
Recent Advances for the Detection of Ochratoxin A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
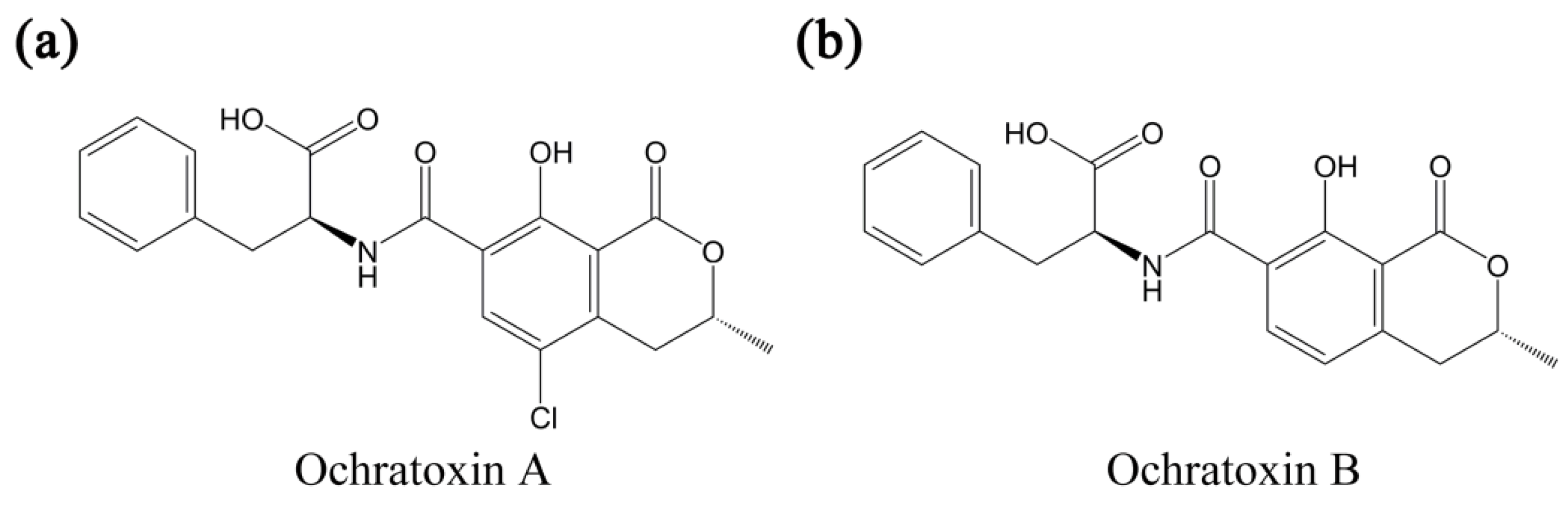
:1. Introduction
2. Conventional Detection Methods for OTA
2.1. Chromatographic and Immunoassays of OTA
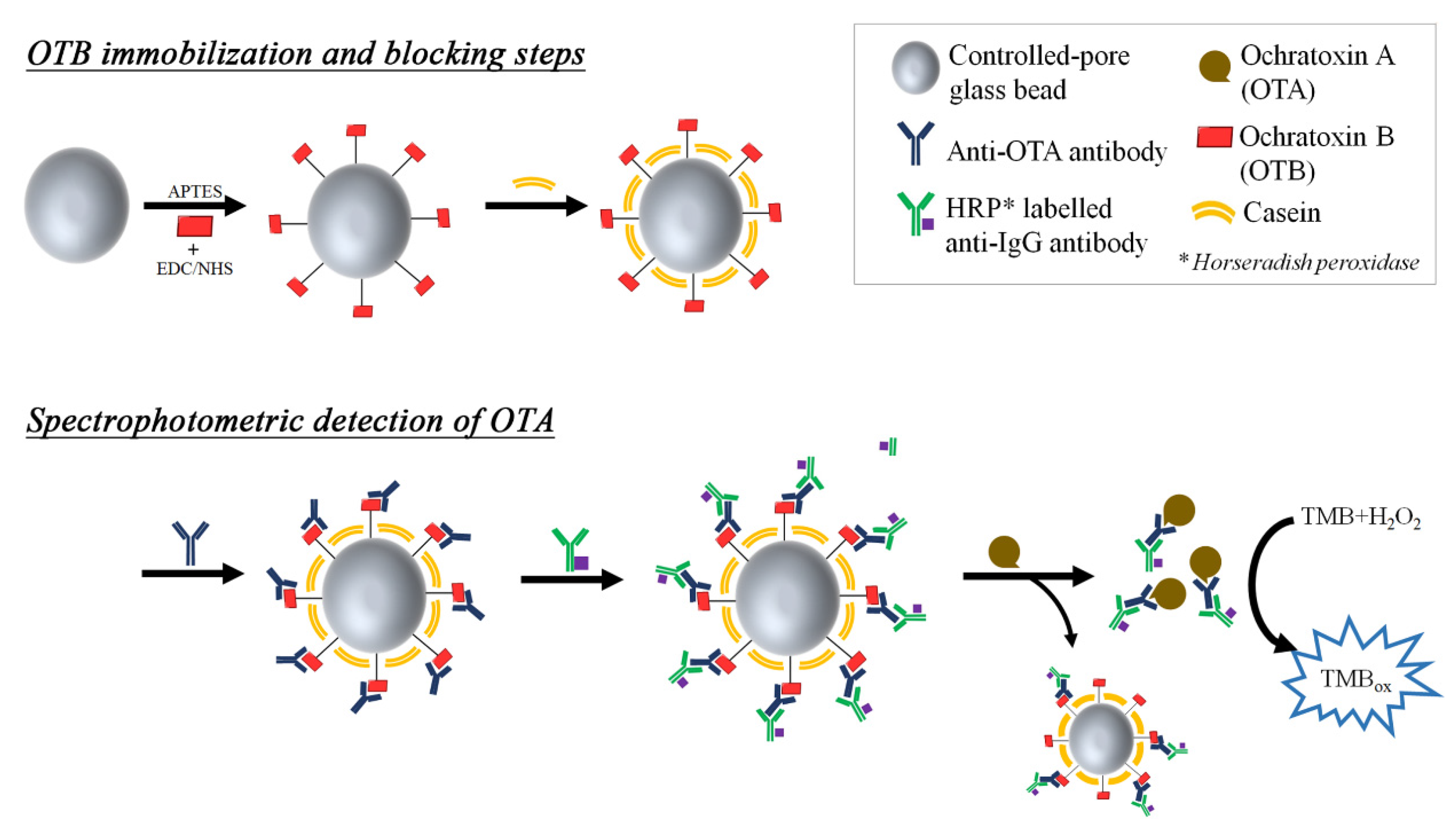
2.2. Pretreatment or Enrichment Process for Real Samples
3. Development of OTA Aptamers and Their Applications
3.1. OTA Aptamers in Affinity Columns and Enzyme Linked Assays
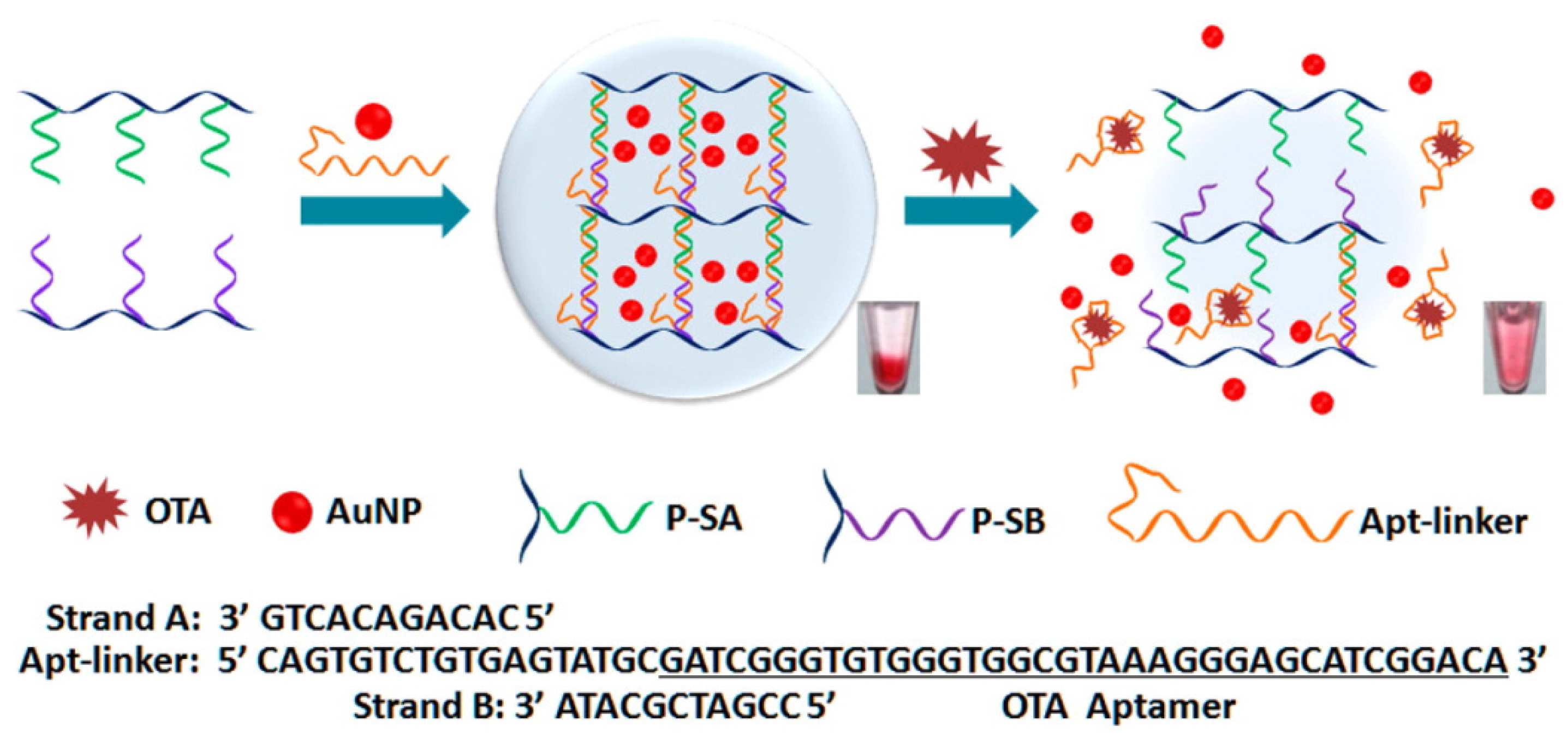
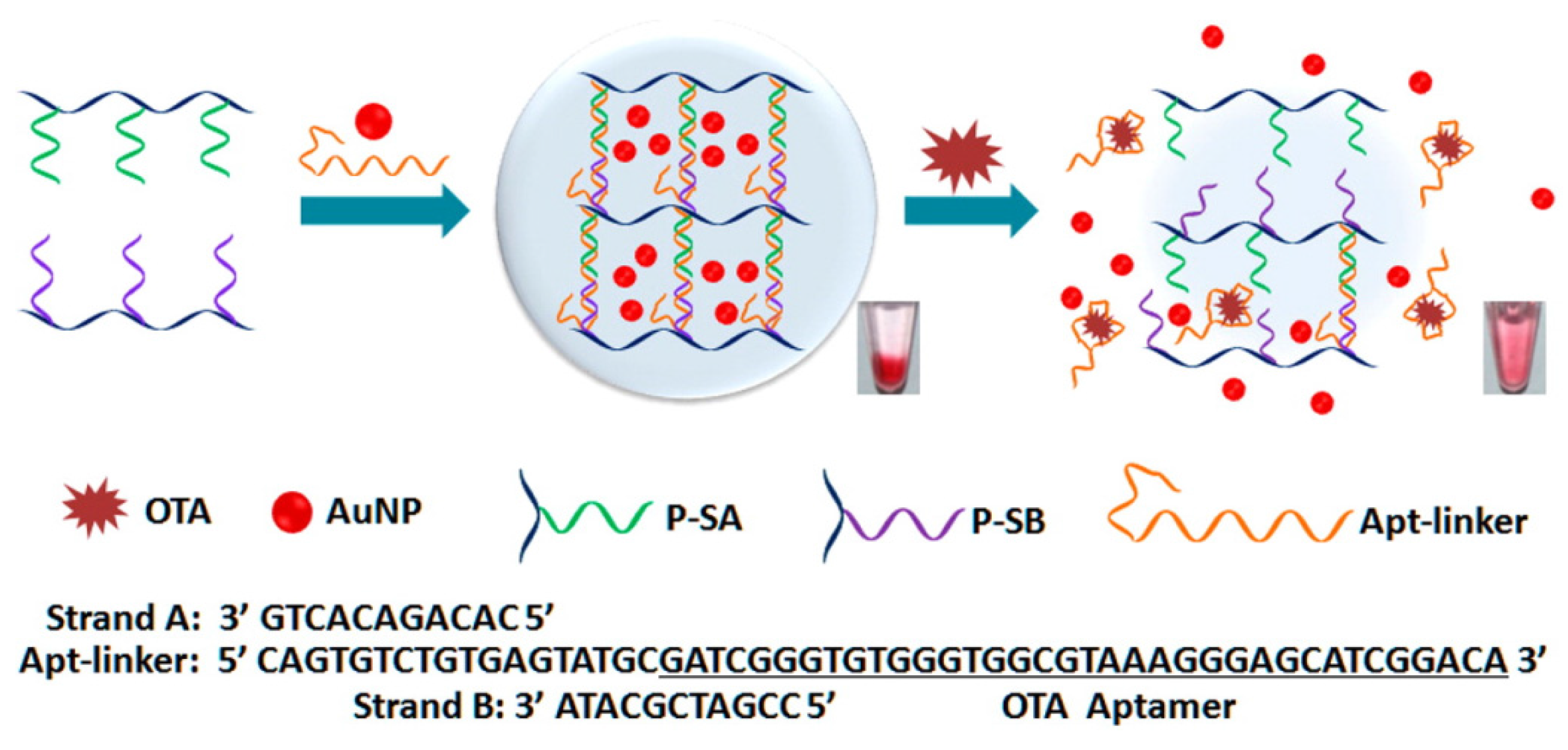
3.2. OTA Aptasensors with Functional Nanomaterials
3.3. Colorimetric and Fluorometric OTA Aptasenosrs
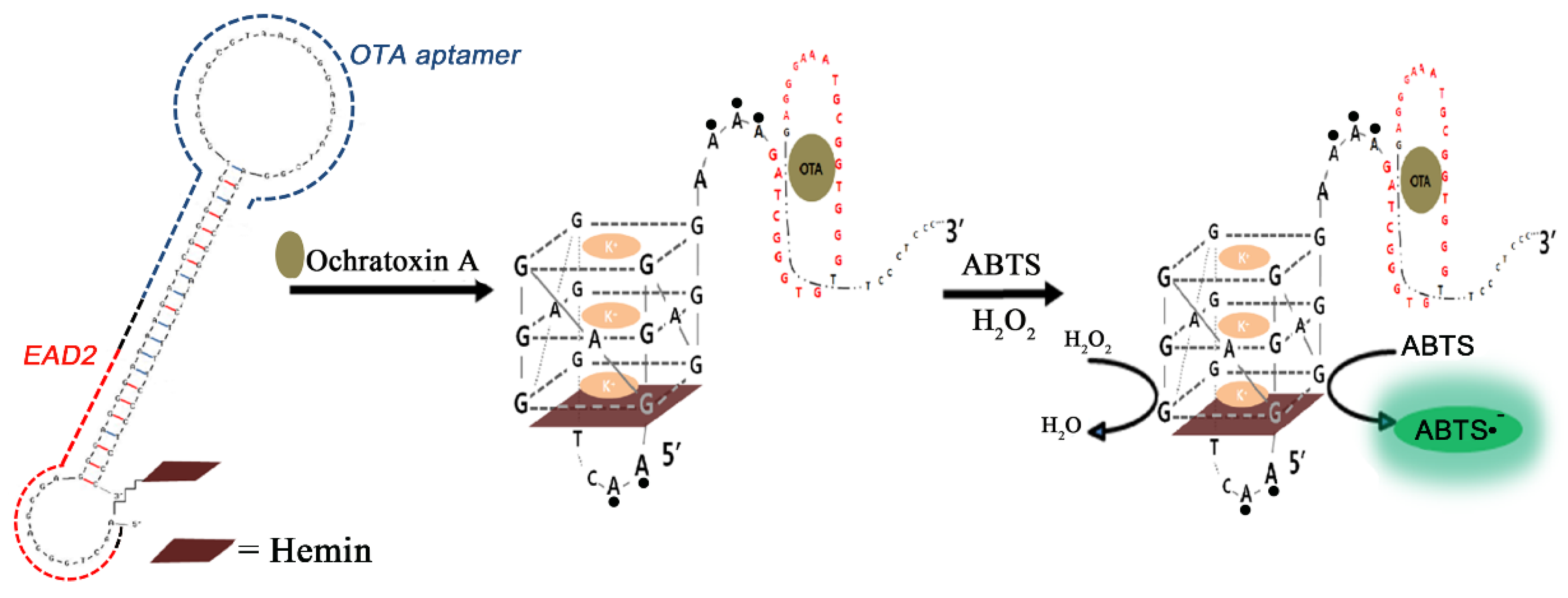
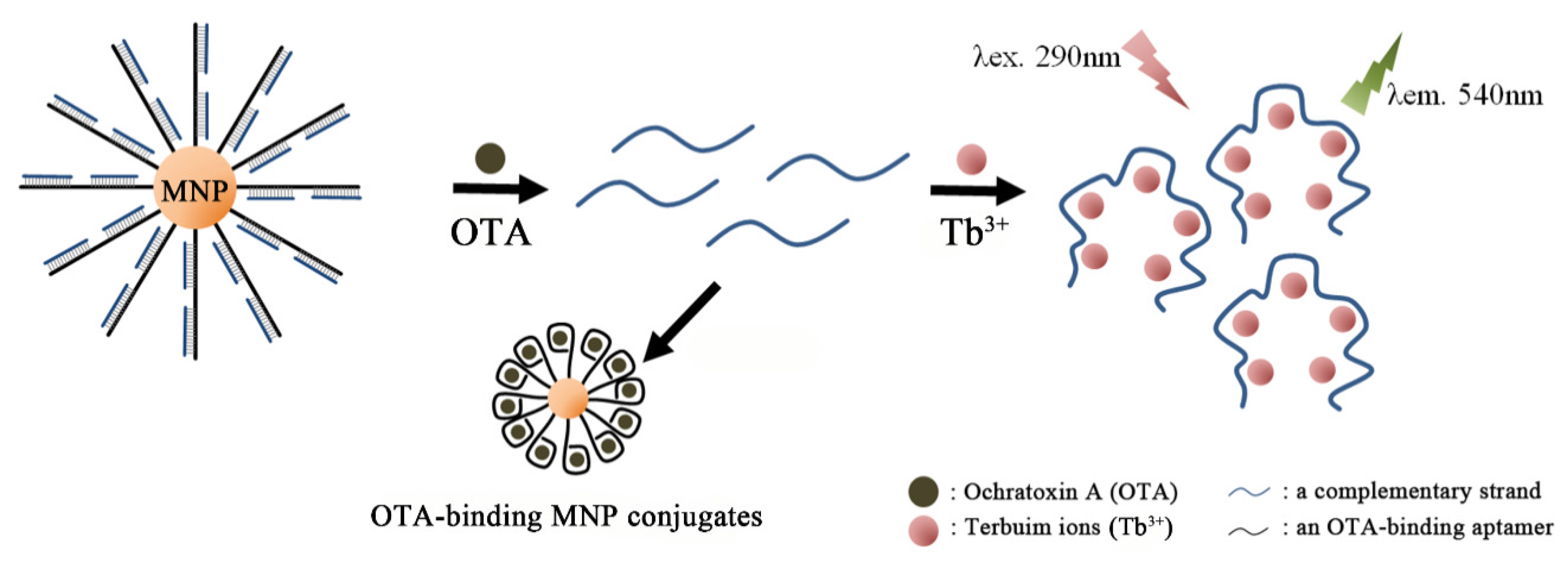
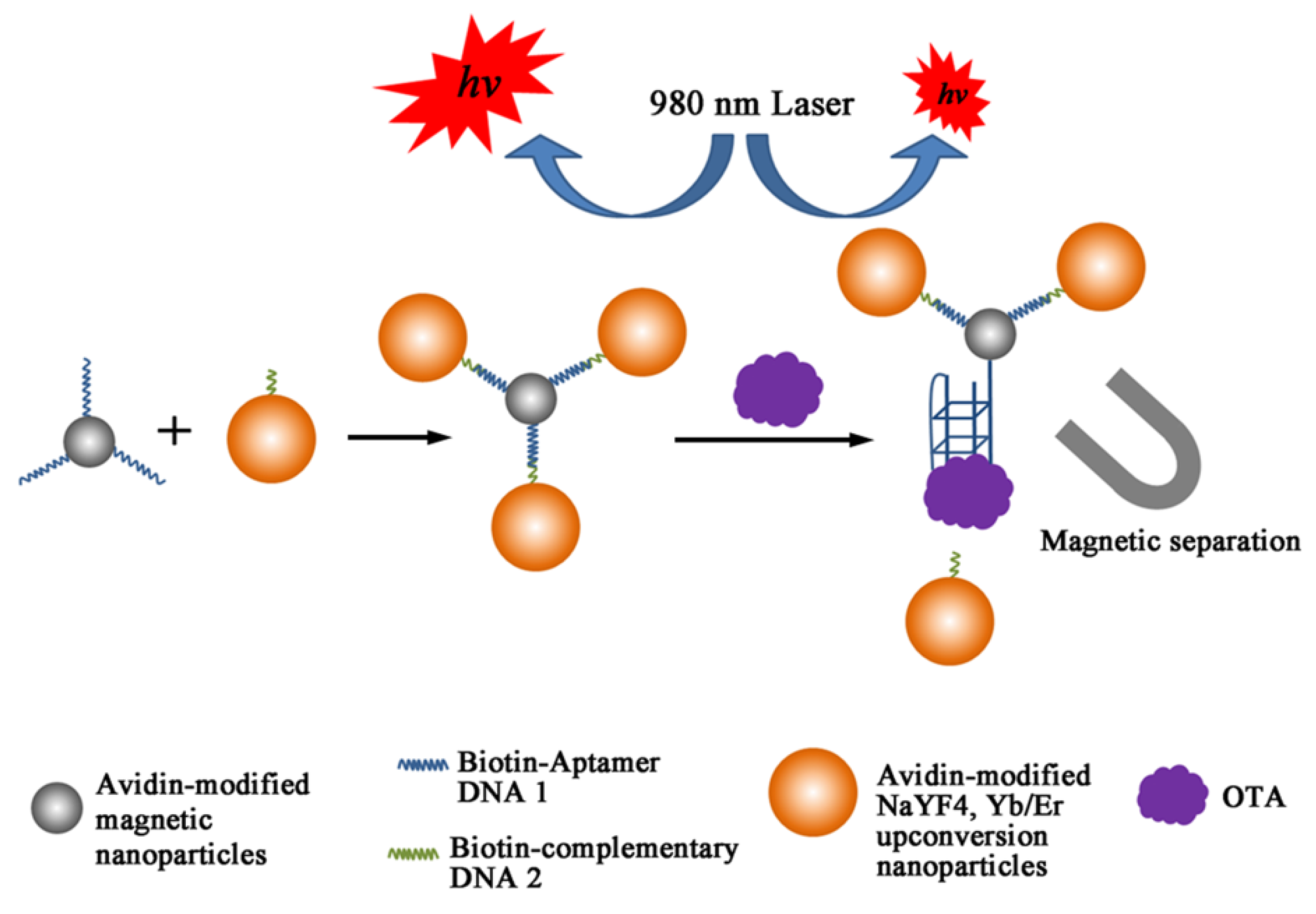
3.4. Electrochemical Detections with OTA Aptamers
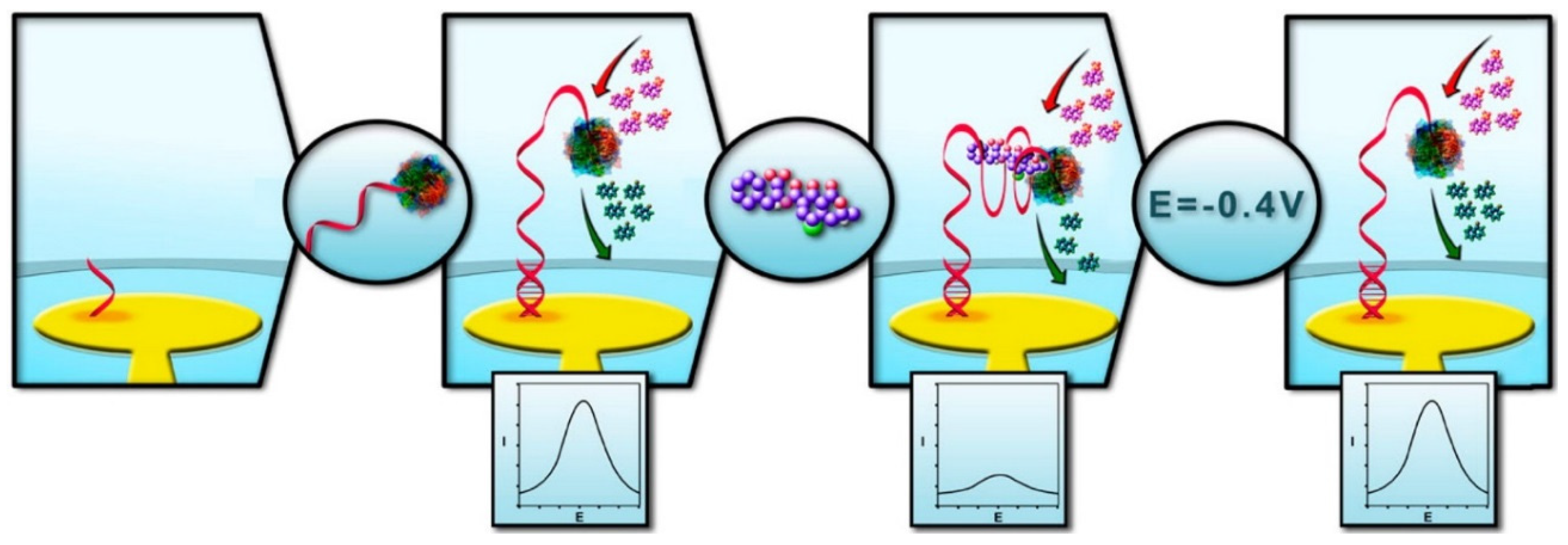
3.5. Signal Amplification Schemes with OTA Aptamers
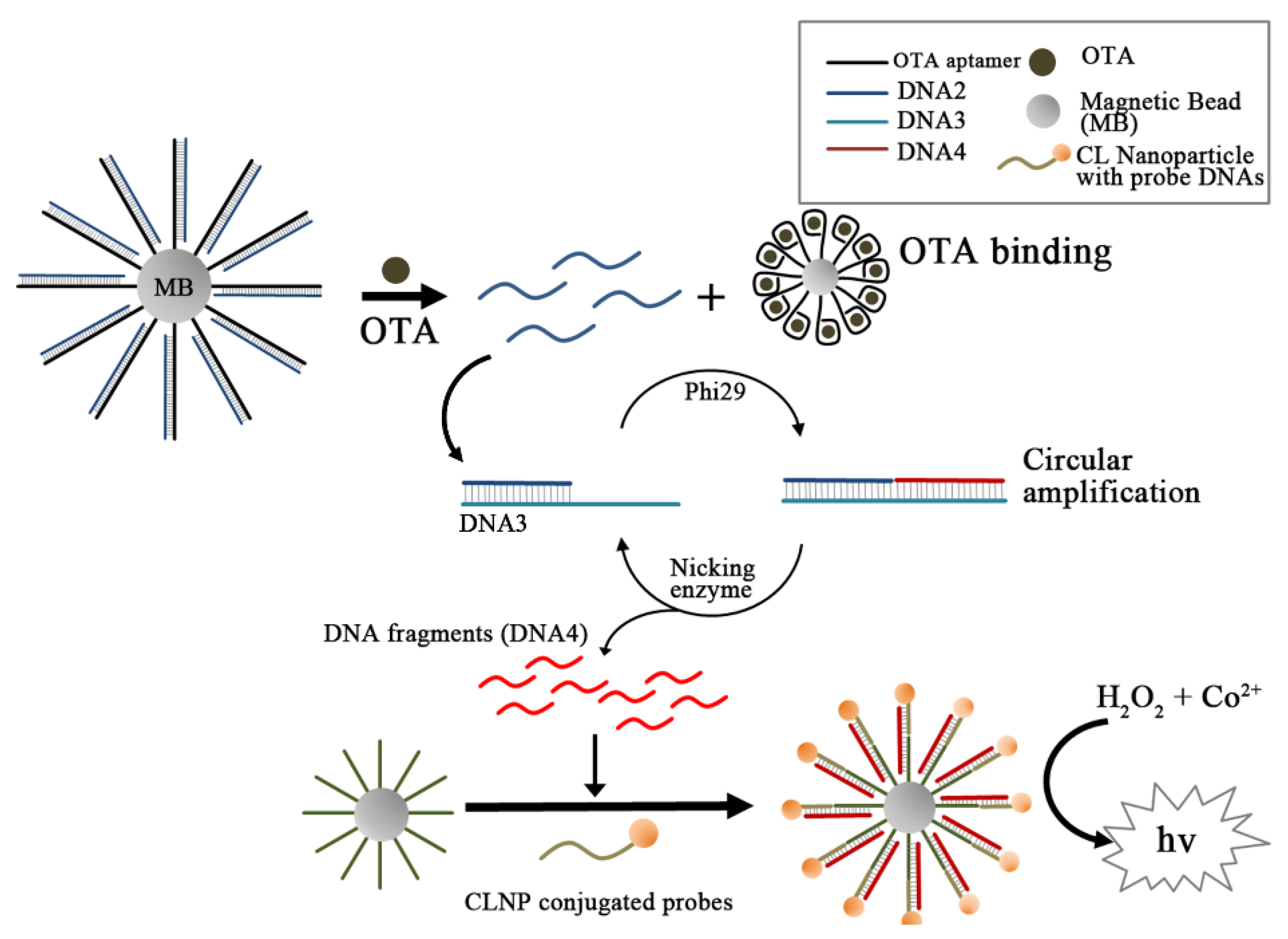
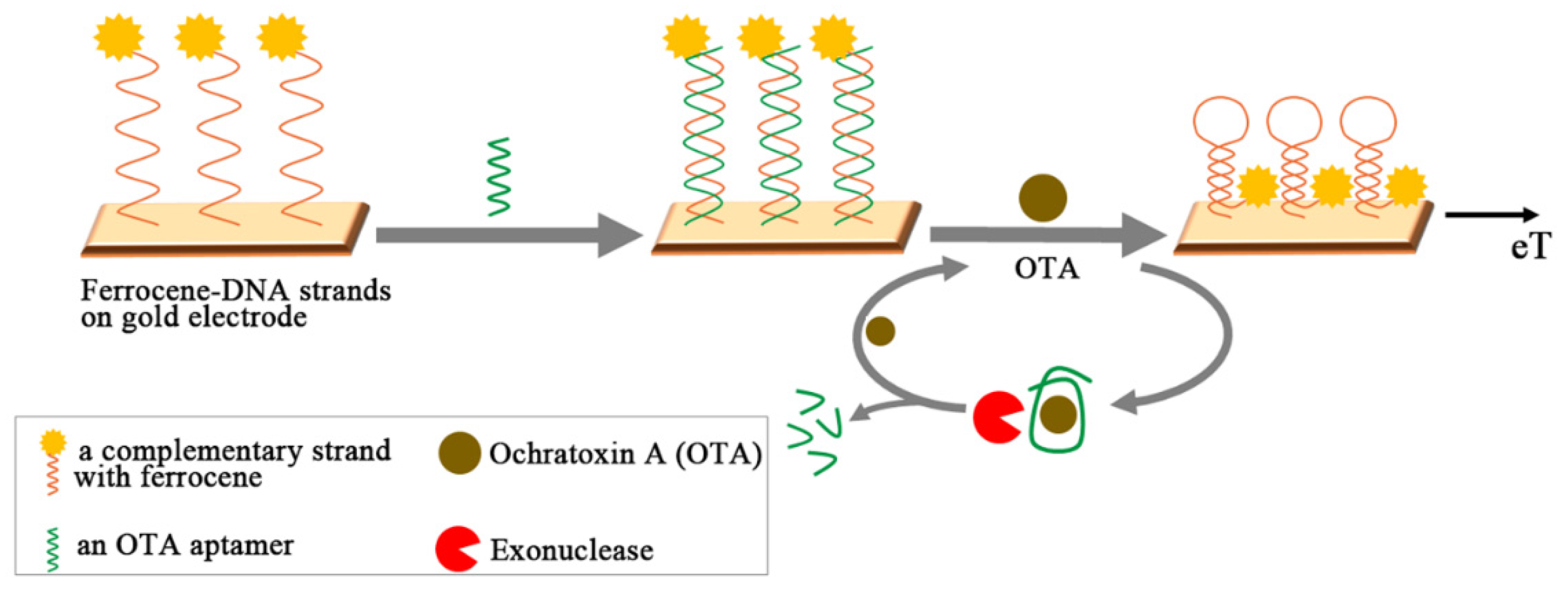
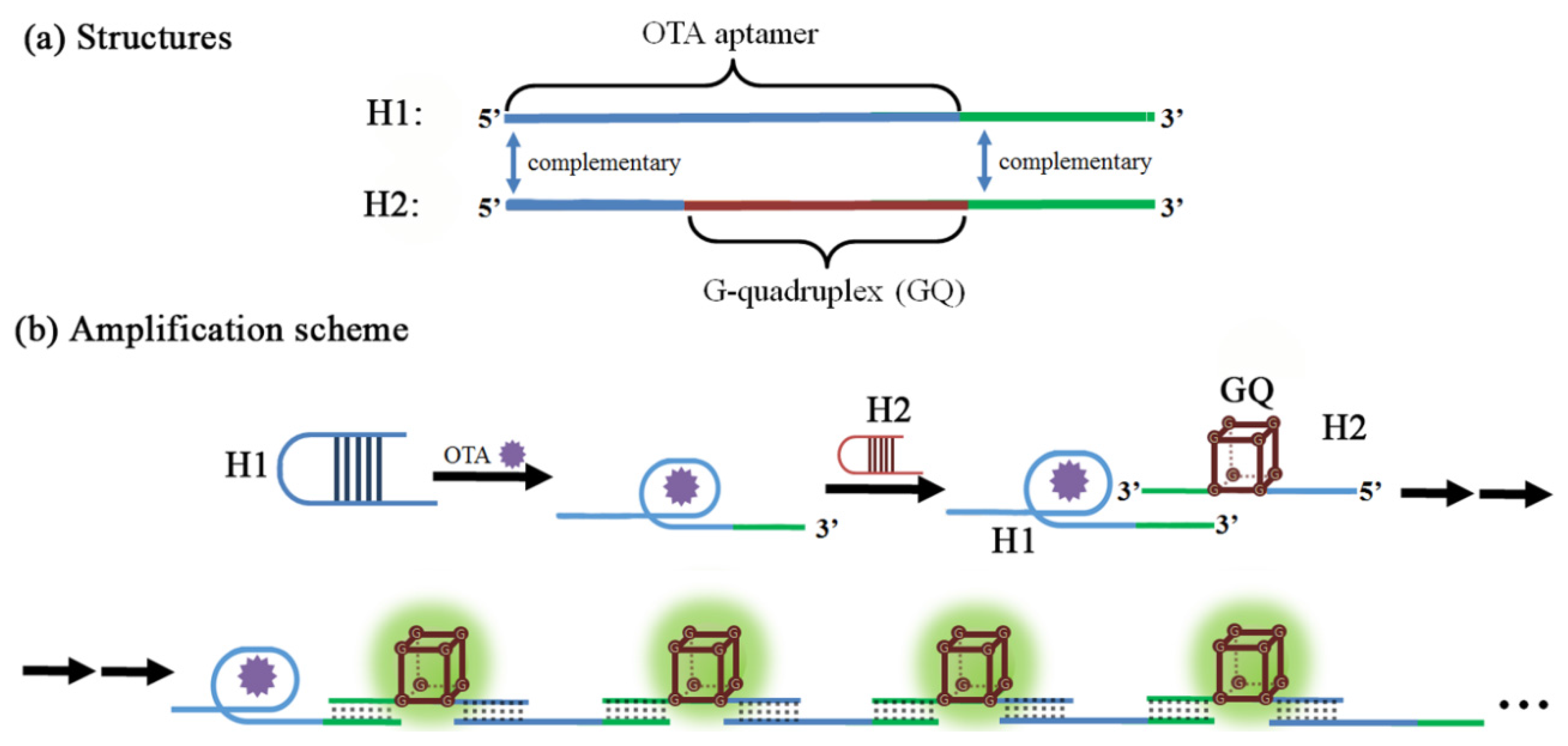
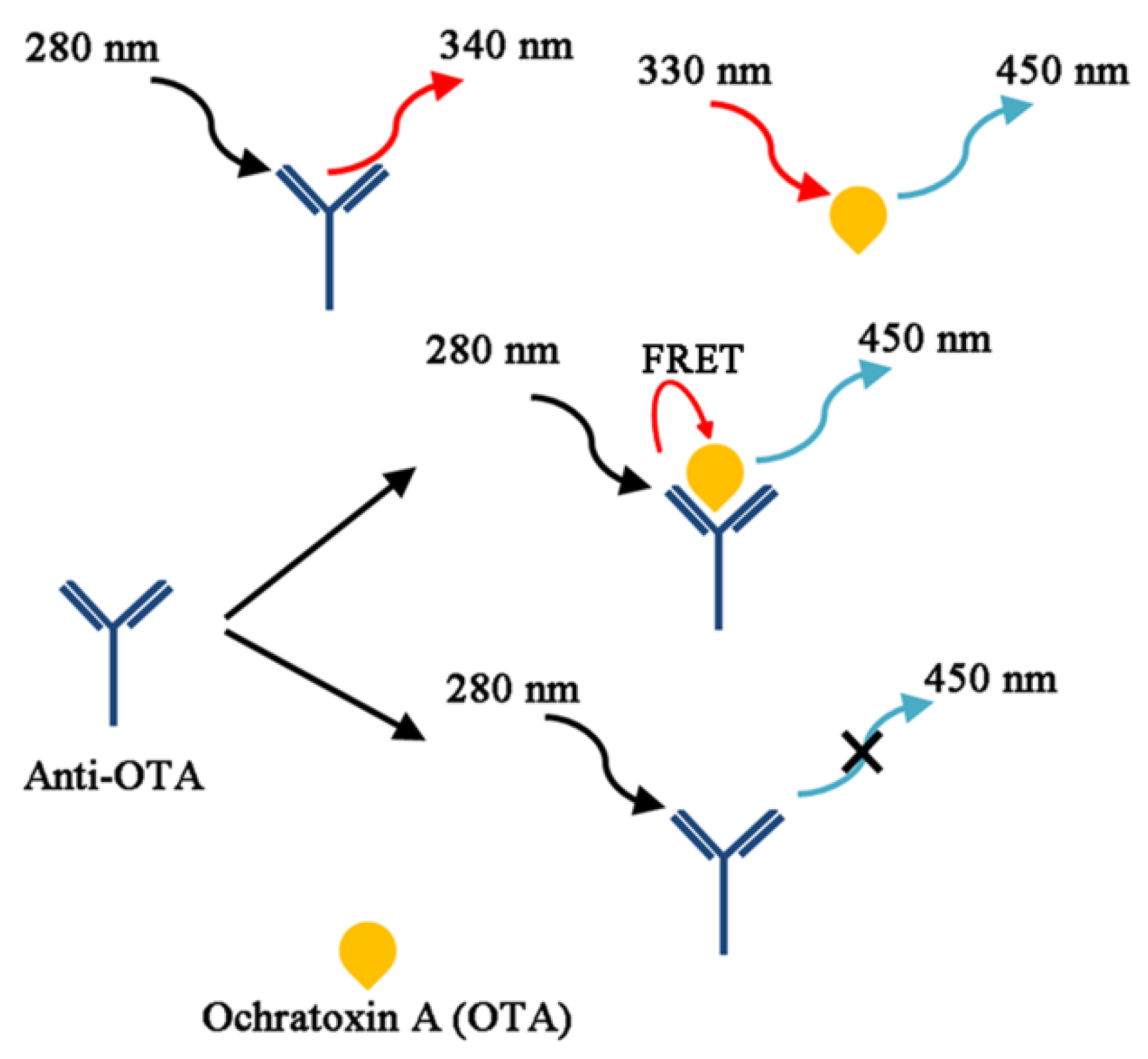
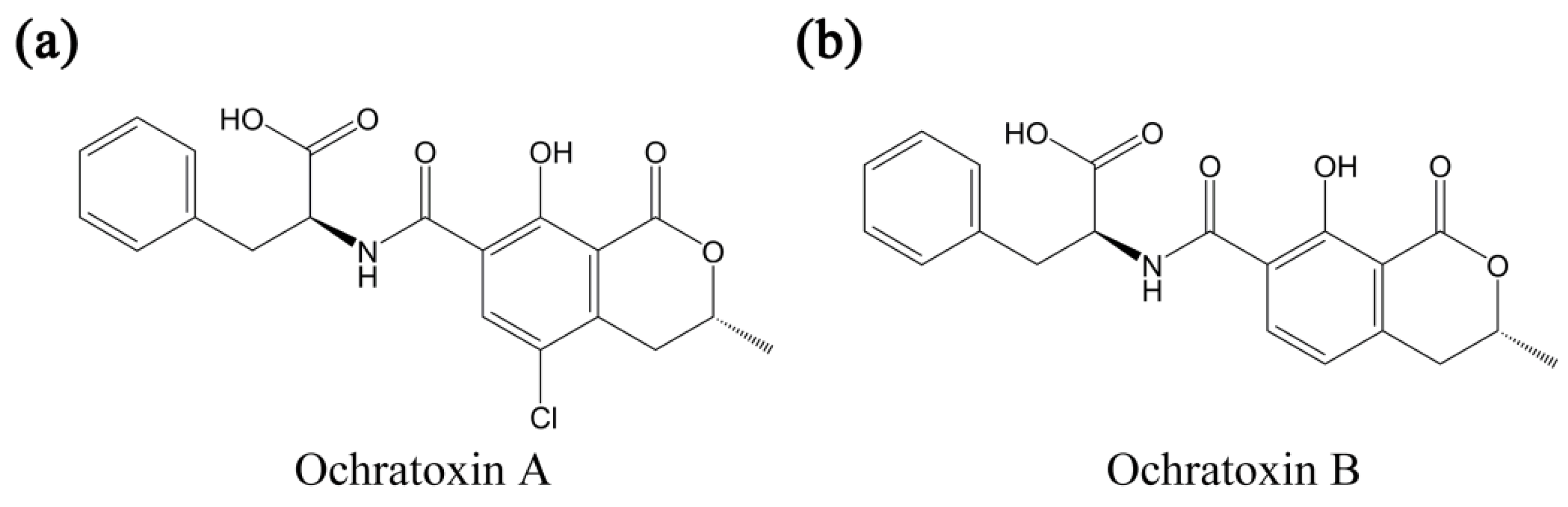
4. OTA Detection Methods Related to Their Physicochemical Characteristics
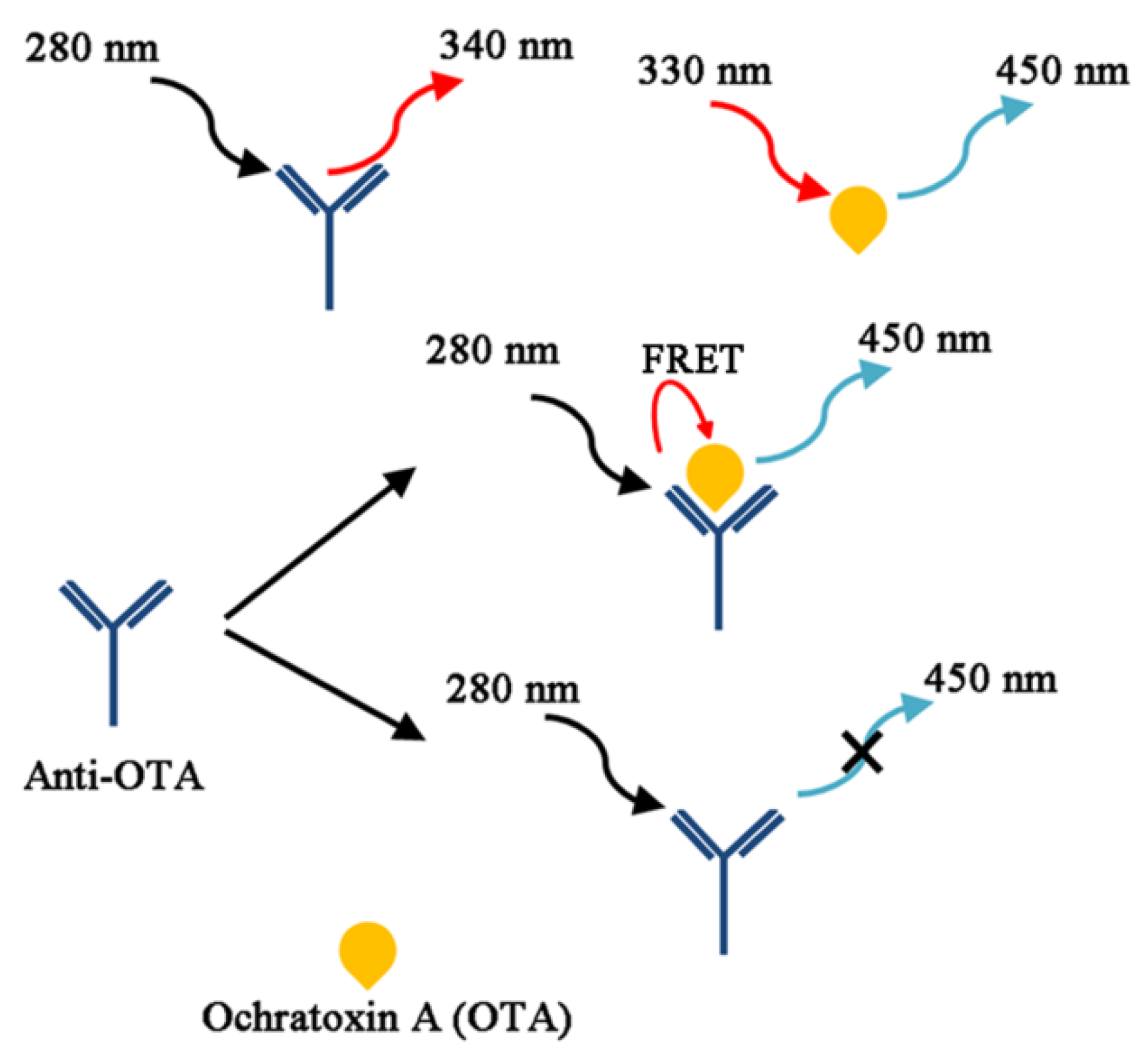

5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Malir, F.; Ostry, V.; Novotna, E. Toxicity of the mycotoxin ochratoxin A in the light of recent data. Toxin Rev. 2013, 32, 19–33. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Manderville, R.A. An update on direct genotoxicity as a molecular mechanism of ochratoxin A carcinogenicity. Chem. Res. Toxicol. 2012, 25, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, H.; Yamamoto, K.; Hisada, K.; Sakabe, Y.; Udagawa, S.-I. Effect of roasting on ochratoxin A level in green coffee beans inoculated with aspergillus ochraceus. Mycopathologia 1987, 97, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Boudra, H.; Le Bars, P.; Le Bars, J. Thermostability of ochratoxin A in wheat under two moisture conditions. Appl. Environ. Microbiol. 1995, 61, 1156–1158. [Google Scholar] [PubMed]
- Battilani, P.; Pietri, A. Ochratoxin A in grapes and wine. Eur. J. Plant Pathol. 2002, 108, 639–643. [Google Scholar] [CrossRef]
- Losito, I.; Monaci, L.; Palmisano, F.; Tantillo, G. Determination of ochratoxin A in meat products by high-performance liquid chromatography coupled to electrospray ionisation sequential mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 1965–1971. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Vallejos, C.; Anaya, I.A.; Broto-Puig, F.; Agut, M.; Comellas, L. Analysis of ochratoxin A in coffee by solid-phase cleanup and narrow-bore liquid chromatography-fluorescence detector-mass spectrometry. J. Agric. Food Chem. 2003, 51, 7564–7567. [Google Scholar] [CrossRef] [PubMed]
- Malir, F.; Ostry, V.; Pfohl-Leszkowicz, A.; Toman, J.; Bazin, I.; Roubal, T. Transfer of ochratoxin A into tea and coffee beverages. Toxins 2014, 6, 3438–3453. [Google Scholar] [CrossRef] [PubMed]
- Studer-Rohr, I.; Schlatter, J.; Dietrich, D.R. Kinetic parameters and intraindividual fluctuations of ochratoxin A plasma levels in humans. Arch. Toxicol. 2000, 74, 499–510. [Google Scholar] [CrossRef] [PubMed]
- IARC (International Agency for Research on Cancer). IARC Monographs on the Evaluation of Carcinogenic Risks to Humonas; IARC: Lyon, France, 1993; Volume 56, p. 489. [Google Scholar]
- Malir, F.; Ostry, V.; Pfohl-Leszkowicz, A.; Novotna, E. Ochratoxin A: Developmental and reproductive toxicity—An overview. Birth Defects Res. B Dev. Reprod. Toxicol. 2013, 98, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Creppy, E.E.; Størmer, F.C.; Kern, D.; Röschenthaler, R.; Dirheimer, G. Effects of ochratoxin A metabolites on yeast phenylalanyl-tRNA synthetase and on the growth and in vivo protein synthesis of hepatoma cells. Chem. Biol. Interact. 1983, 47, 239–247. [Google Scholar] [CrossRef]
- Assaf, H.; Azouri, H.; Pallardy, M. Ochratoxin A induces apoptosis in human lymphocytes through down regulation of BCL-xl. Toxicol. Sci. 2004, 79, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Boorman, G.A.; Hong, H.L.; Dieter, M.P.; Hayes, H.T.; Pohland, A.E.; Stack, M.; Luster, M.I. Myelotoxicity and macrophage alteration in mice exposed to ochratoxin A. Toxicol. Appl. Pharmacol. 1984, 72, 304–312. [Google Scholar] [CrossRef]
