
Computational Studies of Venom Peptides Targeting Potassium Channels

Abstract
:
1. Overview of Ion Channels and Venom Peptides
2. Structure of K+ Channels
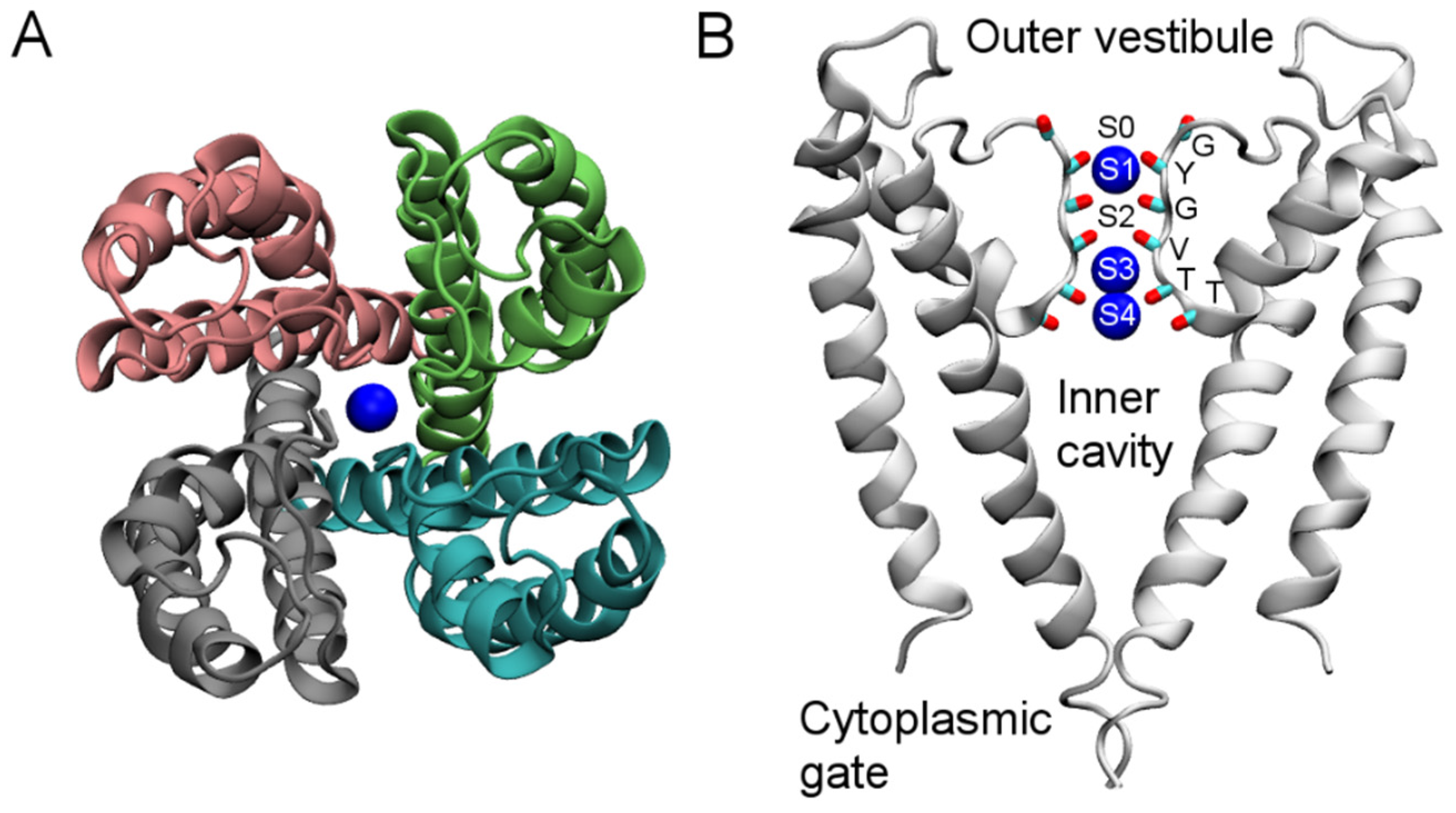
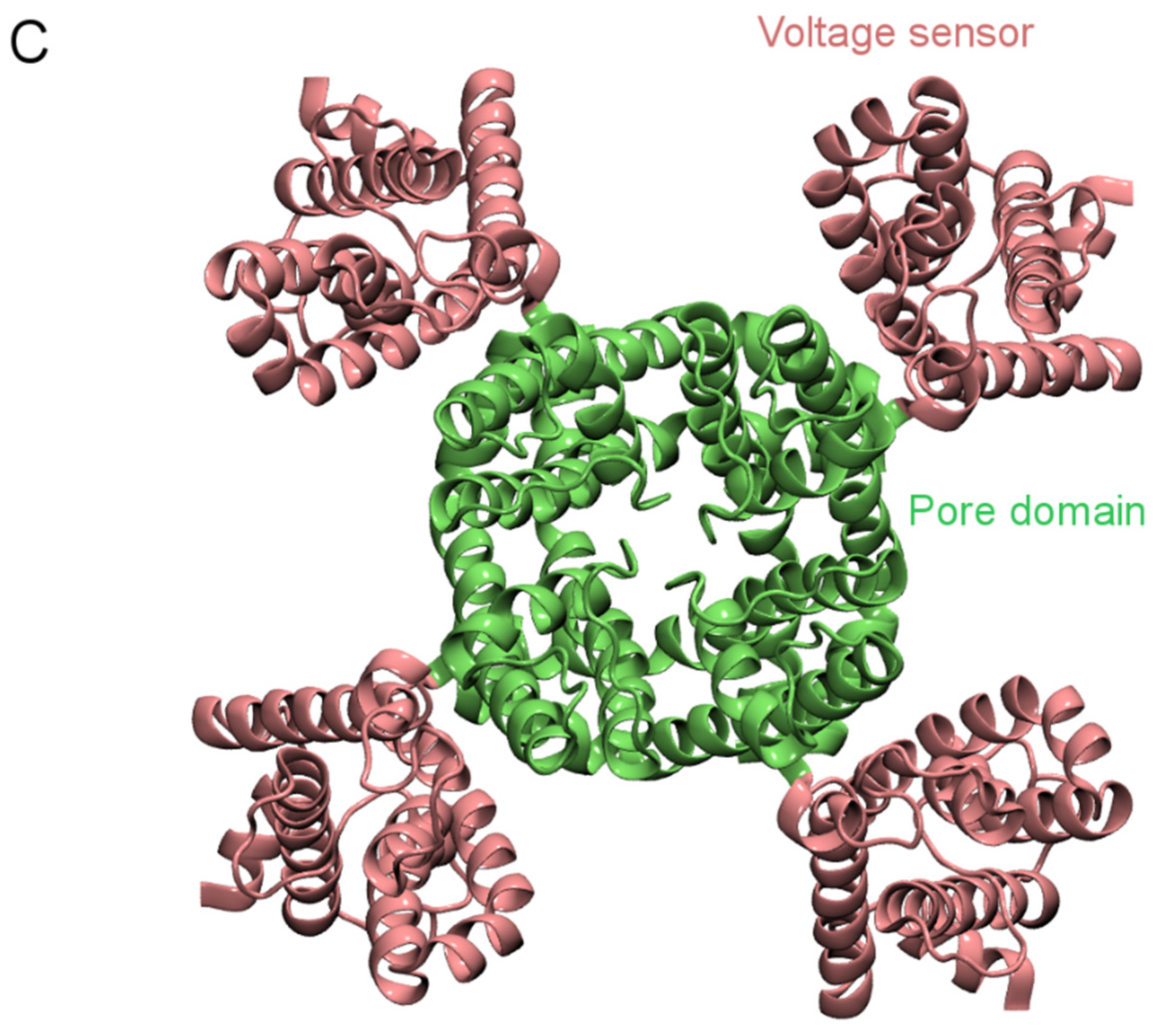
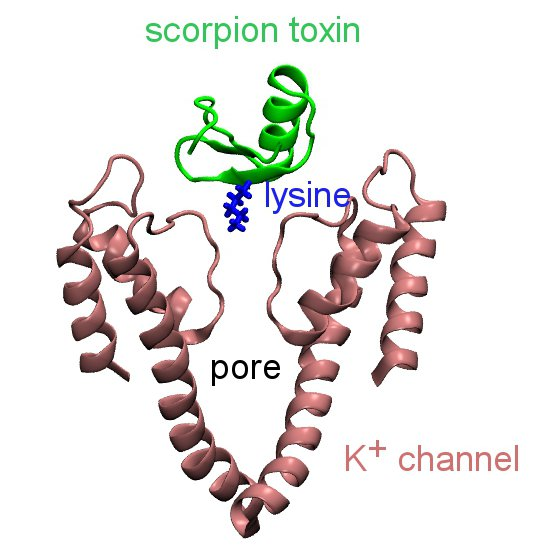
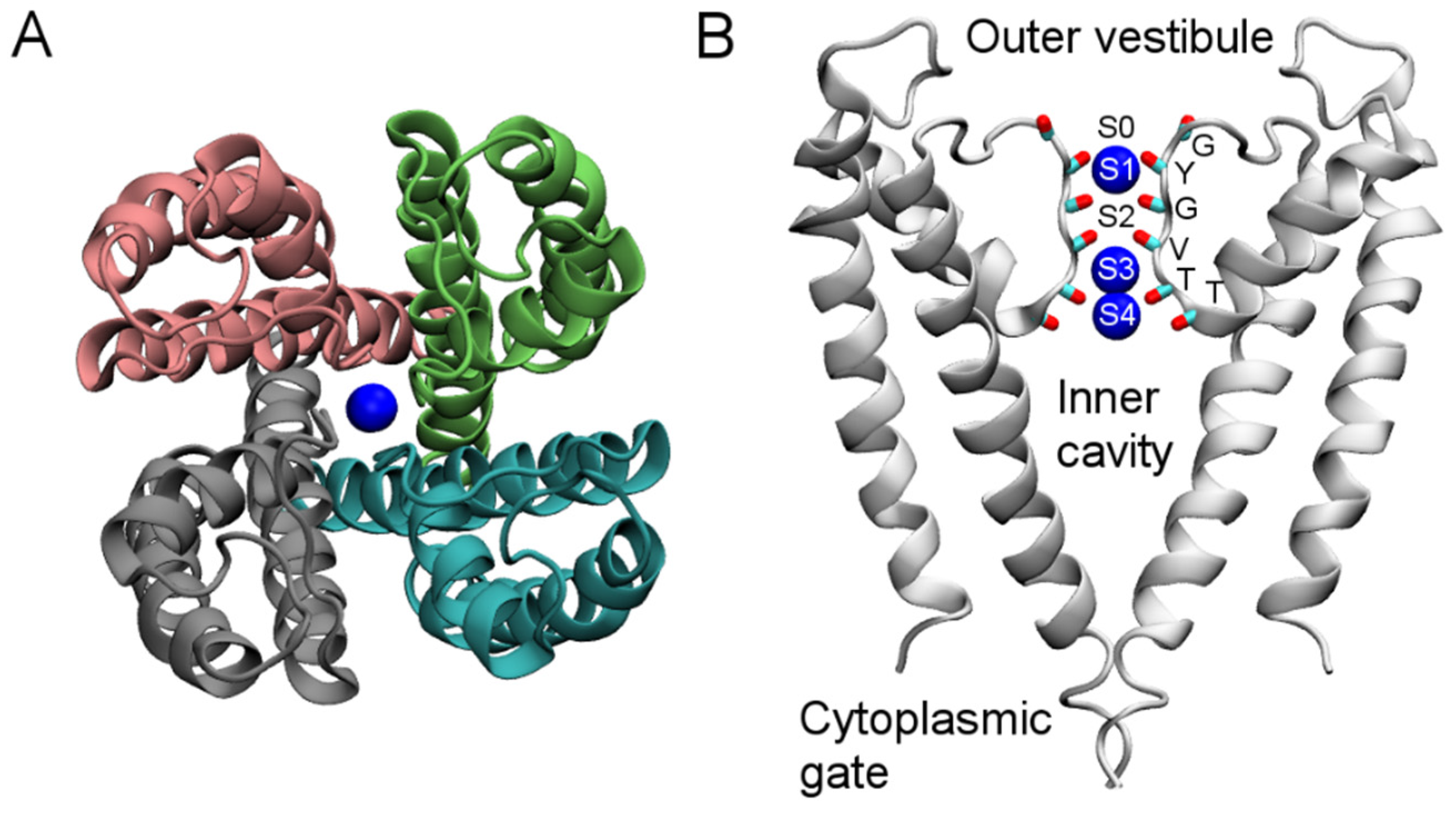
3. Computational Methods
3.1. Prediction of Binding Modes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Docking | BD | MD |
|---|---|---|---|
| Water | Implicit | Implicit | Explicit |
| Ions | Ignored | Explicit | Explicit |
| Membrane | Ignored | Implicit | Explicit |
| Flexibility | Ignored/Limited | Ignored | Allowed |
| Output trajectory | No | Yes | Yes |
| Time scale | - | Microseconds | Nanoseconds |
3.2. Calculation of Kd
4. Venom Peptides
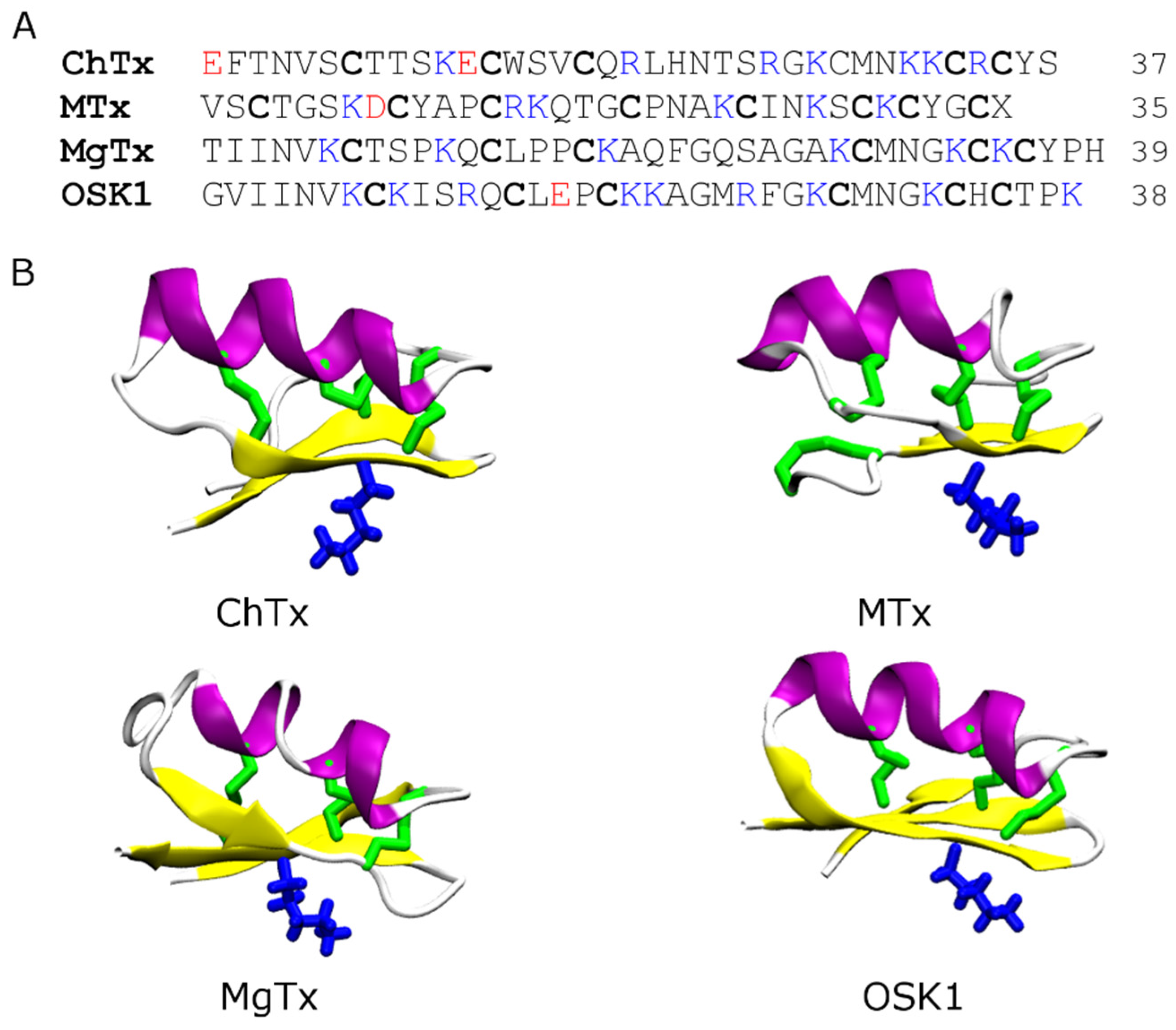
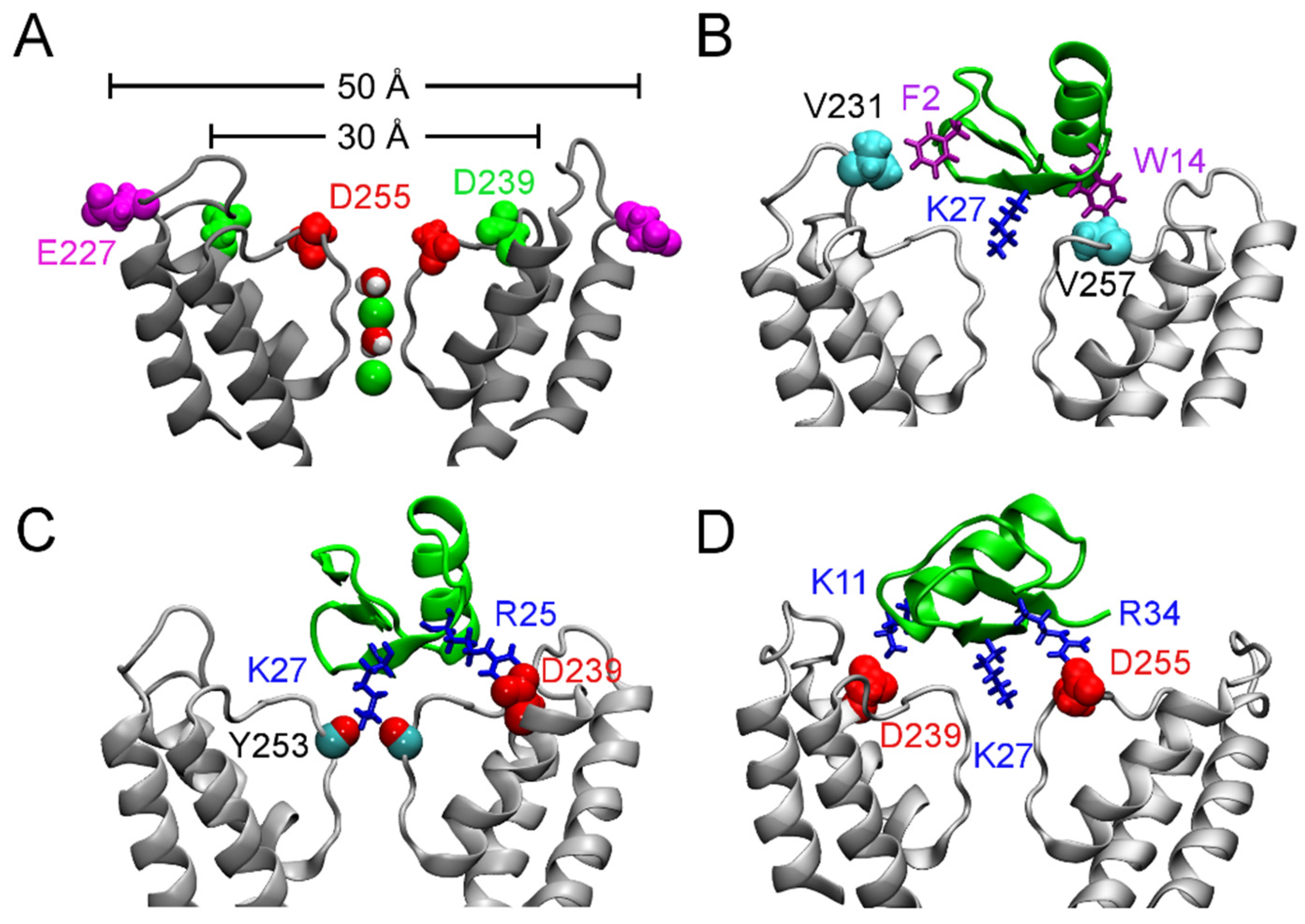
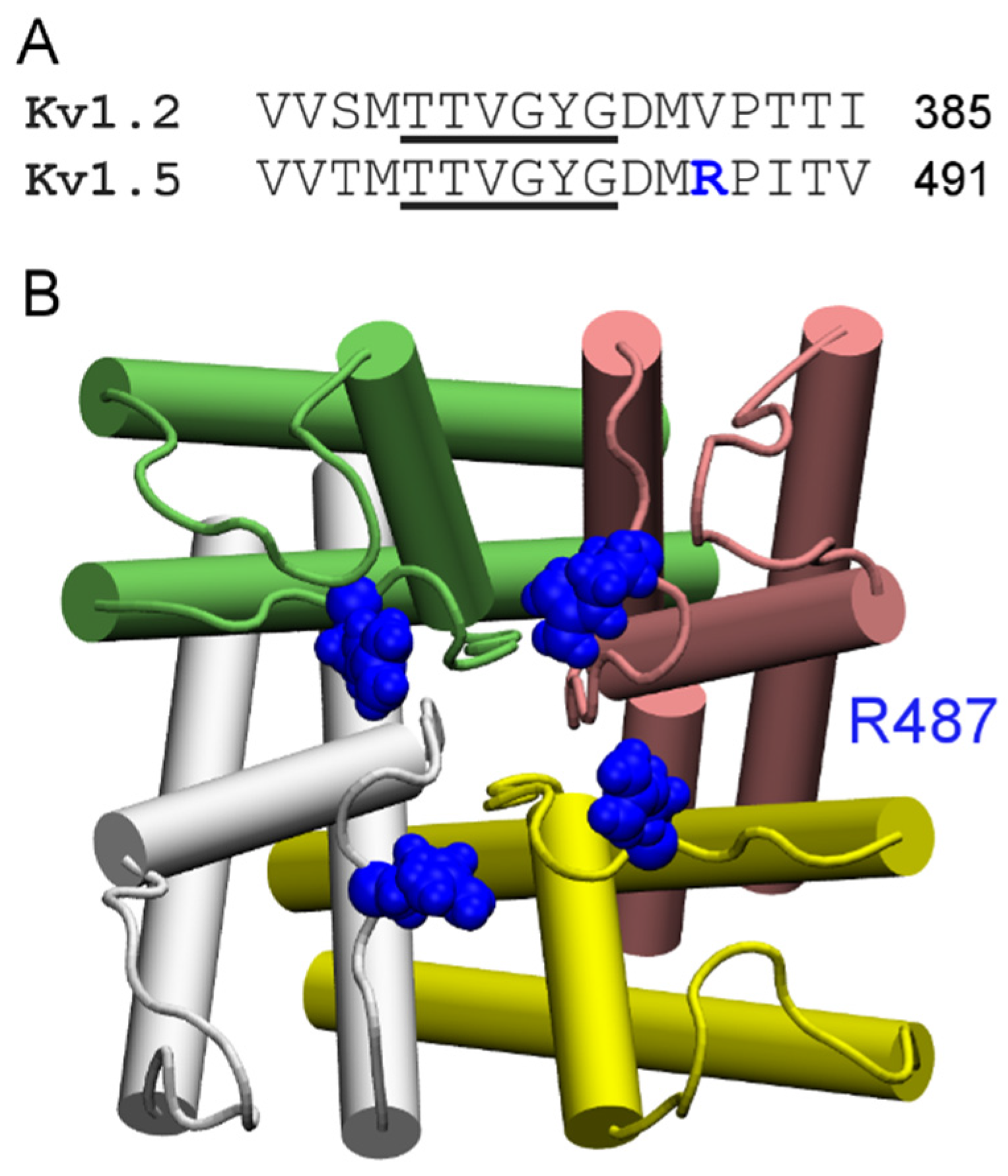
4.1. Scorpion Toxins
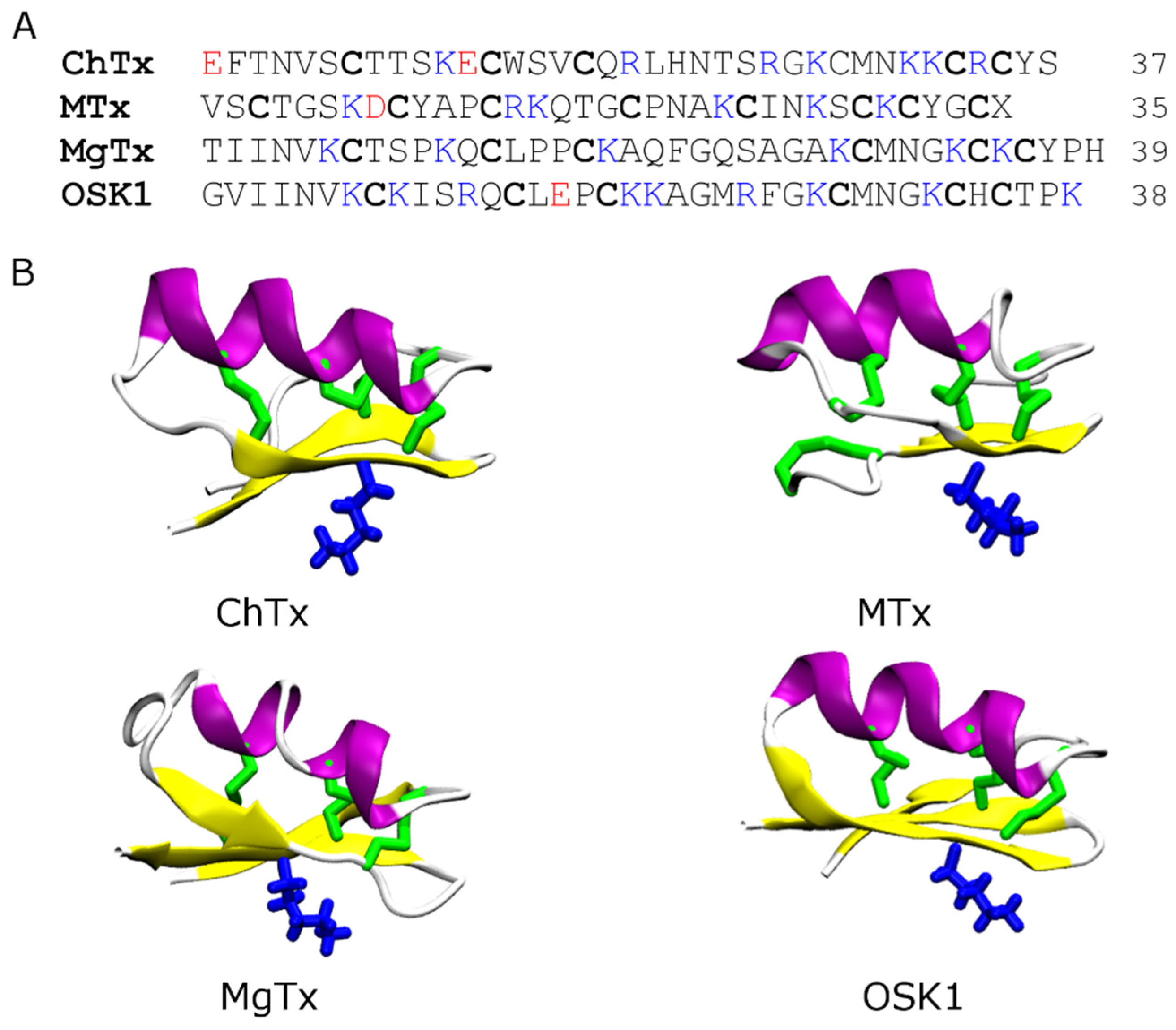
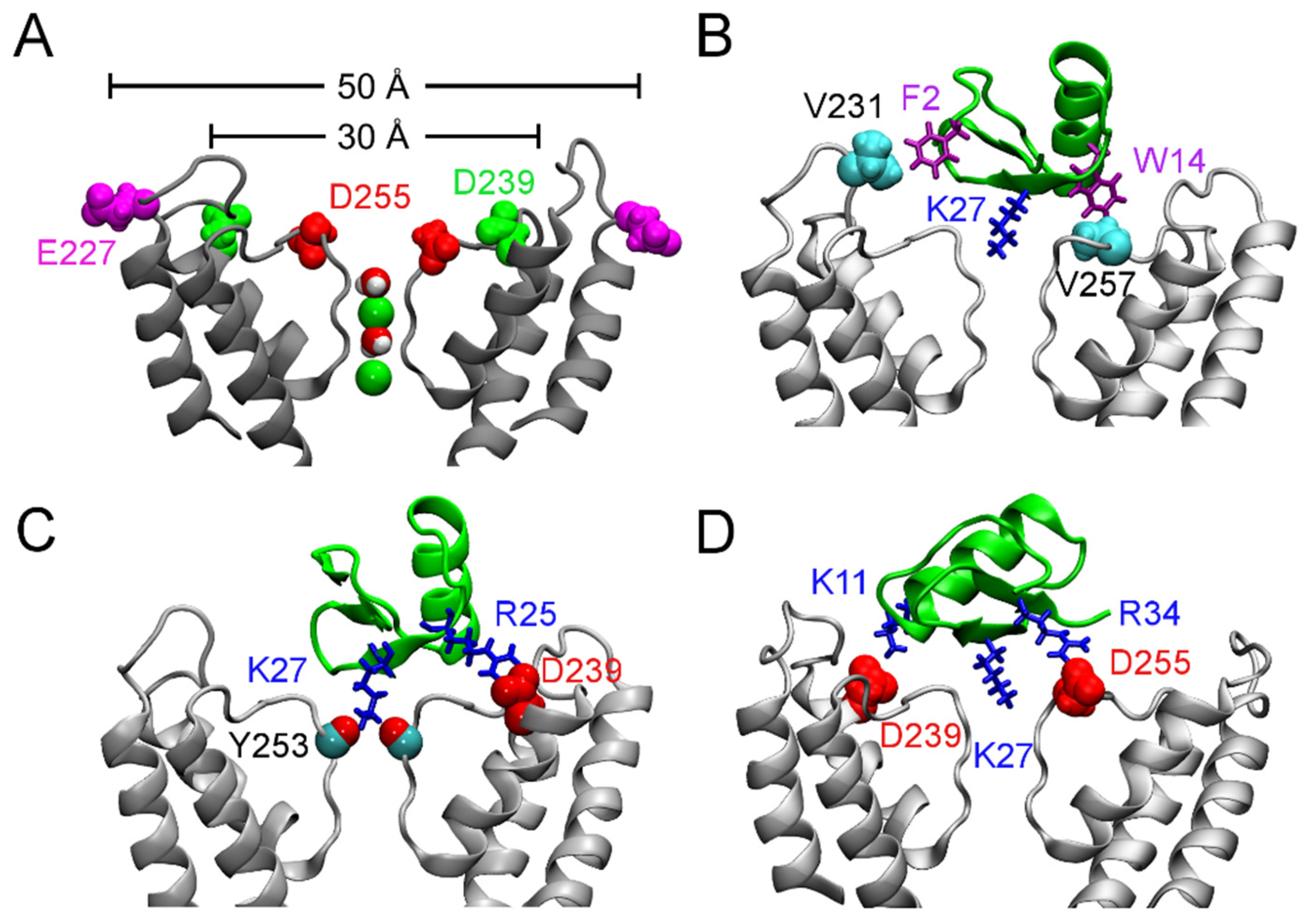
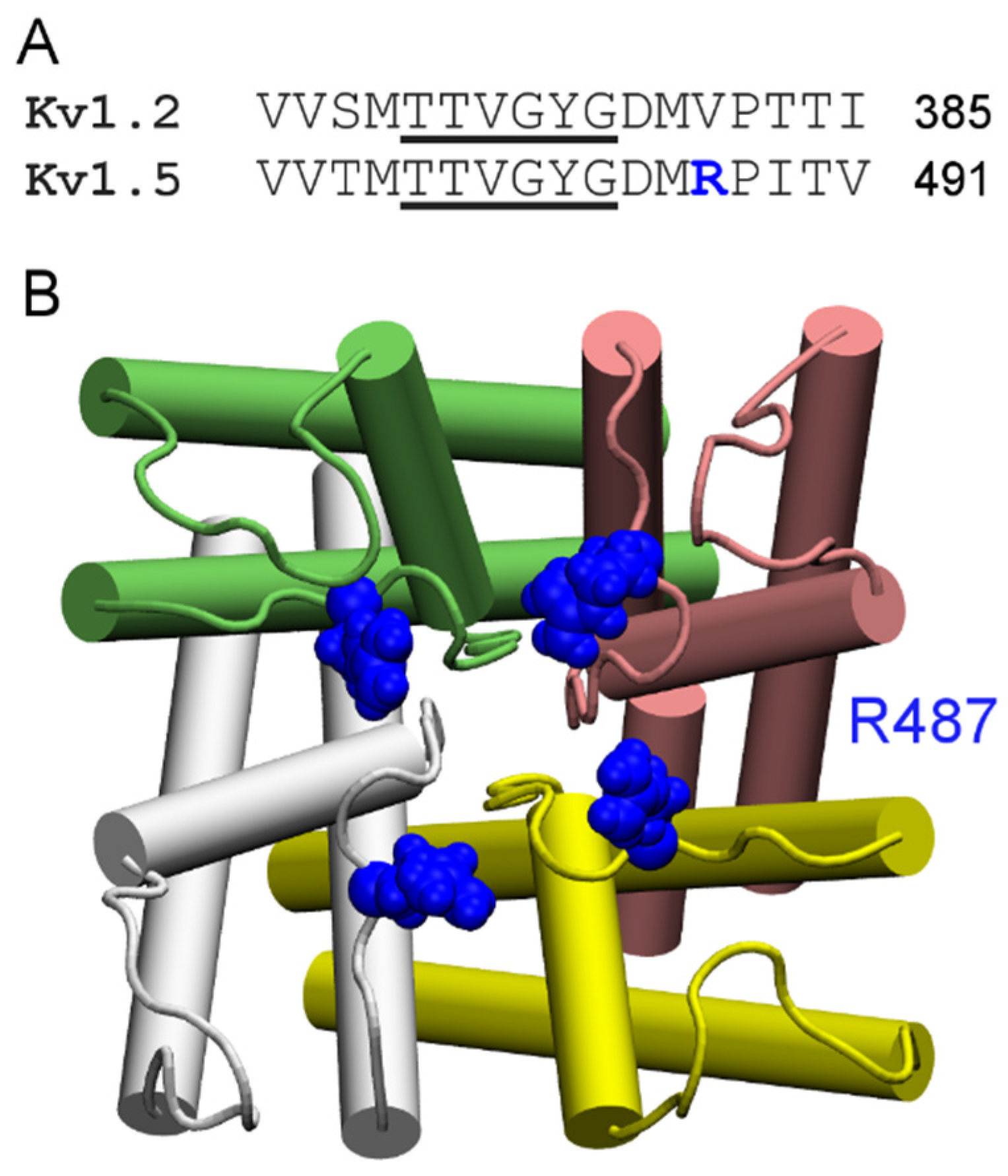
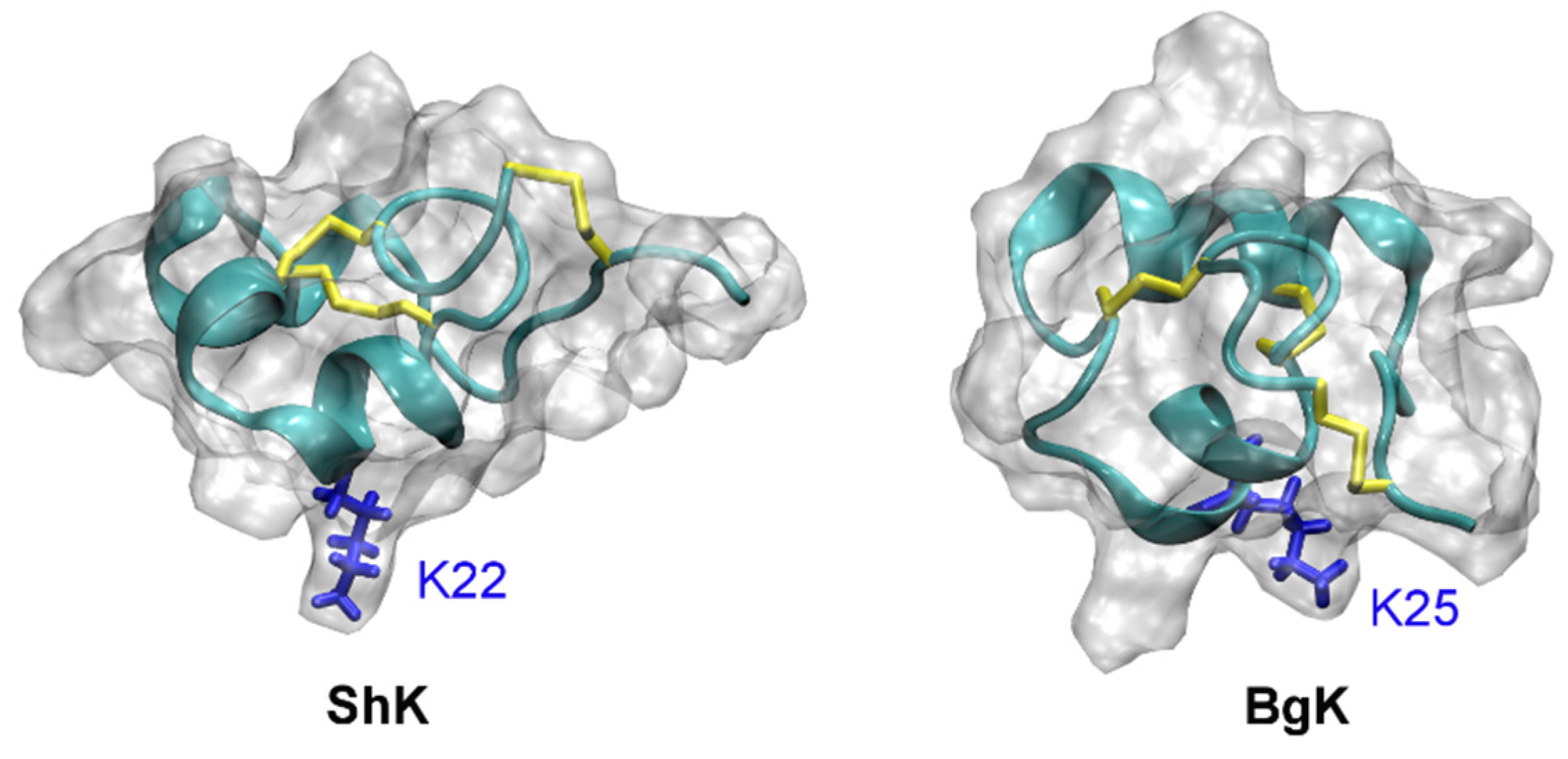
4.2. Conotoxins
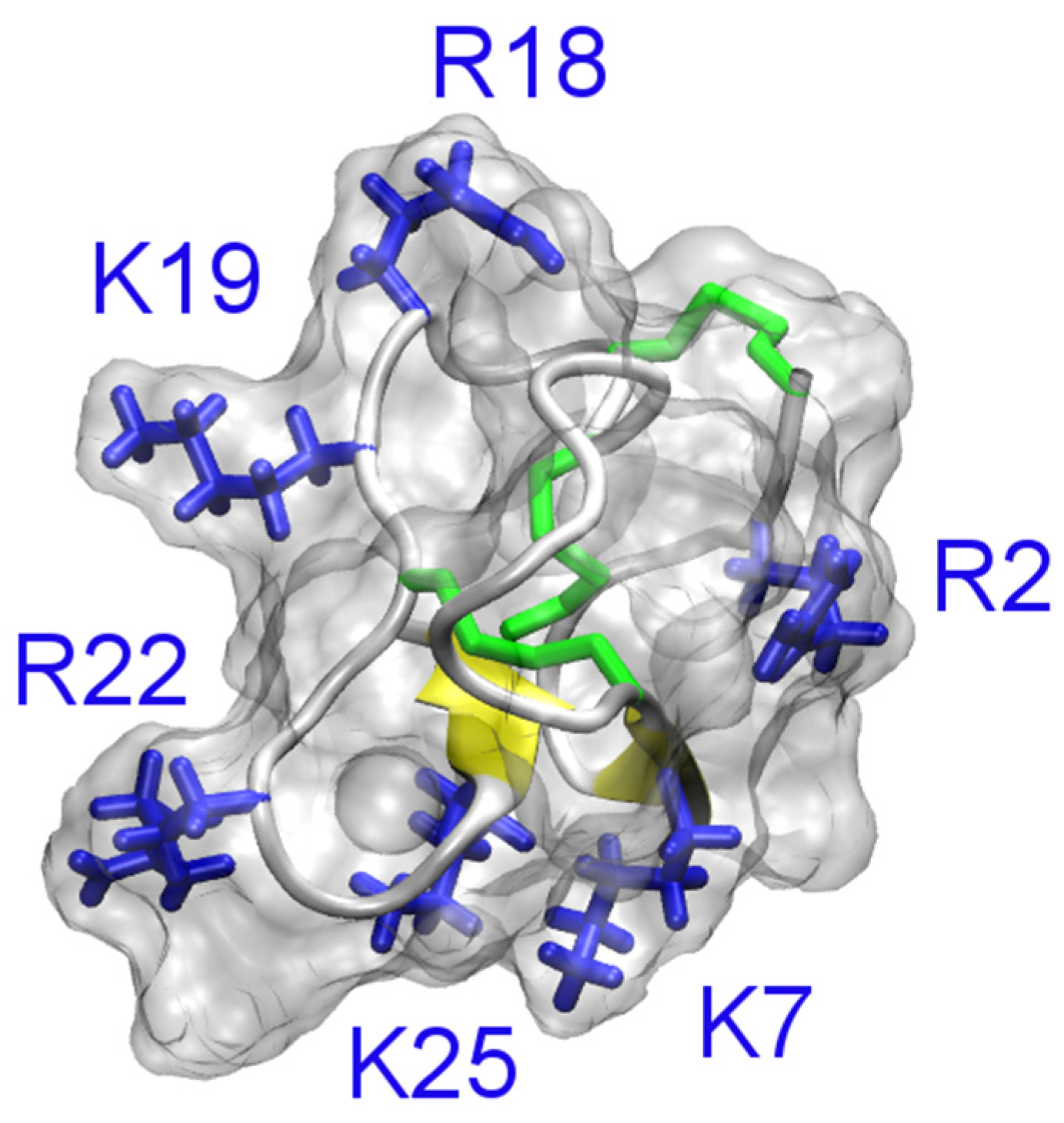
4.3. Spider Toxins
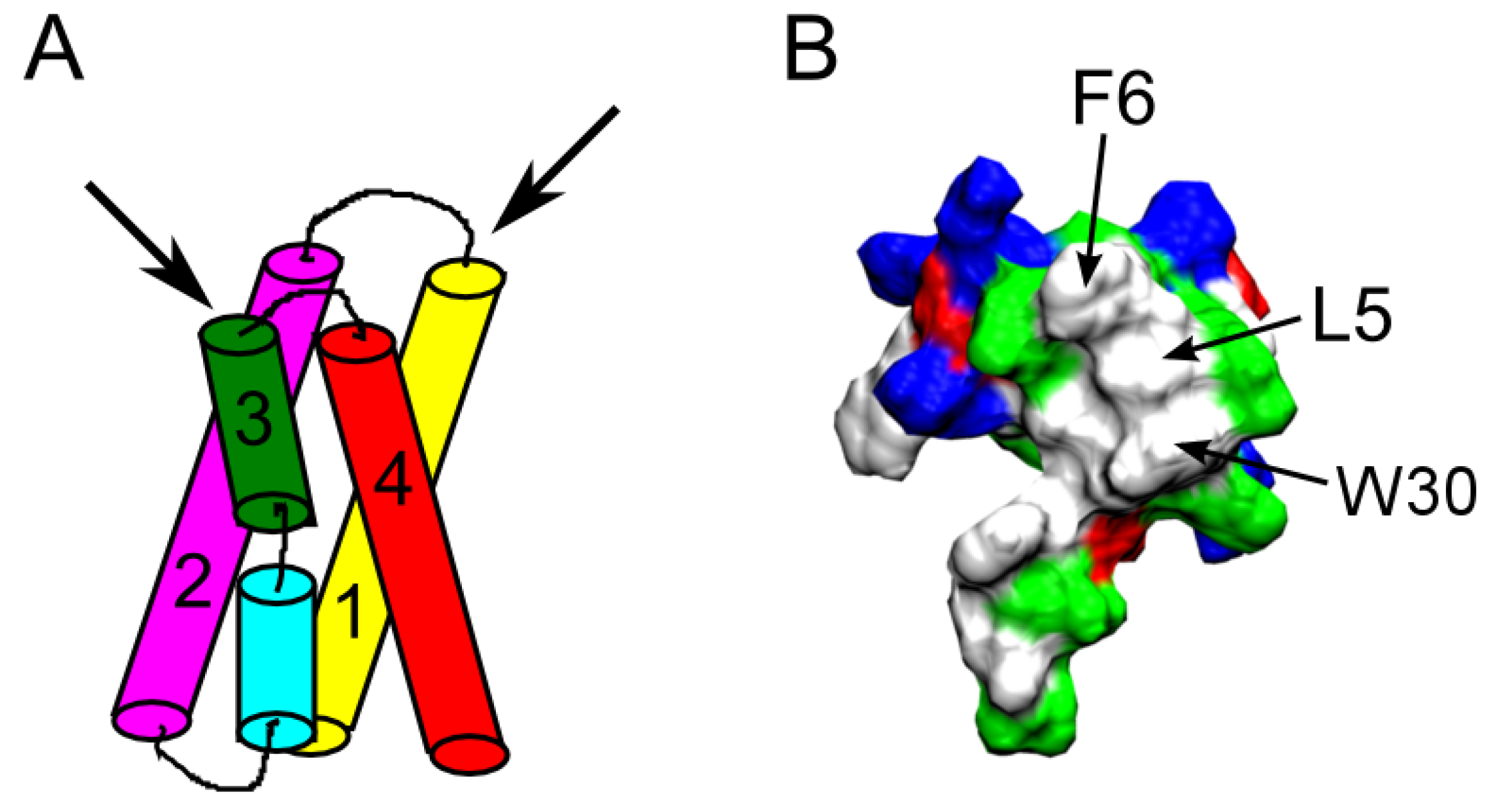
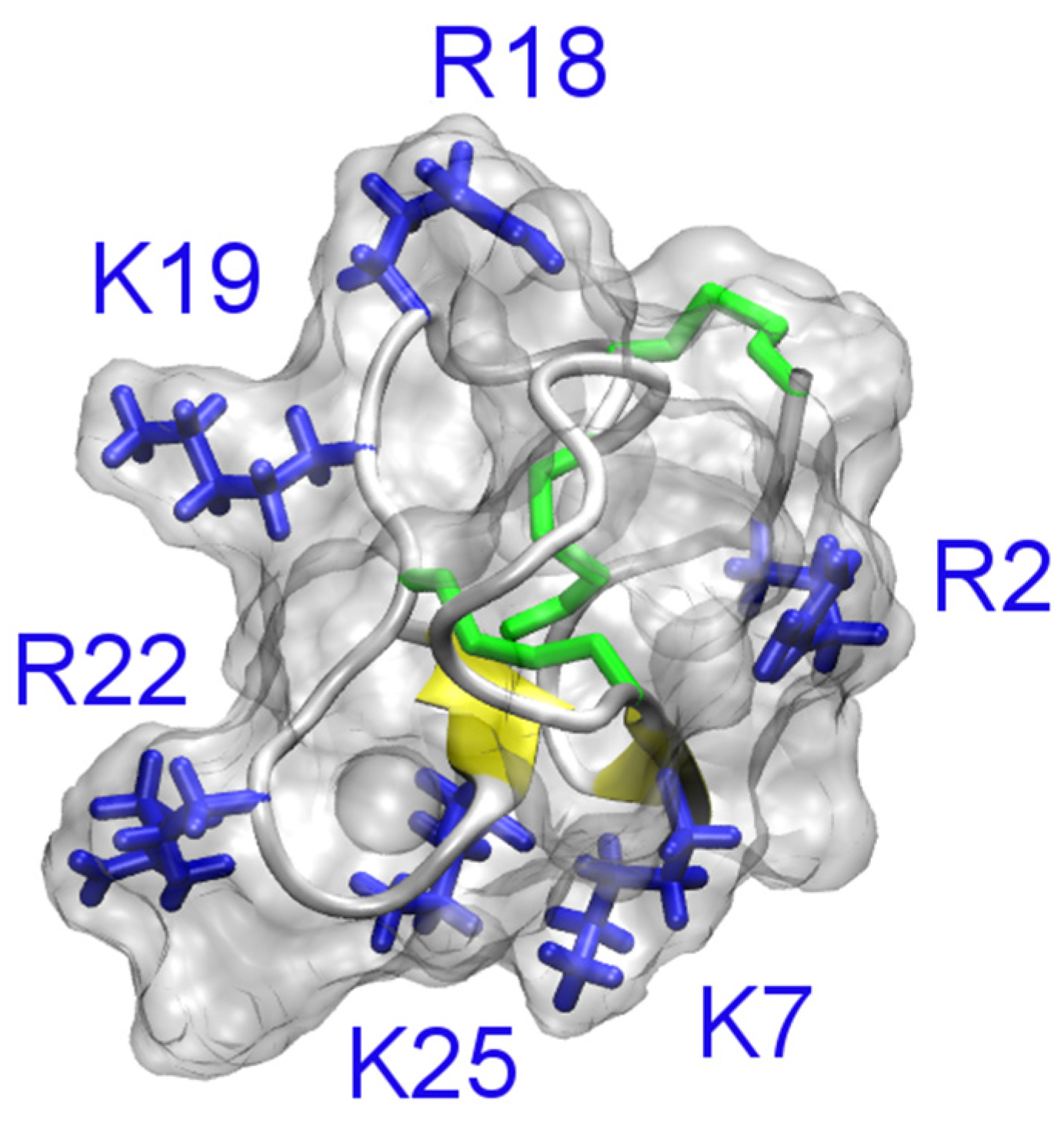
4.4. Other Animal Toxins
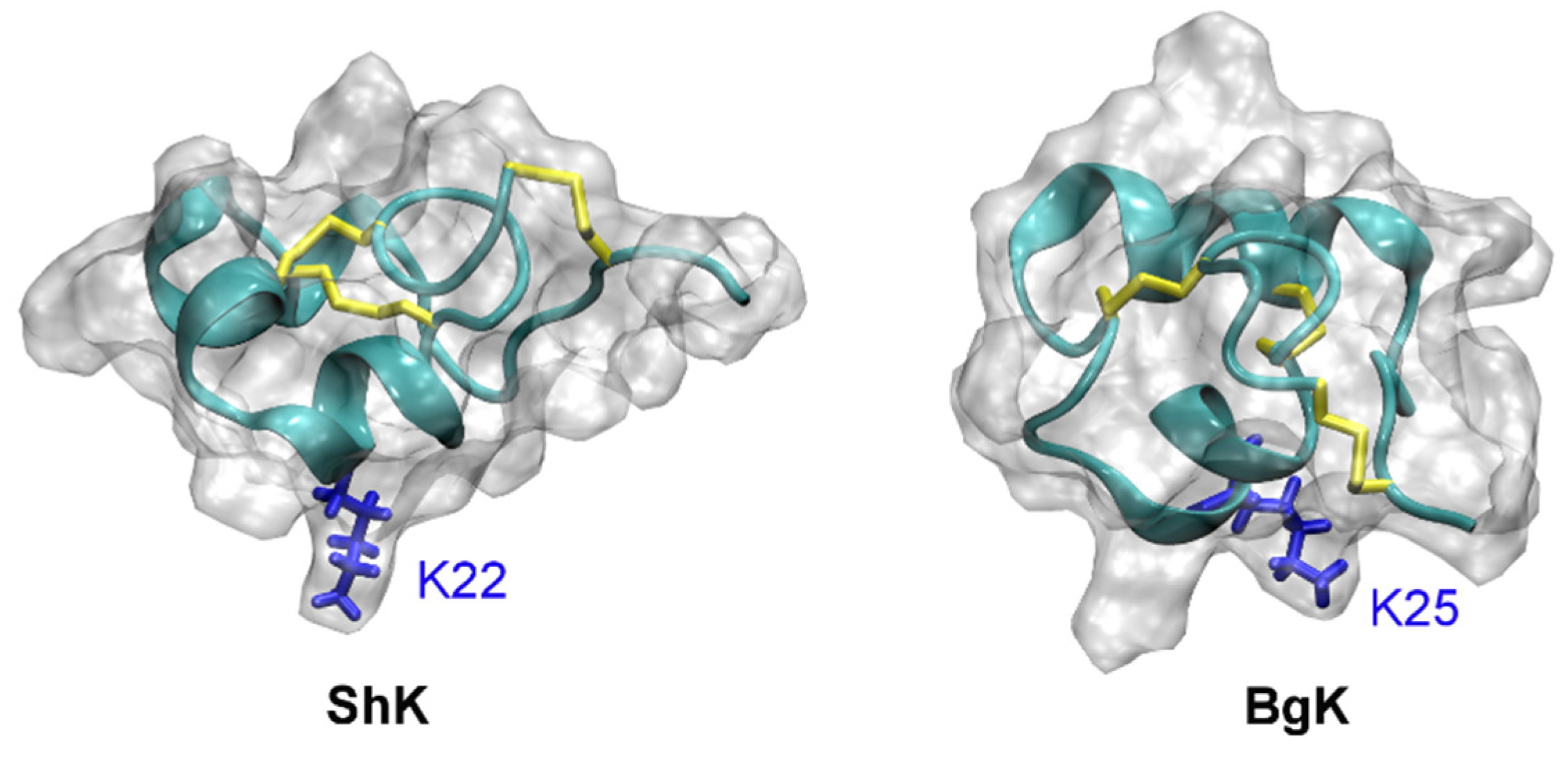
5. Toxin Analogues
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bagal, S.; Brown, A.D.; Cox, P.J.; Omoto, K.; Owen, R.M.; Pryde, D.C.; Sidders, B.; Skerratt, S.E.; Stevens, E.B.; Storer, R.I.; et al. Ion channels as therapeutic targets: A drug discovery perspective. J. Med. Chem. 2013, 56, 593–624. [Google Scholar] [CrossRef] [PubMed]
