
Conotoxins That Could Provide Analgesia through Voltage Gated Sodium Channel Inhibition

Abstract
:
1. Introduction
2. NaV Channels Structure
3. NaV Channel Subtypes
4. Roles of Sodium Channels in Nociception and Chronic Pain
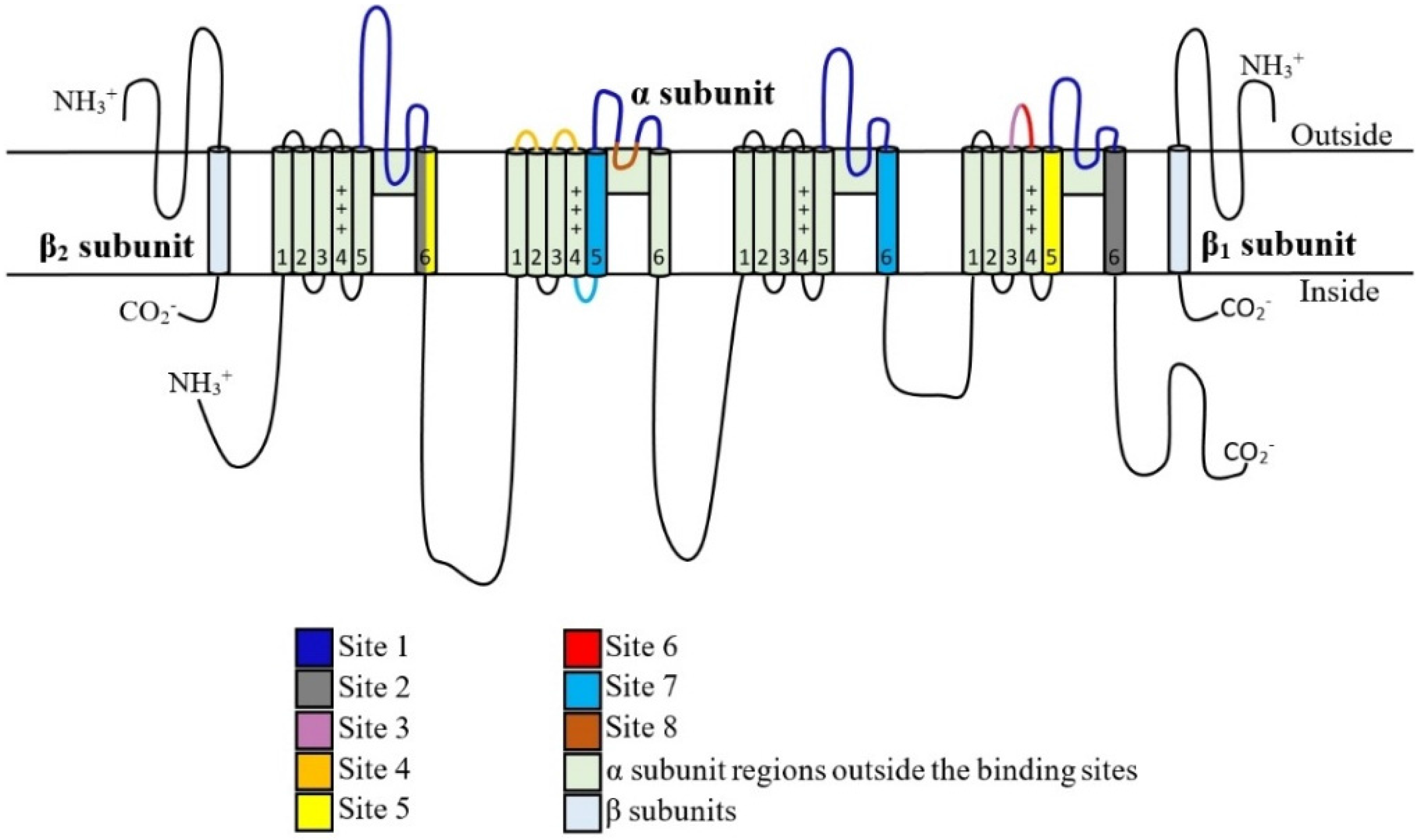
5. Toxin Interaction with NaV Channels
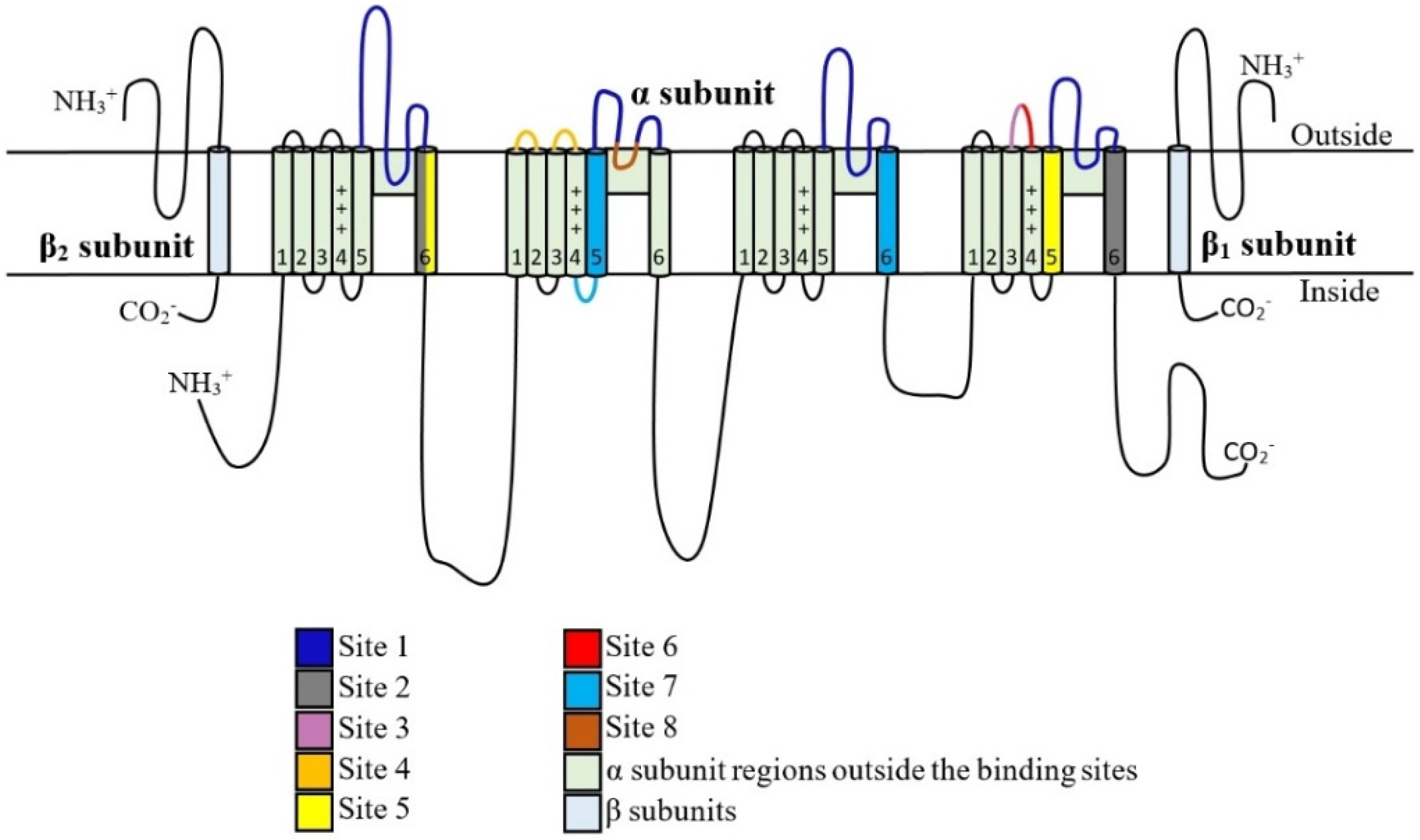
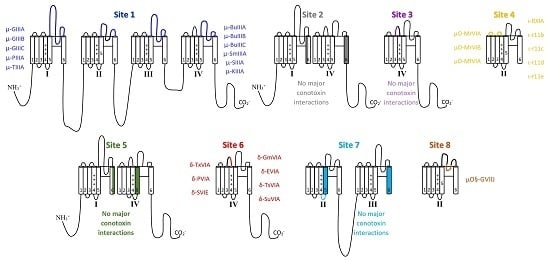
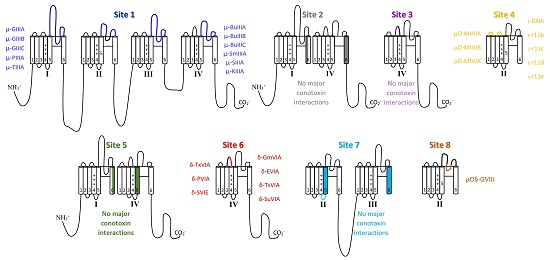
5.1. μ-Conotoxins that Interact with Binding Site 1

{kind=link}
{kind=link}
| μ-Conotoxin Type | Conus Species | Primary NaV Channel Targets |
|---|---|---|
| GIIIA | geographus | NaV1.4 [59,60] |
| GIIIB | geographus | NaV1.4 [61] |
| GIIIC | geographus | NaV1.4 [3,62] |
| PIIIA | purpurascens | NaV1.4, 1.2, 1.7 [63,64] |
| TIIIA | tulipa | NaV1.4, 1.2 [65,66] |
| BuIIIA | bullatus | NaV1.4 [67] |
| BuIIIB | bullatus | NaV1.3, NaV1.4 [67,68,69] |
| BuIIIC | bullatus | NaV1.4 [67] |
| SmIIIA | stercusmuscarum | NaV1.8 [70,71] |
| SIIIA | striatus | NaV1.2, 1.3, 1.4 [72,73] |
| KIIIA | kinoshitai | NaV1.2, 1.3, 1.4, 1.5, 1.6 [74,75] |
5.2. Binding Site 2 and 3 with No Major Conotoxin Interactions
5.3. µO- and Possibly ι-Conotoxin Binding Site 4
5.4. Site 5 Does Not Show Major Conotoxin Interactions
5.5. δ-Conotoxin Binding Site 6
| δ-Conotoxin Type | Conus Species | Primary NaV Channel Targets |
|---|---|---|
| TxVIA | textile | Undetermined (no activity in mammals) [113,114,115] |
| PVIA | purpurascens | NaV1.2, 1.4, 1.7 [64,116] |
| SVIE | striatus | NaV1.4 [112] |
| GmVIA | gloriamaris | NaV1.2, 1.4 [117,118] |
| EVIA | ermineus | NaV1.2, 1.3, 1.6 [119] |
| TsVIA | tessulatus | NaV1.6 [120] |
| SuVIA | suturatus | NaV1.3, 1.4, 1.6 and 1.7 [121] |
5.6. Site 7 Does Not Show Major Conotoxin Interactions
5.7. µO§-GVIIJ Conotoxin Binding Site 8
5.8. Local Anesthetic Binding Site
6. Future of Conotoxins As Potential Analgesics
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gaskin, D.J.; Richard, P. The economic costs of pain in the united states. J. Pain 2012, 13, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; McArthur, J.R.; Adams, D.J. Conotoxins targeting neuronal voltage-gated sodium channel subtypes: Potential analgesics? Toxins 2012, 4, 1236–1260. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Almeida, T.F.; Roizenblatt, S.; Tufik, S. Afferent pain pathways: A neuroanatomical review. Brain Res. 2004, 1000, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Von Hehn, C.A.; Baron, R.; Woolf, C.J. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012, 73, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Rush, A.M.; Cummins, T.R.; Waxman, S.G. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 2007, 579, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, J.; Jayamanne, A.; Vaughan, C.W.; Aslan, S.; Thomas, L.; Mould, J.; Drinkwater, R.; Baker, M.D.; Abrahamsen, B.; Wood, J.N.; et al. μO-conotoxin MrVIB selectively blocks Nav1.8 sensory neuron specific sodium channels and chronic pain behavior without motor deficits. Proc. Natl. Acad. Sci. USA 2006, 103, 17030–17035. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gu, J.; Li, Y.Q.; Tao, Y.X. Are voltage-gated sodium channels on the dorsal root ganglion involved in the development of neuropathic pain? Mol. Pain 2011, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Damir, S.; Sandra, K.; Adriana, B.; Livia, P. Dorsal root ganglion—A potential new therapeutic target for neuropathic pain. J. Pain Res. 2012, 5, 31–38. [Google Scholar]
- Scroggs, R.S.; Fox, A.P. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. J. Physiol. 1992, 445, 639–658. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and function of voltage-gated sodium channels at atomic resolution. Exp. Physiol. 2014, 99, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Beneski, D.A.; Catterall, W.A. Covalent labeling of protein components of the sodium channel with a photoactivable derivative of scorpion toxin. Proc. Natl. Acad. Sci. USA 1980, 77, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International union of pharmacology. XlVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef]
- Dichgans, M.; Freilinger, T.; Eckstein, G.; Babini, E.; Lorez-Depiereux, B.; Biskup, S. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 2005, 371–377. [Google Scholar] [CrossRef]
