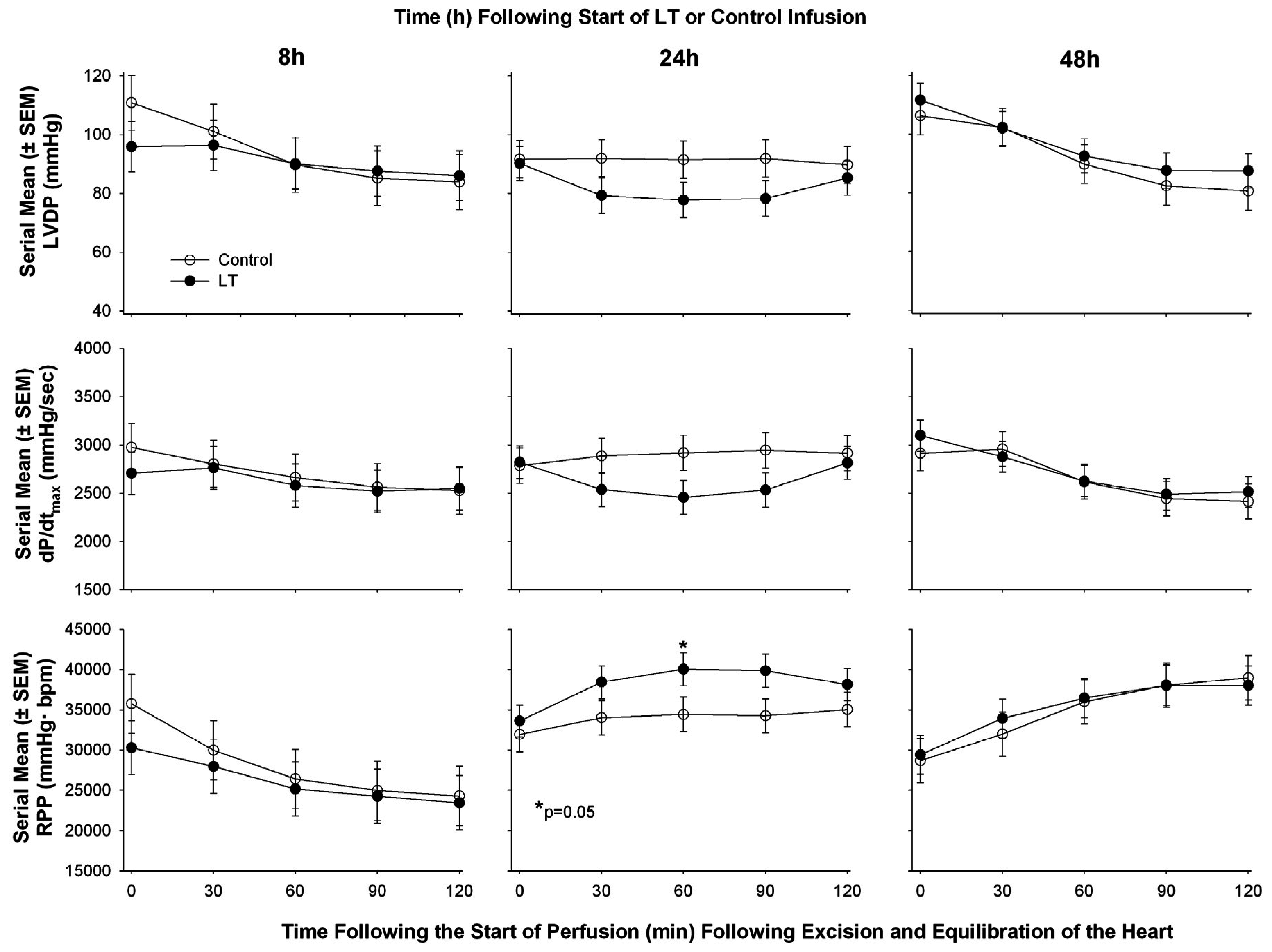
| Li et al. 2015 [60] | Sprague-Dawley rat | Continuous infusion | Echocardiography in vivo followed by ex vivo Langendorff Model | ▪LT decreased CO and decreased LVEF at 8 and 48 h but increased it at 24 h measured with cardiac echo; ▪In isolated hearts following in vivo exposure to LT no consistent change at 8, 24, or 48 h in LVSP, LVDP, RPP, or dP/dt max or min |
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Jernigan, J.A.; Stephens, D.S.; Ashford, D.A.; Omenaca, C.; Topiel, M.S.; Galbraith, M.; Tapper, M.; Fisk, T.L.; Zaki, S.; Popovic, T.; et al. Bioterrorism-related inhalational anthrax: The first 10 cases reported in the united states. Emerg. Infect. Dis. 2001, 7, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.; Donaldson, L.; Cui, X.; Sun, J.; Cole, S.; Dailsey, S.; Hart, A.; Johns, N.; McConnell, P.; McLennan, T.; et al. Confirmed bacillus anthracis infection among persons who inject drugs, scotland, 2009–2010. Emerg. Infect. Dis. 2014, 20, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, E.K.; Rubenstein, A.R.; Radin, G.T.; Wiener, R.S.; Walkey, A.J. Two decades of mortality trends among patients with severe sepsis: A comparative meta-analysis. Crit. Care Med. 2014, 42, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Remy, K.E.; Qiu, P.; Li, Y.; Cui, X.; Eichacker, P.Q.B. Anthracis associated cardiovascular dysfunction and shock: The potential contribution of both non-toxin and toxin components. BMC Med. 2013, 11, 217. [Google Scholar] [CrossRef] [PubMed]
- Rolando, M.; Stefani, C.; Flatau, G.; Auberger, P.; Mettouchi, A.; Mhlanga, M.; Rapp, U.; Galmiche, A.; Lemichez, E. Transcriptome dysregulation by anthrax lethal toxin plays a key role in induction of human endothelial cell cytotoxicity. Cell. Microbiol. 2010, 12, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Guichard, A.; McGillivray, S.M.; Cruz-Moreno, B.; van Sorge, N.M.; Nizet, V.; Bier, E. Anthrax toxins cooperatively inhibit endocytic recycling by the Rab11/Sec15 exocyst. Nature 2010, 467, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Warfel, J.M.; D‘Agnillo, F. Anthrax lethal toxin-mediated disruption of endothelial VE-cadherin is attenuated by inhibition of the Rho-associated kinase pathway. Toxins 2011, 3, 1278–1293. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Moayeri, M.; Leppla, S.H. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol. 2014, 22, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, Y.; Moayeri, M.; Liu, J.; Crown, D.; Fattah, R.J.; Wein, A.N.; Yu, Z.X.; Finkel, T.; Leppla, S.H. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 2013, 501, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Abramova, F.A.; Grinberg, L.M.; Yampolskaya, O.V.; Walker, D.H. Pathology of inhalational anthrax in 42 cases from the sverdlovsk outbreak of 1979. Proc. Natl. Acad. Sci. USA 1993, 90, 2291–2294. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, L.M.; Abramova, F.A.; Yampolskaya, O.V.; Walker, D.H.; Smith, J.H. Quantitative pathology of inhalational anthrax I: Quantitative microscopic findings. Mod. Pathol. 2001, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.E.; Kuo, S.R.; Katki, K.; Dang, T.; Park, S.K.; Dostal, D.E.; Tang, W.J.; Leppla, S.H.; Frankel, A.E. Anthrax toxins induce shock in rats by depressed cardiac ventricular function. PLoS ONE 2007, 2, e466. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Crown, D.; Dorward, D.W.; Gardner, D.; Ward, J.M.; Li, Y.; Cui, X.; Eichacker, P.; Leppla, S.H. The heart is an early target of anthrax lethal toxin in mice: A protective role for neuronal nitric oxide synthase (nNOS). PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Kandadi, M.R.; Frankel, A.E.; Ren, J. Toll-like receptor 4 knockout protects against anthrax lethal toxin-induced cardiac contractile dysfunction: Role of autophagy. Br. J. Pharmacol. 2012, 167, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Golden, H.B.; Watson, L.E.; Nizamutdinov, D.; Feng, H.; Gerilechaogetu, F.; Lal, H.; Verma, S.K.; Mukhopadhyay, S.; Foster, D.M.; Dillmann, W.H.; et al. Anthrax lethal toxin induces acute diastolic dysfunction in rats through disruption of the phospholamban signaling network. Int. J. Cardiol. 2013, 168, 3884–3895. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sweeney, D.A.; Cui, X.; Solomon, S.B.; Vitberg, D.A.; Migone, T.S.; Scher, D.; Danner, R.L.; Natanson, C.; Subramanian, G.M.; Eichacker, P.Q. Anthrax lethal and edema toxins produce different patterns of cardiovascular and renal dysfunction and synergistically decrease survival in canines. J. Infect. Dis. 2010, 202, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Mina, B.; Dym, J.P.; Kuepper, F.; Tso, R.; Arrastia, C.; Kaplounova, I.; Faraj, H.; Kwapniewski, A.; Krol, C.M.; Grosser, M.; et al. Fatal inhalational anthrax with unknown source of exposure in a 61-year-old woman in new york city. JAMA 2002, 287, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.; Pedersen, M.; Jensen, A.V.; Soes, L.M.; Hansen, A.B. Two anthrax cases with soft tissue infection, severe oedema and sepsis in danish heroin users. BMC Infect. Dis. 2013, 13, 408. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, W.S.; Marshall, J.R.; Zavala, D.L.; Weaver, L.E.; Baze, W.B.; Moen, S.T.; Whorton, E.B.; Gourley, R.L.; Peterson, J.W. Hemodynamic effects of anthrax toxins in the rabbit model and the cardiac pathology induced by lethal toxin. Toxins 2011, 3, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Young, J.A.