- Müller, G.; Burkert, B.; Rosner, H.; Köhler, H. Effects of the mycotoxin ochratoxin A and some of its metabolites on human kidney cell lines. Toxicol. In Vitro 2003, 17, 441–448. [Google Scholar] [CrossRef]
- Lea, T.; Steien, K.; Størmer, F. Mechanism of ochratoxin A-induced immunosuppression. Mycopathologia 1989, 107, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Van Egmond, H.; Schothorst, R.; Jonker, M. Regulations relating to mycotoxins in food. Anal. Bioanal. Chem. 2007, 389, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.C.; Lino, C.M.; Pena, A. Mycotoxin food and feed regulation and the specific case of ochratoxin A: A review of the worldwide status. Food Addit. Contam. A 2010, 27, 1440–1450. [Google Scholar] [CrossRef] [PubMed]
- Anklam, E.; Stroka, J.; Boenke, A. Acceptance of analytical methods for implementation of eu legislation with a focus on mycotoxins. Food Control 2002, 13, 173–183. [Google Scholar] [CrossRef]
- European Commission. Commission Regulation (EC) No. 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Union 2006, 364, 5–24. [Google Scholar]
- European Commission. Commission Regulation (EU) No 594/2012 of 5 July 2012 amending Regulation (EC) 1881/2006 as regards the maximum levels of the contaminants ochratoxin A, non dioxin-like PCBs and melamine in foodstuffs. Off. J. Eur. Union 2012, 176, 43–45. [Google Scholar]
- Ostry, V.; Malir, F.; Dofkova, M.; Skarkova, J.; Pfohl-Leszkowicz, A.; Ruprich, J. Ochratoxin A dietary exposure of ten population groups in the Czech Republic: Comparison with data over the world. Toxins 2015, 7, 3608–3635. [Google Scholar] [CrossRef] [PubMed]
- Malir, F.; Ostry, V.; Pfohl-Leszkowicz, A.; Roubal, T. Ochratoxin A exposure biomarkers in the Czech Republic and comparison with foreign countries. Biomarkers 2012, 17, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Barna-Vetró, I.; Solti, L.; Téren, J.; Gyöngyösi, Á.; Szabó, E.; Wölfling, A. Sensitive ELISA test for determination of ochratoxin A. J. Agric. Food Chem. 1996, 44, 4071–4074. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, J.; Wang, P.; Wang, Y.; Chen, J. Thin-layer chromatography of mycotoxins and comparison with other chromatographic methods. J. Chromatogr. A 1998, 815, 3–20. [Google Scholar] [CrossRef]
- Matrella, R.; Monaci, L.; Milillo, M.A.; Palmisano, F.; Tantillo, M.G. Ochratoxin A determination in paired kidneys and muscle samples from swines slaughtered in southern italy. Food Control 2006, 17, 114–117. [Google Scholar] [CrossRef]
- García, A.M.; Fernández, G.S. Contaminating mycoflora in yogurt: General aspects and special reference to the genus penicillium. J. Food Prot. 1984, 47, 629–636. [Google Scholar]
- Pena, A.; Cerejo, F.; Lino, C.; Silveira, I. Determination of ochratoxin A in portuguese rice samples by high performance liquid chromatography with fluorescence detection. Anal. Bioanal. Chem. 2005, 382, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzo, M.; Gambacorta, L.; Lattanzio, V.T.; Powers, S.; Visconti, A. Simultaneous LC–MS/MA determination of aflatoxin M1, ochratoxin A, deoxynivalenol, de-epoxydeoxynivalenol, α and β-zearalenols and fumonisin B1 in urine as a multi-biomarker method to assess exposure to mycotoxins. Anal. Bioanal. Chem. 2011, 401, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Schenzel, J.; Schwarzenbach, R.P.; Bucheli, T.D. Multi-residue screening method to quantify mycotoxins in aqueous environmental samples. J. Agric. Food Chem. 2010, 58, 11207–11217. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-A.; Feng, Y.-N.; Zhu, Y.-Z.; Kim, J.-H. Multi-residue method for determination of 238 pesticides in chinese cabbage and cucumber by liquid chromatography–tandem mass spectrometry: Comparison of different purification procedures. J. Agric. Food Chem. 2014, 62, 11449–11456. [Google Scholar] [CrossRef] [PubMed]
- Anastassiades, M.; Lehotay, S.J.; tajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [PubMed]
- Anastassiades, M.; Maštovská, K.; Lehotay, S.J. Evaluation of analyte protectants to improve gas chromatographic analysis of pesticides. J. Chromatogr. A 2003, 1015, 163–184. [Google Scholar] [CrossRef]