- Kim, C.H.; Oh, Y.; Chung, J.M.; Chung, K. The changes in expression of three subtypes of TTX sensitive sodium channels in sensory neurons after spinal nerve ligation. Brain Res. Mol. Brain Res. 2001, 95, 153–161. [Google Scholar] [CrossRef]
- Waxman, S.G.; Kocsis, J.D.; Black, J.A. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J. Neurophysiol. 1994, 72, 466–470. [Google Scholar] [PubMed]
- Lindia, J.A.; Kohler, M.G.; Martin, W.J.; Abbadie, C. Relationship between sodium channel Nav1.3 expression and neuropathic pain behavior in rats. Pain 2005, 117, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, T.; Kobayashi, K.; Yamanaka, H.; Obata, K.; Dai, Y.; Noguchi, K. Comparative study of the distribution of the α-subunits of voltage-gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. J. Comp. Neurol. 2008, 510, 188–206. [Google Scholar] [CrossRef] [PubMed]
- Nassar, M.A.; Baker, M.D.; Levato, A.; Ingram, R.; Mallucci, G.; McMahon, S.B.; Wood, J.N. Nerve injury induces robust allodynia and ectopic discharges in Nav1.3 null mutant mice. Mol. Pain 2006, 2. [Google Scholar] [CrossRef] [PubMed]
- Berta, T.; Poirot, O.; Pertin, M.; Ji, R.R.; Kellenberger, S.; Decosterd, I. Transcriptional and functional profiles of voltage-gated Na+ channels in injured and non-injured DRG neurons in the SNI model of neuropathic pain. Mol. Cell. Neurosci. 2008, 37, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.A.; Freking, A.R.; Johnson, L.R.; Levinson, S.R. Sodium channel Nav1.6 accumulates at the site of infraorbital nerve injury. BMC Neurosci. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Dib-Hajj, S.; McNabola, K.; Jeste, S.; Rizzo, M.A.; Kocsis, J.D.; Waxman, S.G. Spinal sensory neurons express multiple sodium channel α-subunit mrnas. Brain Res. Mol. Brain Res. 1996, 43, 117–131. [Google Scholar] [CrossRef]
- Cummins, T.R.; Howe, J.R.; Waxman, S.G. Slow closed-state inactivation: A novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 9607–9619. [Google Scholar]
- Black, J.A.; Liu, S.; Tanaka, M.; Cummins, T.R.; Waxman, S.G. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 2004, 108, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Strickland, I.T.; Martindale, J.C.; Woodhams, P.L.; Reeve, A.J.; Chessell, I.P.; McQueen, D.S. Changes in the expression of Nav1.7, Nav1.8 and Nav1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur. J. Pain (Lond. Engl.) 2008, 12, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Nassar, M.A.; Stirling, L.C.; Forlani, G.; Baker, M.D.; Matthews, E.A.; Dickenson, A.H.; Wood, J.N. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. USA 2004, 101, 12706–12711. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Rush, A.M.; Cummins, T.R.; Hisama, F.M.; Novella, S.; Tyrrell, L.; Marshall, L.; Waxman, S.G. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain A J. Neurol. 2005, 128, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Yu, H.S.; Hsieh, S.T.; Stephenson, D.A.; Lu, C.J.; Yang, C.C. Characterization of a familial case with primary erythromelalgia from taiwan. J. Neurol. 2007, 254, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Morrow, T.J.; Paulson, P.E.; Isom, L.L.; Wiley, J.W. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J. Biol. Chem. 2004, 279, 29341–29350. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Oh, Y.; Chung, J.M.; Chung, K. Changes in three subtypes of tetrodotoxin sensitive sodium channel expression in the axotomized dorsal root ganglion in the rat. Neurosci. Lett. 2002, 323, 125–128. [Google Scholar] [CrossRef]
- Coward, K.; Aitken, A.; Powell, A.; Plumpton, C.; Birch, R.; Tate, S.; Bountra, C.; Anand, P. Plasticity of TTX-sensitive sodium channels PN1 and brain III in injured human nerves. Neuroreport 2001, 12, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Nassar, M.A.; Levato, A.; Stirling, L.C.; Wood, J.N. Neuropathic pain develops normally in mice lacking both Nav1.7 and Nav1.8. Mol. Pain 2005, 1. [Google Scholar] [CrossRef] [PubMed]
- Bird, E.V.; Robinson, P.P.; Boissonade, F.M. Nav1.7 sodium channel expression in human lingual nerve neuromas. Arch. Oral Biol. 2007, 52, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Nikolajsen, L.; Kroner, K.; Jensen, T.S.; Waxman, S.G. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann. Neurol. 2008, 64, 644–653. [Google Scholar] [CrossRef] [PubMed]