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Bromberg-White, J.; Lee, C.S.; Duesbery, N. Consequences and utility of the zinc-dependent metalloprotease activity of anthrax lethal toxin. Toxins 2010, 2, 1038–1053. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Sastalla, I.; Leppla, S.H. Anthrax and the inflammasome. Microbes Infect. 2012, 14, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Rolli, J.; Rosenblatt-Velin, N.; Li, J.; Loukili, N.; Levrand, S.; Pacher, P.; Waeber, B.; Feihl, F.; Ruchat, P.; Liaudet, L. Bacterial flagellin triggers cardiac innate immune responses and acute contractile dysfunction. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, X.; Kao, R.; Mele, T.; Kvietys, P.; Martin, C.M.; Rui, T. Cardiac fibroblasts contribute to myocardial dysfunction in mice with sepsis: The role of NLRP3 inflammasome activation. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Natanson, C.; Danner, R.L.; Elin, R.J.; Hosseini, J.M.; Peart, K.W.; Banks, S.M.; MacVittie, T.J.; Walker, R.I.; Parrillo, J.E. Role of endotoxemia in cardiovascular dysfunction and mortality. Escherichia coli and staphylococcus aureus challenges in a canine model of human septic shock. J. Clin. Invest. 1989, 83, 243–251. [Google Scholar]
- Guarner, J.; Jernigan, J.A.; Shieh, W.J.; Tatti, K.; Flannagan, L.M.; Stephens, D.S.; Popovic, T.; Ashford, D.A.; Perkins, B.A.; Zaki, S.R. Pathology and pathogenesis of bioterrorism-related inhalational anthrax. Am. J. Pathol. 2003, 163, 701–709. [Google Scholar] [CrossRef]
- Bush, L.M.; Abrams, B.H.; Beall, A.; Johnson, C.C. Index case of fatal inhalational anthrax due to bioterrorism in the united states. N. Engl. J. Med. 2001, 345, 1607–1610. [Google Scholar] [CrossRef] [PubMed]
- Borio, L.; Frank, D.; Mani, V.; Chiriboga, C.; Pollanen, M.; Ripple, M.; Ali, S.; DiAngelo, C.; Lee, J.; Arden, J.; et al. Death due to bioterrorism-related inhalational anthrax: Report of 2 patients. JAMA 2001, 286, 2554–2559. [Google Scholar] [CrossRef] [PubMed]
- Quintiliani, R., Jr.; Quintiliani, R. Fatal case of inhalational anthrax mimicking intra-abdominal sepsis. Conn. Med. 2002, 66, 261–267. [Google Scholar] [PubMed]
- Barakat, L.A.; Quentzel, H.L.; Jernigan, J.A.; Kirschke, D.L.; Griffith, K.; Spear, S.M.; Kelley, K.; Barden, D.; Mayo, D.; Stephens, D.S.; et al. Fatal inhalational anthrax in a 94-year-old connecticut woman. JAMA 2002, 287, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Albrink, W.S.; Brooks, S.M.; Biron, R.E.; Kopel, M. Human inhalation anthrax. A report of three fatal cases. Am. J. Pathol. 1960, 36, 457–471. [Google Scholar] [PubMed]
- Tabei, S.Z.; Amin, A.; Mowla, A.; Nabavizadeh, S.A.; Razmkon, A. Anthrax: Pathological aspects in autopsy cases in shiraz, islamic republic of iran, 1960–2001. East. Mediterr. Health J. 2004, 10, 27–36. [Google Scholar] [PubMed]