- Maštovská, K.; Lehotay, S.J.; Anastassiades, M. Combination of analyte protectants to overcome matrix effects in routine GC analysis of pesticide residues in food matrixes. Anal. Chem. 2005, 77, 8129–8137. [Google Scholar] [CrossRef] [PubMed]
- Lehotay, S.J.; Son, K.A.; Kwon, H.; Koesukwiwat, U.; Fu, W.; Mastovska, K.; Hoh, E.; Leepipatpiboon, N. Comparison of QuEChERS sample preparation methods for the analysis of pesticide residues in fruits and vegetables. J. Chromatogr. A 2010, 1217, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S.; Zhu, Z.; Fiore, E.; Ravelet, C.; Guieu, V.; Peyrin, E. Capillary gel electrophoresis-coupled aptamer enzymatic cleavage protection strategy for the simultaneous detection of multiple small analytes. Anal. Chem. 2014, 86, 4233–4240. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Jarrosson, F.; Randon, J.; Dugas, V.; Demesmay, C. In-line coupling of an aptamer based miniaturized monolithic affinity preconcentration unit with capillary electrophoresis and laser induced fluorescence detection. J. Chromatogr. A 2015, 1406, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Simón, B.; Campàs, M.; Marty, J.-L.; Noguer, T. Novel highly-performing immunosensor-based strategy for ochratoxin A detection in wine samples. Biosens. Bioelectron. 2008, 23, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Hanneken, J.; Houchins, D.; King, R.; Lee, P.; Richard, J. Validation of an ELISA test kit for the detection of ochratoxin A in several food commodities by comparison with HPLC. Mycopathologia 2005, 159, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-Y.; Vdovenko, M.M.; Wang, J.-J.; Sakharov, I.Y. Comparison of enzyme-linked immunosorbent assays with chemiluminescent and colorimetric detection for the determination of ochratoxin A in food. J. Agric. Food Chem. 2011, 59, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, Y.; Wan, D.-B.; Xiong, Y.-H.; He, Z.-Y.; Wang, X.-X.; Gee, S.J.; Ryu, D.; Hammock, B.D. Development of a nanobody-alkaline phosphatase fusion protein and its application in a highly sensitive direct competitive fluorescence enzyme immunoassay for detection of ochratoxin A in cereal. Anal. Chem. 2015, 87, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Zezza, F.; Longobardi, F.; Pascale, M.; Eremin, S.; Visconti, A. Fluorescence polarization immunoassay for rapid screening of ochratoxin a in red wine. Anal. Bioanal. Chem. 2009, 395, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-J.; Lee, D.-H.; Kim, D.-O.; Min, W.-K.; Bong, K.-T.; Lee, G.-G.; Seo, J.-H. Production of a monoclonal antibody against ochratoxin A and its application to immunochromatographic assay. J. Agric. Food Chem. 2005, 53, 8447–8451. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.-H.; Tsao, Z.-J.; Wang, J.-J.; Yu, F.-Y. Development of a monoclonal antibody against ochratoxin A and its application in enzyme-linked immunosorbent assay and gold nanoparticle immunochromatographic strip. Anal. Chem. 2008, 80, 7029–7035. [Google Scholar] [CrossRef] [PubMed]
- Wanigasuriya, K.P.; Peiris, H.; Ileperuma, N.; Peiris-John, R.J.; Wickremasinghe, R. Could ochratoxin A in food commodities be the cause of chronic kidney disease in Sri Lanka? Trans. R. Soc. Trop. Med. Hyg. 2008, 102, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Bazin, I.; Faucet-Marquis, V.; Monje, M.-C.; El Khoury, M.; Marty, J.-L.; Pfohl-Leszkowicz, A. Impact of pH on the stability and the cross-reactivity of ochratoxin A and citrinin. Toxins 2013, 5, 2324–2340. [Google Scholar] [CrossRef] [PubMed]
- Monaci, L.; Tantillo, G.; Palmisano, F. Determination of ochratoxin A in pig tissues by liquid-liquid extraction and clean-up and high-performance liquid chromatography. Anal. Bioanal. Chem. 2004, 378, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.J.; García-Moreno, M.V.; Durán, E.; Guillén, D.; Barroso, C.G. Validation of two analytical methods for the determination of ochratoxin A by reversed-phased high-performance liquid chromatography coupled to fluorescence detection in musts and sweet wines from Andalusia. Anal. Chim. Acta 2006, 566, 117–121. [Google Scholar] [CrossRef]
- Ratola, N.; Martins, L.S.; Alves, A. Ochratoxin a in wines-assessing global uncertainty associated with the results. Anal. Chim. Acta 2004, 513, 319–324. [Google Scholar] [CrossRef]