- Clare, J.J. Targeting ion channels for drug discovery. Discov. Med. 2010, 9, 253–260. [Google Scholar] [PubMed]
- Xie, M.; Holmqvist, M.H.; Hsia, A.Y. Ion channel drug discovery expands into new disease areas. Curr. Drug Discov. 2004, 31, 31–33. [Google Scholar]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, D.S.; McPhee, J.C.; Scheuer, T.; Catterall, W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994, 265, 1724–1728. [Google Scholar] [CrossRef] [PubMed]
- Chandy, K.G.; Wulff, H.; Beeton, C.; Pennington, M.; Gutman, G.A.; Cahalan, M. K+ channels as targets for specific immunomodulation. Trends Pharmacol. Sci. 2004, 25, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Yi, H.; Yin, S.J.; Chen, Z.Y.; Liu, H.; Cao, Z.J.; Wu, Y.L.; Li, W.X. Structural basis of a potent peptide inhibitor designed for Kv1.3 channel, a therapeutic target of autoimmune disease. J. Biol. Chem. 2008, 283, 19058–19065. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S. µ-Conotoxins as leads in the development of new analgesics. Molecules 2010, 15, 2825–2844. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; McArthur, J.R.; Adams, D.J. Conotoxins targeting neuronal voltage-gated sodium channel subtypes: Potential analgesics? Toxins 2012, 4, 1236–1260. [Google Scholar] [CrossRef] [PubMed]