- Berger, T.; Kassirer, M.; Aran, A.A. Injectional anthrax—New presentation of an old disease. Euro. Surveill. 2014, 19. [Google Scholar] [CrossRef]
- Walsh, J.J.; Pesik, N.; Quinn, C.P.; Urdaneta, V.; Dykewicz, C.A.; Boyer, A.E.; Guarner, J.; Wilkins, P.; Norville, K.J.; Barr, J.R.; et al. A case of naturally acquired inhalation anthrax: Clinical care and analyses of anti-protective antigen immunoglobulin g and lethal factor. Clin. Infect. Dis. 2007, 44, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Anaraki, S.; Addiman, S.; Nixon, G.; Krahe, D.; Ghosh, R.; Brooks, T.; Lloyd, G.; Spencer, R.; Walsh, A.; McCloskey, B.; et al. Investigations and control measures following a case of inhalation anthrax in East London in a drum maker and drummer, October 2008. Euro. Surveill. 2008, 13, 11–13. [Google Scholar]
- Klempner, M.S.; Talbot, E.A.; Lee, S.I.; Zaki, S.; Ferraro, M.J. Case records of the massachusetts general hospital. Case 25–2010. A 24-year-old woman with abdominal pain and shock. N. Engl. J. Med. 2010, 363, 766–777. [Google Scholar] [PubMed]
- Sprenkle, M.D.; Griffith, J.; Marinelli, W.; Boyer, A.E.; Quinn, C.P.; Pesik, N.T.; Hoffmaster, A.; Keenan, J.; Juni, B.A.; Blaney, D.D. Lethal factor and anti-protective antigen IGG levels associated with inhalation anthrax, minnesota, USA. Emerg. Infect. Dis. 2014, 20, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Babamahmoodi, F.; Aghabarari, F.; Arjmand, A.; Ashrafi, G.H. Three rare cases of anthrax arising from the same source. J. Infect. 2006, 53, e175–e179. [Google Scholar] [CrossRef] [PubMed]
- Doganay, M.; Metan, G.; Alp, E. A review of cutaneous anthrax and its outcome. J. Infect. Public Health 2010, 3, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Popescu, R.; Pistol, A.; Miltaru, L.; Caplan, D.; Cucuiu, R.; Popovici, F. Two cases of infection with bacillus anthracis, Romania, October 2011. Euro. Surveill. 2011, 16, 733–735. [Google Scholar]
- Powell, A.G.; Crozier, J.E.; Hodgson, H.; Galloway, D.J. A case of septicaemic anthrax in an intravenous drug user. BMC Infect. Dis. 2011, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Grunow, R.; Verbeek, L.; Jacob, D.; Holzmann, T.; Birkenfeld, G.; Wiens, D.; von Eichel-Streiber, L.; Grass, G.; Reischl, U. Injection anthrax—A new outbreak in heroin users. Dtsch. Arztebl. Int. 2012, 109, 843–848. [Google Scholar] [PubMed]
- Mayer, T.A.; Bersoff-Matcha, S.; Murphy, C.; Earls, J.; Harper, S.; Pauze, D.; Nguyen, M.; Rosenthal, J.; Cerva, D., Jr.; Druckenbrod, G.; et al. Clinical presentation of inhalational anthrax following bioterrorism exposure: Report of 2 surviving patients. JAMA 2001, 286, 2549–2553. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.; Keppie, J. Observations on experimental anthrax; demonstration of a specific lethal factor produced in vivo by bacillus anthracis. Nature 1954, 173, 869–870. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.; Keppie, J.; Stanley, J.L.; Harris-Smith, P.W. The chemical basis of the virulence of bacillus anthracis. IV. Secondary shock as the major factor in death of guinea-pigs from anthrax. Br. J. Exp. Pathol. 1955, 36, 323–335. [Google Scholar] [PubMed]