- Serra, R.; Mendonça, C.; Abrunhosa, L.S.; Pietri, A.; Venâncio, A. Determination of ochratoxin A in wine grapes: Comparison of extraction procedures and method validation. Anal. Chim. Acta 2004, 513, 41–47. [Google Scholar] [CrossRef]
- Giraudi, G.; Anfossi, L.; Baggiani, C.; Giovannoli, C.; Tozzi, C. Solid-phase extraction of ochratoxin A from wine based on a binding hexapeptide prepared by combinatorial synthesis. J. Chromatogr. A 2007, 1175, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Bazin, I.; Andreotti, N.; Hassine, A.I.H.; de Waard, M.; Sabatier, J.M.; Gonzalez, C. Peptide binding to ochratoxin A mycotoxin: A new approach in conception of biosensors. Biosens. Bioelectron. 2013, 40, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Soleri, R.; Demey, H.; Tria, S.A.; Guiseppi-Elie, A.; Ibn Had Hassine, A.; Gonzalez, C.; Bazin, I. Peptide conjugated chitosan foam as a novel approach for capture-purification and rapid detection of hapten—Example of ochratoxin A. Biosens. Bioelectron. 2015, 67, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Aguado, J.A.; Penner, G. Determination of ochratoxin A with a DNA aptamer. J. Agric. Food Chem. 2008, 56, 10456–10461. [Google Scholar] [CrossRef] [PubMed]
- Bonel, L.; Vidal, J.C.; Duato, P.; Castillo, J.R. An electrochemical competitive biosensor for ochratoxin A based on a DNA biotinylated aptamer. Biosens. Bioelectron. 2011, 26, 3254–3259. [Google Scholar] [CrossRef] [PubMed]
- Hun, X.; Liu, F.; Mei, Z.; Ma, L.; Wang, Z.; Luo, X. Signal amplified strategy based on target-induced strand release coupling cleavage of nicking endonuclease for the ultrasensitive detection of ochratoxin A. Biosens. Bioelectron. 2013, 39, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Rhouati, A.; Yang, C.; Hayat, A.; Marty, J.-L. Aptamers: A promising tool for ochratoxin A detection in food analysis. Toxins 2013, 5, 1988–2008. [Google Scholar] [CrossRef] [PubMed]
- Barthelmebs, L.; Jonca, J.; Hayat, A.; Prieto-Simon, B.; Marty, J.-L. Enzyme-linked aptamer assays (ELAAS), based on a competition format for a rapid and sensitive detection of ochratoxin A in wine. Food Control 2011, 22, 737–743. [Google Scholar] [CrossRef]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.S.M.; Papamichael, K.; Guilbault, G.; Schwarzacher, T.; Gariepy, J.; Missailidis, S. DNA aptamers against the muc1 tumour marker: Design of aptamer-antibody sandwich ELISA for the early diagnosis of epithelial tumours. Anal. Bioanal. Chem. 2008, 390, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Burbulis, I.; Yamaguchi, K.; Yu, R.; Resnekov, O.; Brent, R. Quantifying small numbers of antibodies with a “near-universal” protein-DNA chimera. Nat. Meth. 2007, 4, 1011–1013. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.S.M.; Cheung, M.C.; Missailidis, S.; Bisland, S.; Gariépy, J. Phototoxic aptamers selectively enter and kill epithelial cancer cells. Nucleic Acids Res. 2009, 37, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Tong, R.; Mishra, A.; Xu, W.; Wong, G.C.L.; Cheng, J.; Lu, Y. Reversible cell-specific drug delivery with aptamer-functionalized liposomes. Angew. Chem. Int. Ed. 2009, 48, 6494–6498. [Google Scholar] [CrossRef] [PubMed]
- McCauley, T.G.; Hamaguchi, N.; Stanton, M. Aptamer-based biosensor arrays for detection and quantification of biological macromolecules. Anal. Biochem. 2003, 319, 244–250. [Google Scholar] [CrossRef]
- De Girolamo, A.; McKeague, M.; Miller, J.D.; DeRosa, M.C.; Visconti, A. Determination of ochratoxin A in wheat after clean-up through a DNA aptamer-based solid phase extraction column. Food Chem. 2011, 127, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- De Girolamo, A.; Le, L.; Penner, G.; Schena, R.; Visconti, A. Analytical performances of a DNA-ligand system using time-resolved fluorescence for the determination of ochratoxin a in wheat. Anal. Bioanal. Chem. 2012, 403, 2627–2634. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Hu, J.; Zhu, B.; Lu, L.; Huang, X.; Pang, D. Aptamer-targeted magnetic nanospheres as a solid-phase extraction sorbent for determination of ochratoxin A in food samples. J. Chromatogr. A 2011, 1218, 7341–7346. [Google Scholar] [CrossRef] [PubMed]