- King, G.F.; Vetter, I. No gain, no pain: Nav1.7 as an analgesic target. ACS Chem. Neurosci. 2014, 5, 749–751. [Google Scholar] [CrossRef] [PubMed]
- Djouhri, L.; Fang, X.; Okuse, K.; Wood, J.N.; Berry, C.M.; Lawson, S.N. The TTX-resistant sodium channel Nav1.8 (SNS/PN3): Expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J. Physiol. 2003, 550, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Coggeshall, R.E.; Tate, S.; Carlton, S.M. Differential expression of tetrodotoxin-resistant sodium channels Nav1.8 and Nav1.9 in normal and inflamed rats. Neurosci. Lett. 2004, 355, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Akopian, A.N.; Souslova, V.; England, S.; Okuse, K.; Ogata, N.; Ure, J.; Smith, A.; Kerr, B.J.; McMahon, S.B.; Boyce, S.; et al. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat. Neurosci. 1999, 2, 541–548. [Google Scholar] [PubMed]
- Decosterd, I.; Ji, R.R.; Abdi, S.; Tate, S.; Woolf, C.J. The pattern of expression of the voltage-gated sodium channels Nav1.8 and Nav1.9 does not change in uninjured primary sensory neurons in experimental neuropathic pain models. Pain 2002, 96, 269–277. [Google Scholar] [CrossRef]
- Gold, M.S.; Weinreich, D.; Kim, C.S.; Wang, R.; Treanor, J.; Porreca, F.; Lai, J. Redistribution of Nav1.8 in uninjured axons enables neuropathic pain. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 158–166. [Google Scholar]
- He, X.H.; Zang, Y.; Chen, X.; Pang, R.P.; Xu, J.T.; Zhou, X.; Wei, X.H.; Li, Y.Y.; Xin, W.J.; Qin, Z.H.; et al. TNF-alpha contributes to up-regulation of Nav1.3 and Nav1.8 in drg neurons following motor fiber injury. Pain 2010, 151, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M.F.; Honore, P.; Shieh, C.C.; Chapman, M.; Joshi, S.; Zhang, X.F.; Kort, M.; Carroll, W.; Marron, B.; Atkinson, R.; et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. USA 2007, 104, 8520–8525. [Google Scholar] [CrossRef] [PubMed]
- Faber, C.G.; Lauria, G.; Merkies, I.S.; Cheng, X.; Han, C.; Ahn, H.S.; Persson, A.K.; Hoeijmakers, J.G.; Gerrits, M.M.; Pierro, T.; et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 19444–19449. [Google Scholar] [CrossRef] [PubMed]
- Roza, C.; Laird, J.M.A.; Souslova, V.; Wood, J.N.; Cervero, F. The tetrodotoxin-resistant Na+ channel Nav1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J. Physiol. 2003, 550, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.J.; Souslova, V.; McMahon, S.B.; Wood, J.N. A role for the TTX-resistant sodium channel Nav1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport 2001, 12, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Savio-Galimberti, E.; Weeke, P.; Muhammad, R.; Blair, M.; Ansari, S.; Short, L.; Atack, T.C.; Kor, K.; Vanoye, C.G.; Olesen, M.S.; et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc. Res. 2014, 104, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.L. Voltage-gated sodium channel mutations and painful neuropathy: Nav1.9 joins the family. Brain A J. Neurol. 2014, 137, 1574–1576. [Google Scholar] [CrossRef] [PubMed]
- Tate, S.; Benn, S.; Hick, C.; Trezise, D.; John, V.; Mannion, R.J.; Costigan, M.; Plumpton, C.; Grose, D.; Gladwell, Z.; et al. Two sodium channels contribute to the TTX-R sodium current in primary sensory neurons. Nat. Neurosci. 1998, 1, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Priest, B.T.; Murphy, B.A.; Lindia, J.A.; Diaz, C.; Abbadie, C.; Ritter, A.M.; Liberator, P.; Iyer, L.M.; Kash, S.F.; Kohler, M.G.; et al. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel Nav1.9 to sensory transmission and nociceptive behavior. Proc. Natl. Acad. Sci. USA 2005, 102, 9382–9387. [Google Scholar] [CrossRef] [PubMed]