- Sherer, K.; Li, Y.; Cui, X.; Eichacker, P.Q. Lethal and edema toxins in the pathogenesis of bacillus anthracis septic shock: Implications for therapy. Am. J. Respir. Crit. Care Med. 2007, 175, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Leppla, S.H. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects Med. 2009, 30, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Haines, D.; Young, H.A.; Leppla, S.H. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J. Clin. Invest. 2003, 112, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Moayeri, M.; Li, Y.; Li, X.; Haley, M.; Fitz, Y.; Correa-Araujo, R.; Banks, S.M.; Leppla, S.H.; Eichacker, P.Q. Lethality during continuous anthrax lethal toxin infusion is associated with circulatory shock but not inflammatory cytokine or nitric oxide release in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R699–R709. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.R.; Willingham, M.C.; Bour, S.H.; Andreas, E.A.; Park, S.K.; Jackson, C.; Duesbery, N.S.; Leppla, S.H.; Tang, W.J.; Frankel, A.E. Anthrax toxin-induced shock in rats is associated with pulmonary edema and hemorrhage. Microb. Pathog. 2008, 44, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.E.; Mock, J.; Lal, H.; Lu, G.; Bourdeau, R.W.; Tang, W.J.; Leppla, S.H.; Dostal, D.E.; Frankel, A.E. Lethal and edema toxins of anthrax induce distinct hemodynamic dysfunction. Front. Biosci. 2007, 12, 4670–4675. [Google Scholar] [CrossRef] [PubMed]
- Kandadi, M.R.; Hua, Y.; Ma, H.; Li, Q.; Kuo, S.R.; Frankel, A.E.; Ren, J. Anthrax lethal toxin suppresses murine cardiomyocyte contractile function and intracellular Ca2+ handling via a nadph oxidase-dependent mechanism. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Kandadi, M.R.; Yu, X.; Frankel, A.E.; Ren, J. Cardiac-specific catalase overexpression rescues anthrax lethal toxin-induced cardiac contractile dysfunction: Role of oxidative stress and autophagy. BMC Med. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Golden, H.B.; Watson, L.E.; Lal, H.; Verma, S.K.; Foster, D.M.; Kuo, S.R.; Sharma, A.; Frankel, A.; Dostal, D.E. Anthrax toxin: Pathologic effects on the cardiovascular system. Front. Biosci (Landmark Ed.) 2009, 14, 2335–2357. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Kuo, S.R.; Dostal, D.; Watson, L.; Duesbery, N.S.; Cheng, C.P.; Cheng, H.J.; Leppla, S.H. Pathophysiology of anthrax. Front. Biosci. (Landmark Ed.) 2009, 14, 4516–4524. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-P.; Masutani, S.; Cheng, H.-J.; Cross, M.; Zhang, C.-X.; Zhou, P.; Cann, J.; Cline, J.M.; Little, W.C.; Kuo, S.-R.; et al. Progressive left ventricle, myocyte dysfunction, and heart failure in the lethality of anthrax toxin in conscious dogs. Circulation 2007, 116, 758. [Google Scholar]