- Chapuis-Hugon, F.; du Boisbaudry, A.; Madru, B.; Pichon, V. New extraction sorbent based on aptamers for the determination of ochratoxin A in red wine. Anal. Bioanal. Chem. 2011, 400, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Rhouati, A.; Paniel, N.; Meraihi, Z.; Marty, J.-L. Development of an oligosorbent for detection of ochratoxin A. Food Control 2011, 22, 1790–1796. [Google Scholar] [CrossRef]
- Zhao, Q.; Wu, M.; Chris Le, X.; Li, X.-F. Applications of aptamer affinity chromatography. Trac Trends Anal. Chem. 2012, 41, 46–57. [Google Scholar] [CrossRef]
- Liu, L.-H.; Zhou, X.-H.; Shi, H.-C. Portable optical aptasensor for rapid detection of mycotoxin with a reversible ligand-grafted biosensing surface. Biosens. Bioelectron. 2015, 72, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, Y.; Marty, J.-L.; Yang, X. Aptamer-based colorimetric biosensing of ochratoxin A using unmodified gold nanoparticles indicator. Biosens. Bioelectron. 2011, 26, 2724–2727. [Google Scholar] [CrossRef] [PubMed]
- Soh, J.H.; Lin, Y.; Rana, S.; Ying, J.Y.; Stevens, M.M. Colorimetric detection of small molecules in complex matrixes via target-mediated growth of aptamer-functionalized gold nanoparticles. Anal. Chem. 2015, 87, 7644–7652. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Byun, J.-Y.; Mun, H.; Shim, W.-B.; Shin, Y.-B.; Li, T.; Kim, M.-G. A regeneratable, label-free, localized surface plasmon resonance (LSPR) aptasensor for the detection of ochratoxin A. Biosens. Bioelectron. 2014, 59, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Kuang, H.; Xu, C.; Ma, W.; Wang, L.; Kotov, N.A. Regiospecific plasmonic assemblies for in situ Raman spectroscopy in live cells. J. Am. Chem. Soc. 2012, 134, 1699–1709. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hao, C.; Yin, H.; Liu, L.; Ma, W.; Wang, L.; Kuang, H.; Xu, C. Plasmonic core-satellites nanostructures with high chirality and bioproperty. J. Phys. Chem. Lett. 2013, 4, 2379–2384. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, X.; Xu, L.; Ma, W.; Kuang, H.; Wang, L.; Xu, C. Building heterogeneous core–satellite chiral assemblies for ultrasensitive toxin detection. Biosens. Bioelectron. 2015, 66, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Xu, L.; Xu, C.; Ma, W.; Kuang, H.; Wang, L.; Kotov, N.A. Self-assembly of chiral nanoparticle pyramids with strong R/S optical activity. J. Am. Chem. Soc. 2012, 134, 15114–15121. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Huang, Y.; Ma, Y.; Jia, S.; Gao, M.; Li, J.; Zhang, H.; Xu, D.; Wu, M.; Chen, Y.; et al. Design and synthesis of target-responsive aptamer-cross-linked hydrogel for visual quantitative detection of ochratoxin A. ACS Appl. Mater. Interfaces 2015, 7, 6982–6990. [Google Scholar] [CrossRef] [PubMed]
- Battig, M.R.; Soontornworajit, B.; Wang, Y. Programmable release of multiple protein drugs from aptamer-functionalized hydrogels via nucleic acid hybridization. J. Am. Chem. Soc. 2012, 134, 12410–12413. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, C.; Boldt, F.; Wang, Y.; Kuan, S.L.; Tran, T.T.; Mikhalevich, V.; Fortsch, C.; Barth, H.; Yang, Z.; et al. Programmable protein-DNA hybrid hydrogels for the immobilization and release of functional proteins. Chem. Commun. 2014, 50, 14620–14622. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Lates, V.; Prieto-Simón, B.; Marty, J.-L.; Yang, X. Aptamer-DNAzyme hairpins for biosensing of ochratoxin A. Biosens. Bioelectron. 2012, 32, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Teller, C.; Shimron, S.; Willner, I. Aptamer-DNAzyme hairpins for amplified biosensing. Anal. Chem. 2009, 81, 9114–9119. [Google Scholar] [CrossRef] [PubMed]
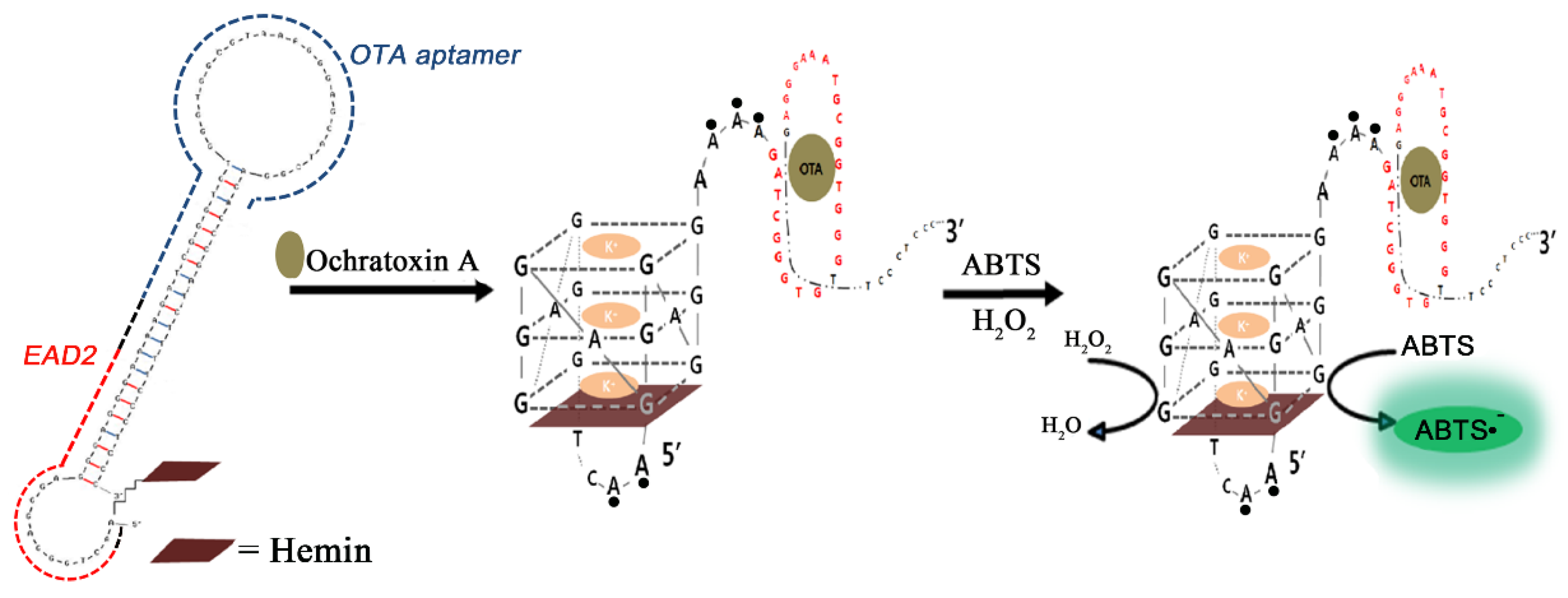