- Estrada, G.; Villegas, E.; Corzo, G. Spider venoms: A rich source of acylpolyamines and peptides as new leads for CNS drugs. Nat. Prod. Rep. 2007, 24, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M. Conus venom peptides, receptor and ion channel targets, and drug design: 50 million years of neuropharmacology. Mol. Biol. Cell 1997, 8, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, E.; Gurrola, G.B.; Schwartz, E.F.; Possani, L.D. Scorpion venom components as potential candidates for drug development. Toxicon 2015, 93, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Nielsen, K.J.; Craik, D.J.; Loughnan, M.L.; Adams, D.A.; Sharpe, I.A.; Luchian, T.; Adams, D.J.; Bond, T.; Thomas, L.; et al. Novel ω-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J. Biol. Chem. 2000, 275, 35335–35344. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Smith, J.J.; Vetter, I.; Rupasinghe, D.B.; Er, S.Y.; Senff, S.; Herzig, V.; Mobli, M.; Lewis, R.J.; Bosmans, F.; et al. Seven novel modulators of the analgesic target NaV1.7 uncovered using a high-throughput venom-based discovery approach. Br. J. Pharmacol. 2015, 172, 2445–2458. [Google Scholar] [PubMed]
- Gur, M.; Kahn, R.; Karbat, I.; Regev, N.; Wang, J.T.; Catterall, W.A.; Gordon, D.; Gurevitz, M. Elucidation of the molecular basis of selective recognition uncovers the interaction site for the core domain of scorpion α-toxins on sodium channels. J. Biol. Chem. 2011, 286, 35209–35217. [Google Scholar] [CrossRef] [PubMed]
- Gurevitz, M. Mapping of scorpion toxin receptor sites at voltage-gated sodium channels. Toxicon 2012, 60, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Yarov-Yarovoy, V.; Scheuer, T.; Karbat, I.; Cohen, L.; Gordon, D.; Gurevitz, M.; Catterall, W.A. Mapping the interaction site for a β-scorpion toxin in the pore module of domain III of voltage-gated Na+ channels. J. Biol. Chem. 2012, 287, 30719–30728. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Yarov-Yarovoy, V.; Kahn, R.; Gordon, D.; Gurevitz, M.; Scheuer, T.; Catterall, W.A. Mapping the receptor site for α-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. USA 2011, 108, 15426–15431. [Google Scholar] [CrossRef] [PubMed]
- Possani, L.D.; Becerril, B.; Delepierre, M.; Tytgat, J. Scorpion toxins specific for Na+-channels. Eur. J. Biochem. 1999, 264, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Tytgat, J. Voltage-gated sodium channel modulation by scorpion α-toxins. Toxicon 2007, 49, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Robertson, B. Dendrotoxins: Structure-activity relationships and effects on potassium ion channels. Curr. Med. Chem. 2004, 11, 3065–3072. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kini, R.M. From snake venom toxins to therapeutics—Cardiovascular examples. Toxicon 2012, 59, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Cao, Z.J.; Li, W.X.; Wu, Y.L. Cloning and characterization of a novel Kunitz-type inhibitor from scorpion with unique cysteine framework. Toxicon 2013, 72, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Hu, Y.T.; Han, S.; Yin, S.J.; He, Y.W.; Wu, Y.L.; Cao, Z.J.; Li, W.X. ImKTx1, a new Kv1.3 channel blocker with a unique primary structure. J. Biochem. Mol. Toxicol. 2011, 25, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Hu, Y.T.; Yang, W.S.; He, Y.W.; Feng, J.; Wang, B.; Zhao, R.M.; Ding, J.P.; Cao, Z.J.; Li, W.X.; et al. Hg1, novel peptide inhibitor specific for Kv1.3 channels from first scorpion Kunitz-type potassium channel toxin family. J. Biol. Chem. 2012, 287, 13813–13821. [Google Scholar] [PubMed]