- Amaya, F.; Wang, H.; Costigan, M.; Allchorne, A.J.; Hatcher, J.P.; Egerton, J.; Stean, T.; Morisset, V.; Grose, D.; Gunthorpe, M.J.; et al. The voltage-gated sodium channel Nav1.9 is an effector of peripheral inflammatory pain hypersensitivity. J. Neurosci. 2006, 26, 12852–12860. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Han, C.; Estacion, M.; Vasylyev, D.; Hoeijmakers, J.G.; Gerrits, M.M.; Tyrrell, L.; Lauria, G.; Faber, C.G.; Dib-Hajj, S.D.; et al. Gain-of-function mutations in sodium channel Nav1.9 in painful neuropathy. Brain A J. Neurol. 2014, 137, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Leipold, E.; Liebmann, L.; Korenke, G.C.; Heinrich, T.; Giesselmann, S.; Baets, J.; Ebbinghaus, M.; Goral, R.O.; Stodberg, T.; Hennings, J.C.; et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 2013, 45, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Wen, J.; Yang, W.; Wang, C.; Gao, L.; Zheng, L.H.; Wang, T.; Ran, K.; Li, Y.; Li, X.; et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am. J. Hum. Genet. 2013, 93, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure-activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Li, R.A.; Tomaselli, G.F. Using the deadly μ-conotoxins as probes of voltage-gated sodium channels. Toxic. Off. J. Int. Soc. Toxinol. 2004, 44, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Fozzard, H.A.; Lipkind, G.M. The tetrodotoxin binding site is within the outer vestibule of the sodium channel. Mar. Drugs 2010, 8, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Aglieco, F.; Dib-Hajj, S.D. Critical molecular determinants of voltage-gated sodium channel sensitivity to μ-conotoxins GIIIA/B. Mol. Pharmacol. 2002, 61, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Kupryszewski, G.; LeCheminant, G.W.; Gray, W.R.; Olivera, B.M.; Rivier, J. μ-conotoxin GIIIA, a peptide ligand for muscle sodium channels: Chemical synthesis, radiolabeling and receptor characterization. Biochemistry 1989, 28, 3437–3442. [Google Scholar] [CrossRef] [PubMed]
- .Hill, J.M.; Alewood, P.F.; Craik, D.J. Three-dimensional solution structure of μ-conotoxin GIIIB, a specific blocker of skeletal muscle sodium channels. Biochemistry 1996, 35, 8824–8835. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Gray, W.R.; Olivera, B.M.; Zeikus, R.D.; Kerr, L.; Yoshikami, D.; Moczydlowski, E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem. 1985, 260, 9280–9288. [Google Scholar] [PubMed]
- Shon, K.J.; Olivera, B.M.; Watkins, M.; Jacobsen, R.B.; Gray, W.R.; Floresca, C.Z.; Cruz, L.J.; Hillyard, D.R.; Brink, A.; Terlau, H.; et al. μ-conotoxin PIIIA, a new peptide for discriminating among tetrodotoxin-sensitive Na channel subtypes. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 4473–4481. [Google Scholar]
- Safo, P.; Rosenbaum, T.; Shcherbatko, A.; Choi, D.-Y.; Han, E.; Toledo-Aral, J.J.; Olivera, B.M.; Brehm, P.; Mandel, G. Distinction among neuronal subtypes of voltage-activated sodium channels by μ-conotoxin PIIIA. J. Neurosci. 2000, 20, 76–80. [Google Scholar] [PubMed]
- Lewis, R.J.; Schroeder, C.I.; Ekberg, J.; Nielsen, K.J.; Loughnan, M.; Thomas, L.; Adams, D.A.; Drinkwater, R.; Adams, D.J.; Alewood, P.F. Isolation and structure-activity of μ-conotoxin TIIIA, a potent inhibitor of tetrodotoxin-sensitive voltage-gated sodium channels. Mol. Pharmacol. 2007, 71, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, J.; Adams, D.J. Neuronal voltage-gated sodium channel subtypes: Key roles in inflammatory and neuropathic pain. Int. J. Biochem. Cell Biol. 2006, 38, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Holford, M.; Zhang, M.M.; Gowd, K.H.; Azam, L.; Green, B.R.; Watkins, M.; Ownby, J.P.; Yoshikami, D.; Bulaj, G.; Olivera, B.M. Pruning nature: Biodiversity-derived discovery of novel sodium channel blocking conotoxins from conus bullatus. Toxic. Off. J. Int. Soc. Toxinol. 2009, 53, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Green, B.R.; Zhang, M.M.; Chhabra, S.; Robinson, S.D.; Wilson, M.J.; Redding, A.; Olivera, B.M.; Yoshikami, D.; Bulaj, G.; Norton, R.S. Interactions of disulfide-deficient selenocysteine analogs of μ-conotoxin BUIIIB with the α-subunit of the voltage-gated sodium channel subtype 1.3. FEBS J. 2014, 281, 2885–2898. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.; Zhang, M.-M.; Gupta, K.; Gajewiak, J.; Gulyas, J.; Balaram, P.; Rivier, J.E.; Olivera, B.M.; Yoshikami, D.; Bulaj, G.; et al. The mammalian neuronal sodium channel blocker μ-conotoxin BuIIIB has a structured N-terminus that influences potency. ACS Chem. Biol. 2013, 8, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- West, P.J.; Bulaj, G.; Garrett, J.E.; Olivera, B.M.; Yoshikami, D. μ-conotoxin smiiia, a potent inhibitor of tetrodotoxin-resistant sodium channels in amphibian sympathetic and sensory neurons. Biochemistry 2002, 41, 15388–15393. [Google Scholar] [CrossRef] [PubMed]