- Lee, J.; Jeon, C.H.; Ahn, S.J.; Ha, T.H. Highly stable colorimetric aptamer sensors for detection of ochratoxin A through optimizing the sequence with the covalent conjugation of hemin. Analyst 2014, 139, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Lates, V.; Prieto-Simón, B.; Marty, J.-L.; Yang, X. Rapid high-throughput analysis of ochratoxin A by the self-assembly of DNAzyme-aptamer conjugates in wine. Talanta 2013, 116, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Mun, H.; Jo, E.-J.; Li, T.; Joung, H.-A.; Hong, D.-G.; Shim, W.-B.; Jung, C.; Kim, M.-G. Homogeneous assay of target molecules based on chemiluminescence resonance energy transfer (CRET) using DNAzyme-linked aptamers. Biosens. Bioelectron. 2014, 58, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.-J.; Mun, H.; Kim, S.-J.; Shim, W.-B.; Kim, M.-G. Detection of ochratoxin a (OTA) in coffee using chemiluminescence resonance energy transfer (CRET) aptasensor. Food Chem. 2016, 194, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Ren, J.; Miao, Y.; Wang, J.; Wang, E. PVP-coated graphene oxide for selective determination of ochratoxin A via quenching fluorescence of free aptamer. Biosens. Bioelectron. 2011, 26, 3494–3499. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Ren, J.; Wang, J.; Wang, E. Single-walled carbon nanotubes based quenching of free FAM-aptamer for selective determination of ochratoxin A. Talanta 2011, 85, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wu, S.; Shu, F.; Liu, Z. An MnO2 nanosheet as a label-free nanoplatform for homogeneous biosensing. Chem. Commun. 2014, 50, 1095–1097. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fang, Z.; Liu, J.; Zeng, L. A simple and rapid biosensor for ochratoxin A based on a structure-switching signaling aptamer. Food Control 2012, 25, 555–560. [Google Scholar] [CrossRef]
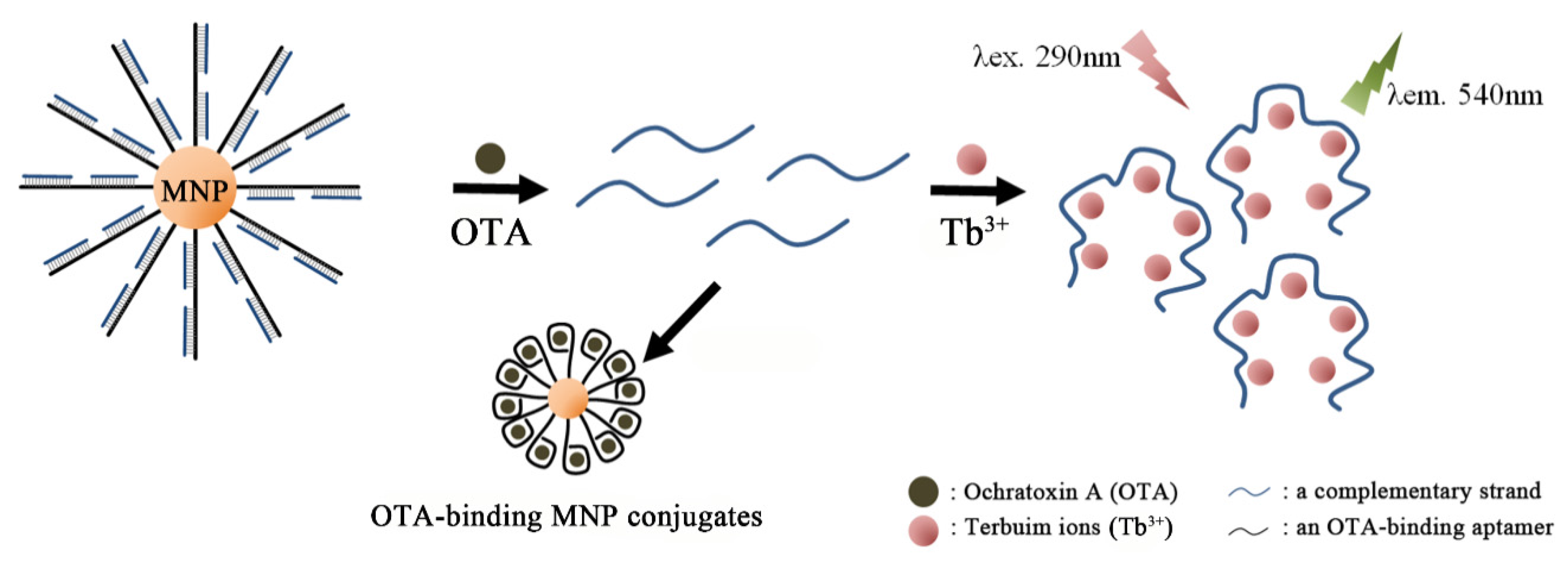
- Zhang, J.; Zhang, X.; Yang, G.; Chen, J.; Wang, S. A signal-on fluorescent aptasensor based on Tb3+ and structure-switching aptamer for label-free detection of ochratoxin A in wheat. Biosens. Bioelectron. 2013, 41, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.K.L.; Turro, C. Energy transfer from nucleic acids to Tb(III): Selective emission enhancement by single DNA mismatches. J. Am. Chem. Soc. 1999, 121, 1–7. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, X.; Cai, S.; Wu, D.; Chen, M.; Wang, S.; Zhang, J. A fluorescent aptasensor based on DNA-scaffolded silver-nanocluster for ochratoxin A detection. Biosens. Bioelectron. 2014, 57, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chen, A.; Liu, J.; Guan, Z.; Zhou, Y.; Xu, S.; Yang, S.; Li, C. A simple and sensitive approach for ochratoxin A detection using a label-free fluorescent aptasensor. PLoS ONE 2014, 9, e85968. [Google Scholar] [CrossRef] [PubMed]
- McKeague, M.; Velu, R.; Hill, K.; Bardóczy, V.; Mészáros, T.; DeRosa, M. Selection and characterization of a novel DNA aptamer for label-free fluorescence biosensing of ochratoxin A. Toxins 2014, 6, 2435–2452. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Wang, M.; Liu, L.-J.; Leung, C.-H.; Ma, D.-L. Label-free luminescent switch-on probe for ochratoxin A detection using a G-quadruplex-selective iridium(III) complex. ACS Appl. Mater. Interfaces 2015, 7, 8313–8318. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Geng, X.; Wang, H. Fluorescent sensing ochratoxin A with single fluorophore-labeled aptamer. Anal. Bioanal. Chem. 2013, 405, 6281–6286. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Duan, N.; Wang, Z.; Wang, H. Aptamer-functionalized magnetic nanoparticle-based bioassay for the detection of ochratoxin A using upconversion nanoparticles as labels. Analyst 2011, 136, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
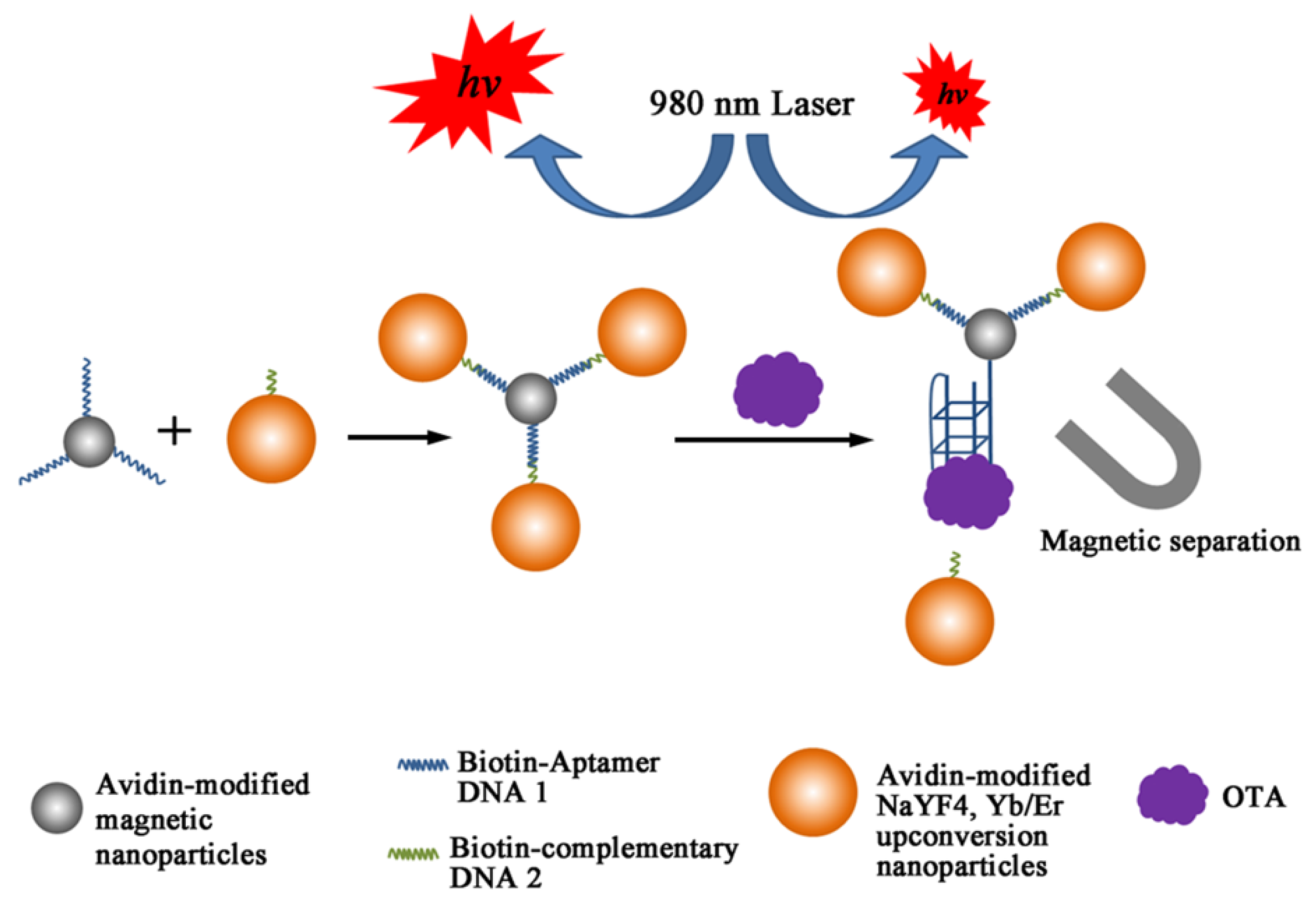
- Wang, C.; Qian, J.; Wang, K.; Wang, K.; Liu, Q.; Dong, X.; Wang, C.; Huang, X. Magnetic-fluorescent-targeting multifunctional aptasensorfor highly sensitive and one-step rapid detection of ochratoxin A. Biosens. Bioelectron. 2015, 68, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Wang, K.; Wang, C.; Hua, M.; Yang, Z.; Liu, Q.; Mao, H.; Wang, K. A FRET-based ratiometric fluorescent aptasensor for rapid and onsite visual detection of ochratoxin A. Analyst 2015, 140, 7434–7442. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.K.; Hayat, A.; Catanante, G.; Ocaña, C.; Marty, J.-L. A label free aptasensor for ochratoxin A detection in cocoa beans: An application to chocolate industries. Anal. Chim. Acta 2015, 889, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Jiang, L.; Yang, X.; Yan, Y.; Mao, H.; Wang, K. Highly sensitive impedimetric aptasensor based on covalent binding of gold nanoparticles on reduced graphene oxide with good dispersity and high density. Analyst 2014, 139, 5587–5593. [Google Scholar] [CrossRef] [PubMed]