- Barochia, A.V.; Cui, X.; Sun, J.; Li, Y.; Solomon, S.B.; Migone, T.S.; Subramanian, G.M.; Bolmer, S.D.; Eichacker, P.Q. Protective antigen antibody augments hemodynamic support in anthrax lethal toxin shock in canines. J. Infect. Dis. 2012, 205, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.W.; Li, Y.; Okugawa, S.; Solomon, S.B.; Moayeri, M.; Leppla, S.H.; Mohanty, A.; Subramanian, G.M.; Mignone, T.S.; Fitz, Y.; et al. Anthrax edema toxin has camp-mediated stimulatory effects and high-dose lethal toxin has depressant effects in an isolated perfused rat heart model. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1108–H1118. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Abu-Asab, M.; Su, J.; Qiu, P.; Feng, J.; Ohanjanian, L.; Kumar, H.S.; Fitz, Y.; Eichacker, P.Q.; Cui, X. Bacillus anthracis edema but not lethal toxin challenge in rats is associated with depressed myocardial function in hearts isolated and tested in a langendorff system. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1592–H1602. [Google Scholar] [CrossRef] [PubMed]
- Brojatsch, J.; Casadevall, A.; Goldman, D.L. Molecular determinants for a cardiovascular collapse in anthrax. Front. Biosci. (Elite Ed.) 2014, 6, 139–147. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suffredini, D.A.; Sampath-Kumar, H.; Li, Y.; Ohanjanian, L.; Remy, K.E.; Cui, X.; Eichacker, P.Q. Does Bacillus anthracis Lethal Toxin Directly Depress Myocardial Function? A Review of Clinical Cases and Preclinical Studies. Toxins 2015, 7, 5417-5434. https://doi.org/10.3390/toxins7124891
Suffredini DA, Sampath-Kumar H, Li Y, Ohanjanian L, Remy KE, Cui X, Eichacker PQ. Does Bacillus anthracis Lethal Toxin Directly Depress Myocardial Function? A Review of Clinical Cases and Preclinical Studies. Toxins. 2015; 7(12):5417-5434. https://doi.org/10.3390/toxins7124891
Chicago/Turabian StyleSuffredini, Dante A., Hanish Sampath-Kumar, Yan Li, Lernik Ohanjanian, Kenneth E. Remy, Xizhong Cui, and Peter Q. Eichacker. 2015. "Does Bacillus anthracis Lethal Toxin Directly Depress Myocardial Function? A Review of Clinical Cases and Preclinical Studies" Toxins 7, no. 12: 5417-5434. https://doi.org/10.3390/toxins7124891
APA StyleSuffredini, D. A., Sampath-Kumar, H., Li, Y., Ohanjanian, L., Remy, K. E., Cui, X., & Eichacker, P. Q. (2015). Does Bacillus anthracis Lethal Toxin Directly Depress Myocardial Function? A Review of Clinical Cases and Preclinical Studies. Toxins, 7(12), 5417-5434. https://doi.org/10.3390/toxins7124891