- Keizer, D.W.; West, P.J.; Lee, E.F.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structural basis for tetrodotoxin-resistant sodium channel binding by μ-conotoxin SmIIIA. J. Biol. Chem. 2003, 278, 46805–46813. [Google Scholar] [CrossRef] [PubMed]
- Bulaj, G.; West, P.J.; Garrett, J.E.; Watkins, M.; Zhang, M.M.; Norton, R.S.; Smith, B.J.; Yoshikami, D.; Olivera, B.M. Novel conotoxins from conus striatus and conus kinoshitai selectively block TTX-resistant sodium channels. Biochemistry 2005, 44, 7259–7265. [Google Scholar] [CrossRef] [PubMed]
- Green, B.R.; Catlin, P.; Zhang, M.M.; Fiedler, B.; Bayudan, W.; Morrison, A.; Norton, R.S.; Smith, B.J.; Yoshikami, D.; Olivera, B.M.; et al. Conotoxins containing nonnatural backbone spacers: Cladistic-based design, chemical synthesis, and improved analgesic activity. Chem. Biol. 2007, 14, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-M.; Han, T.S.; Olivera, B.M.; Bulaj, G.; Yoshikami, D. µ-conotoxin KIIIA derivatives with divergent affinities versus efficacies in blocking voltage-gated sodium channels. Biochemistry 2010, 49, 4804–4812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Green, B.R.; Catlin, P.; Fiedler, B.; Azam, L.; Chadwick, A.; Terlau, H.; McArthur, J.R.; French, R.J.; Gulyas, J.; et al. Structure/function characterization of μ-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J. Biol. Chem. 2007, 282, 30699–30706. [Google Scholar] [CrossRef] [PubMed]
- Moczydlowski, E.; Olivera, B.M.; Gray, W.R.; Strichartz, G.R. Discrimination of muscle and neuronal Na-channel subtypes by binding competition between [3H]saxitoxin and mu-conotoxins. Proc. Natl. Acad. Sci. USA 1986, 83, 5321–5325. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Prusak-Sochaczewski, E.; Zamponi, G.; Beck-Sickinger, A.G.; Gordon, R.D.; French, R.J. Action of derivatives of μ-conotoxin GIIIA on sodium channels. Single amino acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry 1992, 31, 8229–8238. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Kohda, D.; Hatanaka, H.; Lancelin, J.M.; Ishida, Y.; Oya, M.; Nakamura, H.; Inagaki, F.; Sato, K. Structure-activity relationships of μ-conotoxin GIIIA: Structure determination of active and inactive sodium channel blocker peptides by nmr and simulated annealing calculations. Biochemistry 1992, 31, 12577–12584. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, S.; Kuyucak, S. Molecular dynamics study of binding of µ-conotoxin GIIIA to the voltage-gated sodium channel Nav1.4. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- McArthur, J.R.; Singh, G.; O'Mara, M.L.; McMaster, D.; Ostroumov, V.; Tieleman, D.P.; French, R.J. Orientation of μ-conotoxin PIIIA in a sodium channel vestibule, based on voltage dependence of its binding. Mol. Pharmacol. 2011, 80, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Zhang, M.M.; Yoshikami, D.; Azam, L.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure, dynamics, and selectivity of the sodium channel blocker μ-conotoxin SIIIA. Biochemistry 2008, 47, 10940–10949. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, C.I.; Ekberg, J.; Nielsen, K.J.; Adams, D.; Loughnan, M.L.; Thomas, L.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Neuronally selective μ-conotoxins from conus striatus utilize an α-helical motif to target mammalian sodium channels. J. Biol. Chem. 2008, 283, 21621–21628. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.A.; Fisher, K.M.; Lapointe, B.; du Souich, P.; Chary, S.; Moulin, D.; Sellers, E.; Ngoc, A.H. An open-label, multi-dose efficacy and safety study of intramuscular tetrodotoxin in patients with severe cancer-related pain. J. Pain Symptom Manag. 2007, 34, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.A.; du Souich, P.; Lapointe, B.; Ong-Lam, M.; Dubuc, B.; Walde, D.; Love, R.; Ngoc, A.H. Tetrodotoxin for moderate to severe cancer pain: A randomized, double blind, parallel design multicenter study. J. Pain Symptom Manag. 2008, 35, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.A.; Lapointe, B.; Ong–Lam, M.; Dubuc, B.; Walde, D.; Gagnon, B.; Love, R.; Goel, R.; Hawley, P.; Ngoc, A.H.; et al. A multicentre open-label safety and efficacy study of tetrodotoxin for cancer pain. Curr. Oncol. 2011, 18, e109–e116. [Google Scholar] [CrossRef] [PubMed]