- Rivas, L.; Mayorga-Martinez, C.C.; Quesada-González, D.; Zamora-Gálvez, A.; de la Escosura-Muñiz, A.; Merkoçi, A. Label-free impedimetric aptasensor for ochratoxin-A detection using iridium oxide nanoparticles. Anal. Chem. 2015, 87, 5167–5172. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Qian, J.; Jiang, L.; Yan, Y.; Wang, K.; Liu, Q.; Wang, K. Ultrasensitive electrochemical aptasensor for ochratoxin A based on two-level cascaded signal amplification strategy. Bioelectrochemistry 2014, 96, 7–13. [Google Scholar] [CrossRef] [PubMed]
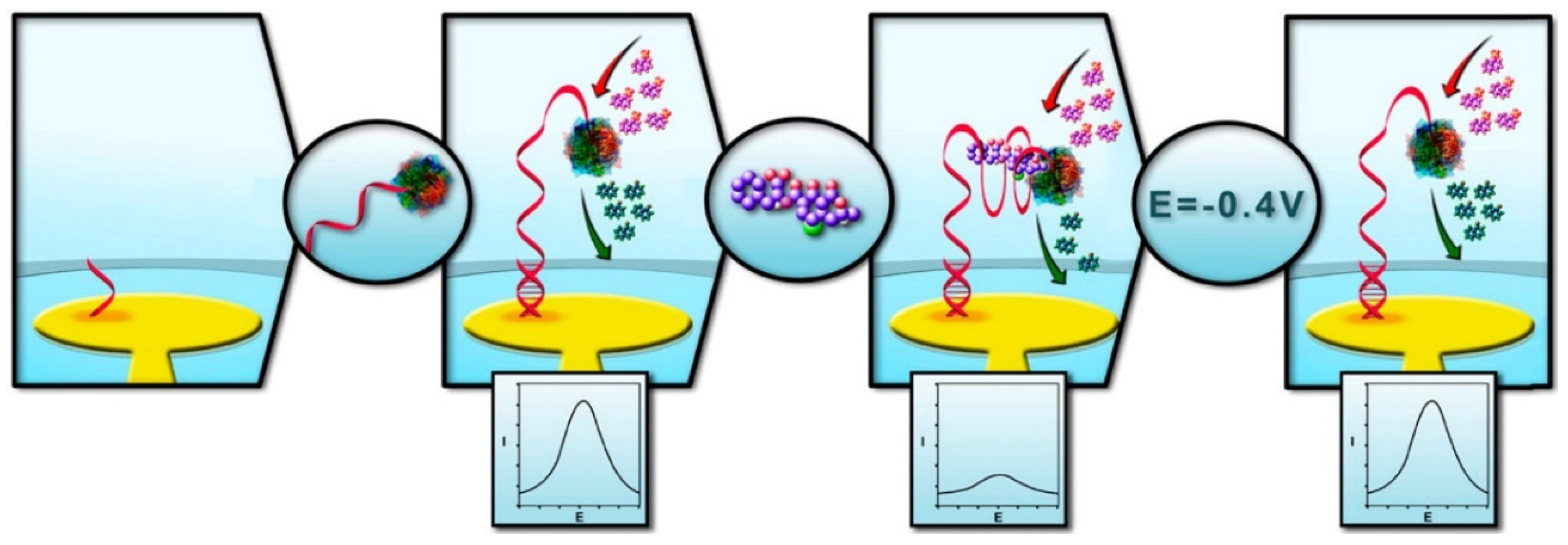
- Prieto-Simón, B.; Samitier, J. “Signal off” aptasensor based on enzyme inhibition induced by conformational switch. Anal. Chem. 2014, 86, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.K.; Hayat, A.; Catanante, G.; Istamboulie, G.; Marty, J.-L. Sensitive quantitation of ochratoxin A in cocoa beans using differential pulse voltammetry based aptasensor. Food Chem. 2016, 192, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Bulbul, G.; Hayat, A.; Andreescu, S. A generic amplification strategy for electrochemical aptasensors using a non-enzymatic nanoceria tag. Nanoscale 2015, 7, 13230–13238. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, J.; Li, J.; Yang, H.-H.; Fu, F.; Chen, G. An ultrasensitive signal-on electrochemical aptasensor via target-induced conjunction of split aptamer fragments. Biosens. Bioelectron. 2010, 25, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Duan, N.; Hun, X.; Wu, S. Electrochemiluminescent aptamer biosensor for the determination of ochratoxin A at a gold-nanoparticles-modified gold electrode using N-(aminobutyl)-N-ethylisoluminol as a luminescent label. Anal. Bioanal. Chem. 2010, 398, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Yin, H.; Xu, L.; Xu, Z.; Kuang, H.; Wang, L.; Xu, C. Femtogram ultrasensitive aptasensor for the detection of ochratoxin A. Biosens. Bioelectron. 2013, 42, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Nutiu, R.; Li, Y. Aptamers with fluorescence-signaling properties. Methods 2005, 37, 16–25. [Google Scholar] [CrossRef] [PubMed]
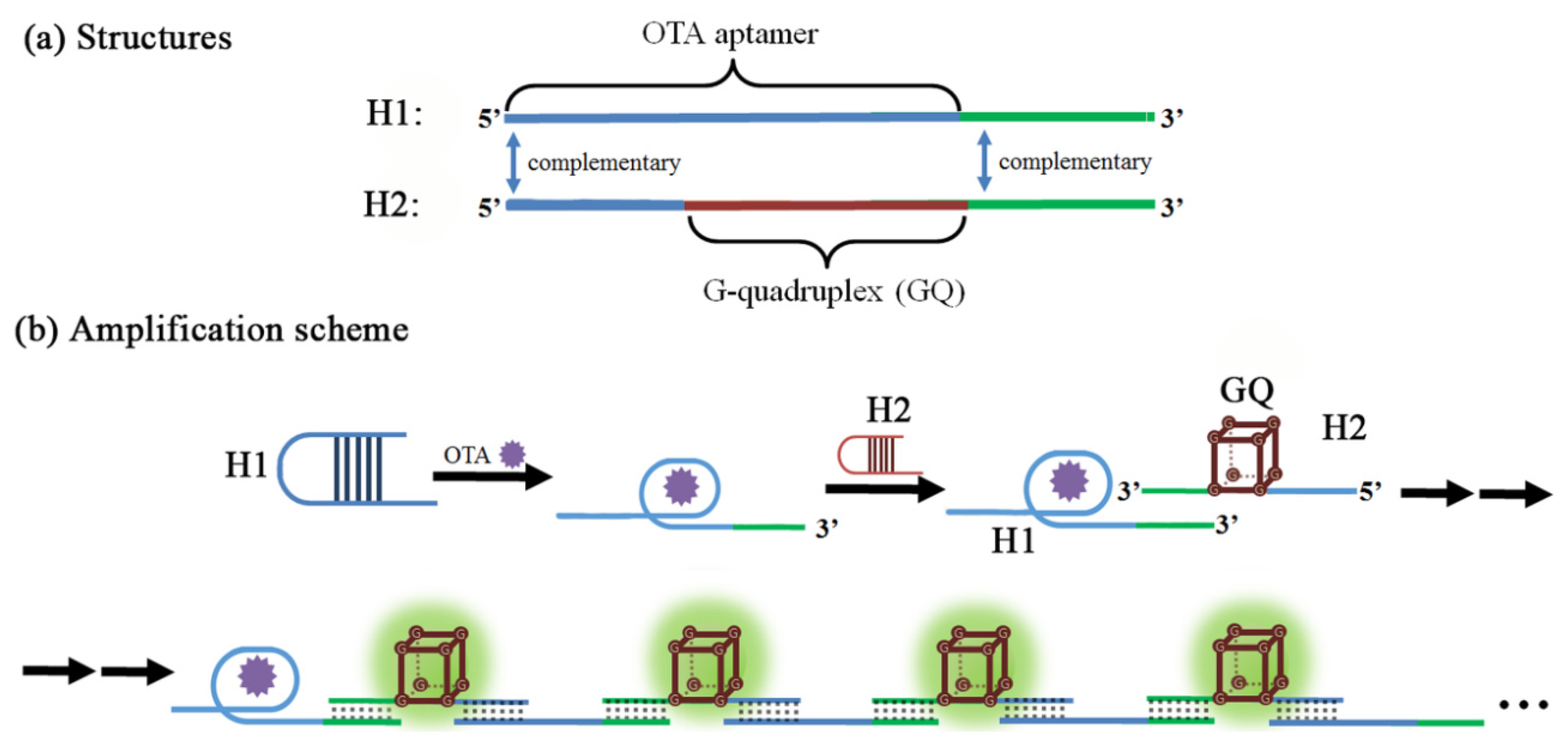
- Zhang, Y.; Yang, L.; Lin, C.; Guo, L.; Qiu, B.; Lin, Z.; Chen, G. Fluorescence aptasensor for ochratoxin A in food samples based on hyperbranched rolling circle amplification. Anal. Methods 2015, 7, 6109–6113. [Google Scholar] [CrossRef]
- Yao, L.; Chen, Y.; Teng, J.; Zheng, W.; Wu, J.; Adeloju, S.B.; Pan, D.; Chen, W. Integrated platform with magnetic purification and rolling circular amplification for sensitive fluorescent detection of ochratoxin A. Biosens. Bioelectron. 2015, 74, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, J.; Zheng, L.; Qian, H.; Xue, F.; Wu, Y.; Pan, D.; Adeloju, S.B.; Chen, W. Rolling chain amplification based signal-enhanced electrochemical aptasensor for ultrasensitive detection of ochratoxin A. Anal. Chem. 2013, 85, 10842–10849. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wei, S.; Liu, G.; Xie, S.; Chai, Y.; Yuan, R. Ultrasensitive electrochemiluminescent aptasensor for ochratoxin A detection with the loop-mediated isothermal amplification. Anal. Chim. Acta 2014, 811, 70–75. [Google Scholar] [CrossRef] [PubMed]
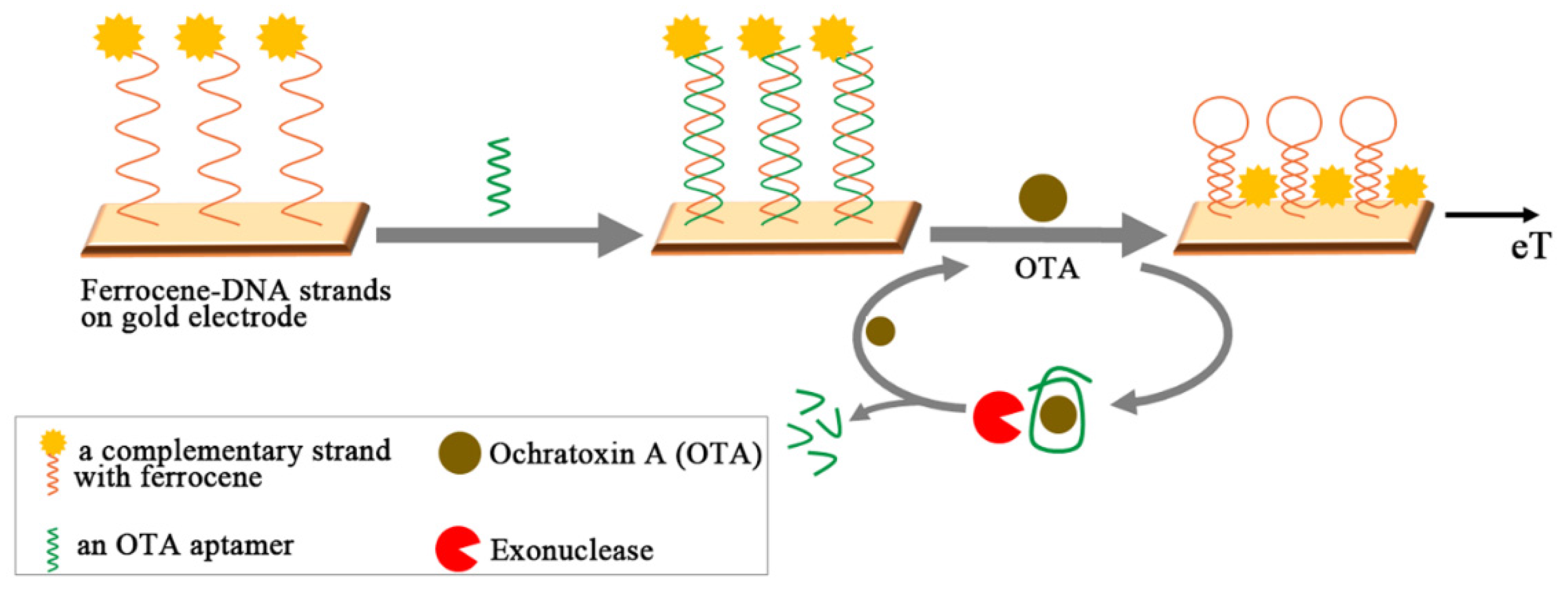
- Tong, P.; Zhang, L.; Xu, J.-J.; Chen, H.-Y. Simply amplified electrochemical aptasensor of ochratoxin a based on exonuclease-catalyzed target recycling. Biosens. Bioelectron. 2011, 29, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, J.; Wang, X.; Duan, Y. Amplified fluorescent aptasensor through catalytic recycling for highly sensitive detection of ochratoxin A. Biosens. Bioelectron. 2015, 65, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Jiang, B.; Xie, J.; Xiang, Y.; Yuan, R.; Chai, Y. Electrochemiluminescence recovery-based aptasensor for sensitive ochratoxin a detection via exonuclease-catalyzed target recycling amplification. Talanta 2014, 125, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Dong, X.; Liu, Q.; Wang, K. Label-free colorimetric aptasensor for sensitive detection of ochratoxin A utilizing hybridization chain reaction. Anal. Chim. Acta 2015, 860, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, M.; Xiang, Y.; Yuan, R.; Chai, Y. Binding-induced autonomous disassembly of aptamer-DNAzyme supersandwich nanostructures for sensitive electrochemiluminescence turn-on detection of ochratoxin A. Nanoscale 2014, 6, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jeon, K.-S.; Suh, Y.D.; Kim, M.-G. A label-free, direct and noncompetitive fret immunoassay for ochratoxin A based on intrinsic fluorescence of an antigen and antibody complex. Chem. Commun. 2011, 47, 9098–9100. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jo, E.-J.; Kim, M.-G. A label-free fluorescence immunoassay system for the sensitive detection of the mycotoxin, ochratoxin A. Chem. Commun. 2012, 48, 2304–2306. [Google Scholar] [CrossRef] [PubMed]
- Cheikhousman, R.; Zude, M.; Bouveresse, D.-R.; Léger, C.; Rutledge, D.; Birlouez-Aragon, I. Fluorescence spectroscopy for monitoring deterioration of extra virgin olive oil during heating. Anal. Bioanal. Chem. 2005, 382, 1438–1443. [Google Scholar] [CrossRef] [PubMed]
- Dridi, F.; Marrakchi, M.; Gargouri, M.; Saulnier, J.; Jaffrezic-Renault, N.; Lagarde, F. Comparison of carboxypeptidase Y and thermolysin for ochratoxin A electrochemical biosensing. Anal. Methods 2015, 7, 8954–8960. [Google Scholar] [CrossRef]
- Golightly, R.S.; Doering, W.E.; Natan, M.J. Surface-enhanced Raman spectroscopy and homeland security: A perfect match? ACS Nano 2009, 3, 2859–2869. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Delwiche, S.R.; Dong, Y. Feasibility of FT-Raman spectroscopy for rapid screening for DON toxin in ground wheat and barley. Food Addit. Contam. A 2009, 26, 1396–1401. [Google Scholar] [CrossRef]
- Sohn, M.; Himmelsbach, D.S.; Barton, F.E. A comparative study of fourier transform Raman and NIR spectroscopic methods for assessment of protein and apparent amylose in rice. Cereal Chem. J. 2004, 81, 429–433. [Google Scholar] [CrossRef]
- Lee, K.-M.; Herrman, T.J.; Bisrat, Y.; Murray, S.C. Feasibility of surface-enhanced Raman spectroscopy for rapid detection of aflatoxins in maize. J. Agric. Food Chem. 2014, 62, 4466–4474. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; He, L. Surface-enhanced Raman spectroscopy for the chemical analysis of food. Compr. Rev. Food Sci. Food Saf. 2014, 13, 317–328. [Google Scholar] [CrossRef]
- Ganbold, E.-O.; Lee, C.M.; Cho, E.-M.; Son, S.J.; Kim, S.; Joo, S.-W.; Yang, S.I. Subnanomolar detection of ochratoxin A using aptamer-attached silver nanoparticles and surface-enhanced Raman scattering. Anal. Methods 2014, 6, 3573–3577. [Google Scholar] [CrossRef]
- Galarreta, B.; Tabatabaei, M.; Guieu, V.; Peyrin, E.; Lagugné-Labarthet, F. Microfluidic channel with embedded SERS 2D platform for the aptamer detection of ochratoxin A. Anal. Bioanal. Chem. 2013, 405, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, Y.; Luo, Y.; Yang, X.; Li, M.; Song, Q. Double detection of mycotoxins based on SERS labels embedded Ag@Au core-shell nanoparticles. ACS Appl. Mater. Interfaces 2015, 7, 21780–21786. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.P.; Mantle, P.G. Biosynthesis of ochratoxins by aspergillus ochraceus. Phytochemistry 2001, 58, 709–716. [Google Scholar] [CrossRef]
- Chung, S.W.C.; Kwong, K.P. Determination of ochratoxin A at parts-per-trillion levels in cereal products by immunoaffinity column cleanup and high-performance liquid chromatography/mass spectrometry. J. AOAC Int. 2007, 90, 773–777. [Google Scholar] [PubMed]
- Lates, V.; Yang, C.; Popescu, I.; Marty, J.-L. Displacement immunoassay for the detection of ochratoxin A using ochratoxin B modified glass beads. Anal. Bioanal. Chem. 2012, 402, 2861–2870. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, T.H. Recent Advances for the Detection of Ochratoxin A. Toxins 2015, 7, 5276-5300. https://doi.org/10.3390/toxins7124882
Ha TH. Recent Advances for the Detection of Ochratoxin A. Toxins. 2015; 7(12):5276-5300. https://doi.org/10.3390/toxins7124882
Chicago/Turabian StyleHa, Tai Hwan. 2015. "Recent Advances for the Detection of Ochratoxin A" Toxins 7, no. 12: 5276-5300. https://doi.org/10.3390/toxins7124882
APA StyleHa, T. H. (2015). Recent Advances for the Detection of Ochratoxin A. Toxins, 7(12), 5276-5300. https://doi.org/10.3390/toxins7124882