- Khoo, K.K.; Feng, Z.P.; Smith, B.J.; Zhang, M.M.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure of the analgesic μ-conotoxin KIIIA and effects on the structure and function of disulfide deletion. Biochemistry 2009, 48, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; McArthur, J.R.; Azam, L.; Bulaj, G.; Olivera, B.M.; French, R.J.; Yoshikami, D. Synergistic and antagonistic interactions between tetrodotoxin and μ-conotoxin in blocking voltage-gated sodium channels. Channels (Austin) 2009, 3, 32–38. [Google Scholar] [CrossRef] [PubMed]
- French, R.J.; Yoshikami, D.; Sheets, M.F.; Olivera, B.M. The tetrodotoxin receptor of voltage-gated sodium channels—Perspectives from interactions with μ-conotoxins. Mar. Drugs 2010, 8, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Wang, G.K. Voltage-gated sodium channels as primary targets of diverse lipid-soluble neurotoxins. Cell. Signal. 2003, 15, 151–159. [Google Scholar] [CrossRef]
- Cestèle, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef]
- Zhu, H.-L.; Wassall, R.D.; Takai, M.; Morinaga, H.; Nomura, M.; Cunnane, T.C.; Teramoto, N. Actions of veratridine on tetrodotoxin-sensitive voltage-gated Na+ currents, Nav1.6, in murine vas deferens myocytes. Br. J. Pharmacol. 2009, 157, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.V.; Chanda, B.; Beirao, P.S.; Bezanilla, F. α-scorpion toxin impairs a conformational change that leads to fast inactivation of muscle sodium channels. J. Gen. Physiol. 2008, 132, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Gurevitz, M. Mapping of scorpion toxin receptor sites at voltage-gated sodium channels. Toxic. Off. J. Int. Soc. Toxinol. 2012, 60, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Leipold, E.; Hansel, A.; Borges, A.; Heinemann, S.H. Subtype specificity of scorpion β-toxin Tz1 interaction with voltage-gated sodium channels is determined by the pore loop of domain 3. Mol. Pharmacol. 2006, 70, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Zorn, S.; Leipold, E.; Hansel, A.; Bulaj, G.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. The μO-conotoxin MrVIA inhibits voltage-gated sodium channels by associating with domain-3. FEBS Lett. 2006, 580, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Fainzilber, M.; van der Schors, R.; Lodder, J.C.; Li, K.W.; Geraerts, W.P.; Kits, K.S. New sodium channel-blocking conotoxins also affect calcium currents in lymnaea neurons. Biochemistry 1995, 34, 5364–5371. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Hasson, A.; Spira, M.E.; Gray, W.R.; Li, W.; Marsh, M.; Hillyard, D.R.; Olivera, B.M. A new family of conotoxins that blocks voltage-gated sodium channels. J. Biol. Chem. 1995, 270, 16796–16802. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, S.H.; Leipold, E. Conotoxins of the O-superfamily affecting voltage-gated sodium channels. Cell. Mol. Life Sci. 2007, 64, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Leipold, E.; DeBie, H.; Zorn, S.; Borges, A.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. μO-conotoxins inhibit Nav channels by interfering with their voltage sensors in domain-2. Channels (Austin) 2007, 1, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Peigneur, S.; Tytgat, J. Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.J.; Zhang, M.-M.; Azam, L.; Olivera, B.M.; Bulaj, G.; Yoshikami, D. Navβ subunits modulate the inhibition of Nav1.8 by the analgesic gating modifier μO-conotoxin MrVIB. J. Pharmacol. Exp. Ther. 2011, 338, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Daly, N.L.; Ekberg, J.A.; Thomas, L.; Adams, D.J.; Lewis, R.J.; Craik, D.J. Structures of μO-conotoxins from conus marmoreus. I nhibitors of tetrodotoxin (TTX)-sensitive and TTX-resistant sodium channels in mammalian sensory neurons. J. Biol. Chem. 2004, 279, 25774–25782. [Google Scholar] [CrossRef] [PubMed]
- Pertin, M.; Ji, R.R.; Berta, T.; Powell, A.J.; Karchewski, L.; Tate, S.N.; Isom, L.L.; Woolf, C.J.; Gilliard, N.; Spahn, D.R.; et al. Upregulation of the voltage-gated sodium channel β2 subunit in neuropathic pain models: Characterization of expression in injured and non-injured primary sensory neurons. J. Neurosci. 2005, 25, 10970–10980. [Google Scholar] [CrossRef] [PubMed]
- Bulaj, G.; Zhang, M.-M.; Green, B.R.; Fiedler, B.; Layer, R.T.; Wei, S.; Nielsen, J.S.; Low, S.J.; Klein, B.D.; Wagstaff, J.D.; et al. Synthetic μO-conotoxin MrVIB blocks TTX-resistant sodium channel Nav1.8 and has a long-lasting analgesic activity. Biochemistry 2006, 45, 7404–7414. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, A.D.; Callaghan, B.; Nevin, S.T.; Daly, N.L.; Craik, D.J.; Moretta, M.; Hopping, G.; Christie, M.J.; Adams, D.J.; Alewood, P.F. Total synthesis of the analgesic conotoxin MrVIB through selenocysteine-assisted folding. Angew. Chem. 2011, 50, 6527–6529. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Dekan, Z.; Knapp, O.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Isolation, characterization and total regioselective synthesis of the novel μO-conotoxin MfVIA from conus magnificus that targets voltage-gated sodium channels. Biochem. Pharmacol. 2012, 84, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, B.; Zhang, M.-M.; Buczek, O.; Azam, L.; Bulaj, G.; Norton, R.S.; Olivera, B.M.; Yoshikami, D. Specificity, affinity and efficacy of iota-conotoxin RXIA, an agonist of voltage-gated sodium channels Nav1.2, 1.6 and 1.7. Biochem. Pharmacol. 2008, 75, 2334–2344. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, E.C.; Shetty, R.P.; Lirazan, M.; Rivier, J.; Walker, C.; Abogadie, F.C.; Yoshikami, D.; Cruz, L.J.; Olivera, B.M. Novel excitatory conus peptides define a new conotoxin superfamily. J. Neurochem. 2003, 85, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Buczek, O.; Wei, D.; Babon, J.J.; Yang, X.; Fiedler, B.; Chen, P.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure and sodium channel activity of an excitatory I1-superfamily conotoxin. Biochemistry 2007, 46, 9929–9940. [Google Scholar] [CrossRef] [PubMed]
- Schiavon, E.; Sacco, T.; Cassulini, R.R.; Gurrola, G.; Tempia, F.; Possani, L.D.; Wanke, E. Resurgent current and voltage sensor trapping enhanced activation by a β-scorpion toxin solely in Nav1.6 channel. Significance in mice purkinje neurons. J. Biol. Chem. 2006, 281, 20326–20337. [Google Scholar] [CrossRef] [PubMed]
- Cestele, S.; Qu, Y.; Rogers, J.C.; Rochat, H.; Scheuer, T.; Catterall, W.A. Voltage sensor-trapping: Enhanced activation of sodium channels by β-scorpion toxin bound to the S3-S4 loop in domain II. Neuron 1998, 21, 919–931. [Google Scholar] [CrossRef]
- Leipold, E.; Hansel, A.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. Molecular interaction of δ-conotoxins with voltage-gated sodium channels. FEBS Lett. 2005, 579, 3881–3884. [Google Scholar] [CrossRef] [PubMed]
- Hillyard, D.R.; Olivera, B.M.; Woodward, S.; Corpuz, G.P.; Gray, W.R.; Ramilo, C.A.; Cruz, L.J. A molluscivorous conus toxin: Conserved frameworks in conotoxins. Biochemistry 1989, 28, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Bruce, C.; Fitches, E.C.; Chougule, N.; Bell, H.A.; Gatehouse, J.A. Recombinant conotoxin, TxVIA, produced in yeast has insecticidal activity. Toxicon 2011, 58, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Fainzilber, M.; Kofman, O.; Zlotkin, E.; Gordon, D. A new neurotoxin receptor site on sodium channels is identified by a conotoxin that affects sodium channel inactivation in molluscs and acts as an antagonist in rat brain. J. Biol. Chem. 1994, 269, 2574–2580. [Google Scholar] [PubMed]
- Buczek, P.; Buczek, O.; Bulaj, G. Total chemical synthesis and oxidative folding of δ-conotoxin PVIA containing an N-terminal propeptide. Biopolymers 2005, 80, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Hasson, A.; Shon, K.J.; Olivera, B.M.; Spira, M.E. Alterations of voltage-activated sodium current by a novel conotoxin from the venom of conus gloriamaris. J. Neurophysiol. 1995, 73, 1295–1301. [Google Scholar] [PubMed]
- Shon, K.-J.; Hasson, A.; Spira, M.E.; Cruz, L.J.; Gray, W.R.; Olivera, B.M. δ-conotoxin GmVIA, a novel peptide from the venom of conus gloriamaris. Biochemistry 1994, 33, 11420–11425. [Google Scholar] [CrossRef] [PubMed]
- Barbier, J.; Lamthanh, H.; Le Gall, F.; Favreau, P.; Benoit, E.; Chen, H.; Gilles, N.; Ilan, N.; Heinemann, S.H.; Gordon, D.; et al. A δ-conotoxin from conus ermineus venom inhibits inactivation in vertebrate neuronal Na+ channels but not in skeletal and cardiac muscles. J. Biol. Chem. 2004, 279, 4680–4685. [Google Scholar] [CrossRef] [PubMed]
- Aman, J.W.; Imperial, J.S.; Ueberheide, B.; Zhang, M.-M.; Aguilar, M.; Taylor, D.; Watkins, M.; Yoshikami, D.; Showers-Corneli, P.; Safavi-Hemami, H.; et al. Insights into the origins of fish hunting in venomous cone snails from studies of conus tessulatus. Proc. Natl. Acad. Sci. USA 2015, 112, 5087–5092. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.-H.; Israel, M.R.; Inserra, M.C.; Smith, J.J.; Lewis, R.J.; Alewood, P.F.; Vetter, I.; Dutertre, S. δ-conotoxin SuVIA suggests an evolutionary link between ancestral predator defence and the origin of fish-hunting behaviour in carnivorous cone snails. Proc. R. Soc. Lond. B Biol. Sci. 2015, 282. [Google Scholar] [CrossRef] [PubMed]
- West, P.J.; Bulaj, G.; Yoshikami, D. Effects of δ-conotoxins PVIA and SVIE on sodium channels in the amphibian sympathetic nervous system. J. Neurophysiol. 2005, 94, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- O'Reilly, A.O.; Khambay, B.P.S.; Williamson, M.S.; Field, L.M.; Wallace, B.A.; Davies, T.G.E. Modelling insecticide-binding sites in the voltage-gated sodium channel. Biochem. J. 2006, 396, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Gajewiak, J.; Azam, L.; Imperial, J.; Walewska, A.; Green, B.R.; Bandyopadhyay, P.K.; Raghuraman, S.; Ueberheide, B.; Bern, M.; Zhou, H.M.; et al. A disulfide tether stabilizes the block of sodium channels by the conotoxin μO§-GVIIJ. Proc. Natl. Acad. Sci. USA 2014, 111, 2758–2763. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, D.S.; McPhee, J.C.; Scheuer, T.; Catterall, W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science (New York N.Y.) 1994, 265, 1724–1728. [Google Scholar] [CrossRef]
- Kalso, E.; Tramer, M.R.; McQuay, H.J.; Moore, R.A. Systemic local-anaesthetic-type drugs in chronic pain: A systematic review. Eur. J. Pain (Lond. Engl.) 1998, 2, 3–14. [Google Scholar] [CrossRef]
- Clare, J.J. Targeting voltage-gated sodium channels for pain therapy. Expert Opin. Investig. Drugs 2010, 19, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.S.; Dyck, J.B.; Rossi, S.S.; Yaksh, T.L. Computer-controlled lidocaine infusion for the evaluation of neuropathic pain after peripheral nerve injury. Pain 1996, 66, 69–77. [Google Scholar] [CrossRef]
- Sorensen, J.; Bengtsson, A.; Backman, E.; Henriksson, K.G.; Bengtsson, M. Pain analysis in patients with fibromyalgia. Effects of intravenous morphine, lidocaine, and ketamine. Scand. J. Rheumatol. 1995, 24, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M.; Cruz, L.J.; de Santos, V.; LeCheminant, G.; Griffin, D.; Zeikus, R.; McIntosh, J.M.; Galyean, R.; Varga, J. Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using ω-conotoxin from conus magus venom. Biochemistry 1987, 26, 2086–2090. [Google Scholar] [CrossRef] [PubMed]
- Snutch, T. Targeting chronic and neuropathic pain: The N-type calcium channel comes of age. Neurotherapeutics 2005, 2, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Chabal, C.; Jacobson, L.; Mariano, A.; Chaney, E.; Britell, C.W. The use of oral mexiletine for the treatment of pain after peripheral nerve injury. Anesthesiology 1992, 76, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Dejgard, A.; Petersen, P.; Kastrup, J. Mexiletine for treatment of chronic painful diabetic neuropathy. Lancet (Lond. Engl.) 1988, 1, 9–11. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munasinghe, N.R.; Christie, M.J. Conotoxins That Could Provide Analgesia through Voltage Gated Sodium Channel Inhibition. Toxins 2015, 7, 5386-5407. https://doi.org/10.3390/toxins7124890
Munasinghe NR, Christie MJ. Conotoxins That Could Provide Analgesia through Voltage Gated Sodium Channel Inhibition. Toxins. 2015; 7(12):5386-5407. https://doi.org/10.3390/toxins7124890
Chicago/Turabian StyleMunasinghe, Nehan R., and MacDonald J. Christie. 2015. "Conotoxins That Could Provide Analgesia through Voltage Gated Sodium Channel Inhibition" Toxins 7, no. 12: 5386-5407. https://doi.org/10.3390/toxins7124890
APA StyleMunasinghe, N. R., & Christie, M. J. (2015). Conotoxins That Could Provide Analgesia through Voltage Gated Sodium Channel Inhibition. Toxins, 7(12), 5386-5407. https://doi.org/10.3390/toxins7124890