- Chen, Z.Y.; Zeng, D.Y.; Hu, Y.T.; He, Y.W.; Pan, N.; Ding, J.P.; Cao, Z.J.; Liu, M.L.; Li, W.X.; Yi, H.; et al. Structural and functional diversity of acidic scorpion potassium channel toxins. PLoS ONE 2012, 7, e35154. [Google Scholar] [PubMed]
- Han, S.; Yin, S.J.; Yi, H.; Mouhat, S.; Qiu, S.; Cao, Z.J.A.; Sabatier, J.M.; Wu, Y.L.; Li, W.X. Protein-protein recognition control by modulating electrostatic interactions. J. Proteome Res. 2010, 9, 3118–3125. [Google Scholar] [PubMed]
- Wu, Y.L.; Cao, Z.J.; Yi, H.; Jiang, D.H.; Mao, X.; Liu, H.; Li, W.X. Simulation of the interaction between ScyTx and small conductance calcium-activated potassium channel by docking and MM-PBSA. Biophys. J. 2004, 87, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Cao, Z.J.; Yin, S.J.; Dai, C.; Wu, Y.L.; Li, W.X. Interaction simulation of hERG K+ channel with its specific BeKm-1 peptide: Insights into the selectivity of molecular recognition. J. Proteome Res. 2007, 6, 611–620. [Google Scholar] [PubMed]
- Yi, H.; Qiu, S.; Wu, Y.; Li, W.X.; Wang, B. Differential molecular information of maurotoxin peptide recognizing IKCa and Kv1.2 channels explored by computational simulation. BMC Struct. Biol. 2011, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.J.; Jiang, L.; Yi, H.; Han, S.; Yang, D.W.; Liu, M.L.; Liu, H.; Cao, Z.J.; Wu, Y.L.; Li, W.X. Different residues in channel turret determining the selectivity of ADWX-1 inhibitor peptide between Kv1.1 and Kv1.3 channels. J. Proteome Res. 2008, 7, 4890–4897. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Chen, R.; Chung, S.H. Computational methods of studying the binding of toxins from venomous animals to biological ion channels: Theory and applications. Physiol. Rev. 2013, 93, 767–802. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Mahdavi, S.; Kuyucak, S. Computational studies of marine toxins targeting ion channels. Mar. Drugs 2013, 11, 848–869. [Google Scholar] [CrossRef] [PubMed]
- Kuyucak, S.; Norton, R.S. Computational approaches for designing potent and selective analogs of peptide toxins as novel therapeutics. Future Med. Chem. 2014, 6, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lee, A.; Chen, J.; Cadene, M.; Chait, B.T.; MacKinnon, R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature 2002, 417, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lee, A.; Chen, J.; Ruta, V.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray structure of a voltage-dependent K+ channel. Nature 2003, 423, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science 2005, 309, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Avalos, J.L.; Chen, J.Y.; MacKinnon, R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 Å resolution. Science 2009, 326, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Lee, A.; Campbell, E.; Mackinnon, R. Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K+ channel. eLife 2013, 2, e00594. [Google Scholar] [CrossRef] [PubMed]
- Long, S.B.; Tao, X.; Campbell, E.B.; MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Shen, J.H.; Briggs, J.M.; Luo, X.M.; Tan, X.J.; Jiang, H.L.; Chen, K.X.; Ji, R.Y. Brownian dynamics simulations of interaction between scorpion toxin Lq2 and potassium ion channel. Biophys. J. 2001, 80, 1659–1669. [Google Scholar] [CrossRef]
- Eriksson, M.A.; Roux, B. Modeling the structure of agitoxin in complex with the Shaker K+ channel: A computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys. J. 2002, 83, 2595–2609. [Google Scholar] [CrossRef]
- Wang, C.; Bradley, P.; Baker, D. Protein-protein docking with backbone flexibility. J. Mol. Biol. 2007, 373, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F.; Bakowies, D.; Baron, R.; Chandrasekhar, I.; Christen, M.; Daura, X.; Gee, P.; Geerke, D.P.; Glattli, A.; Hunenberger, P.H.; et al. Biomolecular modeling: Goals, problems, perspectives. Angew. Chem. Int. Ed. 2006, 45, 4064–4092. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.W.; Andersen, O.S.; Roux, B. Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. USA 2004, 101, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Jarzynski, C. Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 1997, 78, 2690–2693. [Google Scholar] [CrossRef]
- Baştuğ, T.; Chen, P.C.; Patra, S.M.; Kuyucak, S. Potential of mean force calculations of ligand binding to ion channels from Jarzynski‘s equality and umbrella sampling. J. Chem. Phys. 2008, 128, 155104. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenberg, J.M. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Anderson, C.S.; MacKinnon, R.; Smith, C.; Miller, C. Charybdotoxin block of single Ca2+-activated K+ channels. Effects of channel gating, voltage, and ionic strength. J. Gen. Physiol. 1988, 91, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Kharrat, R.; Mabrouk, K.; Crest, M.; Darbon, H.; Oughideni, R.; MartinEauclaire, M.F.; Jacquet, G.; ElAyeb, M.; VanRietschoten, J.; Rochat, H.; et al. Chemical synthesis and characterization of maurotoxin, a short scorpion toxin with four disulfide bridges that acts on K+ channels. Eur. J. Biochem. 1996, 242, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Garciacalvo, M.; Leonard, R.J.; Novick, J.; Stevens, S.P.; Schmalhofer, W.; Kaczorowski, G.J.; Garcia, M.L. Purification, characterization, and biosynthesis of margatoxin, a component of Centruroides Margaritatus venom that selectively inhibits voltage-dependent potassium channels. J. Biol. Chem. 1993, 268, 18866–18874. [Google Scholar]
- Mouhat, S.; Visan, V.; Ananthakrishnan, S.; Wulff, H.; Andreotti, N.; Grissmer, S.; Darbon, H.; de Waard, M.; Sabatier, J.-M. K+ channel types targeted by synthetic OSK1, a toxin from Orthochirus scrobiculosus scorpion venom. Biochem. J. 2005, 385, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Nirthanan, S.; Pil, J.; Abdel-Mottaleb, Y.; Sugahara, Y.; Gopalakrishnakone, P.; Joseph, J.S.; Sato, K.; Tytgat, J. Assignment of voltage-gated potassium channel blocking activity to κ-KTx1.3, a non-toxic homologue of κ-hefutoxin-1, from Heterometrus spinifer venom. Biochem. Pharmacol. 2005, 69, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Bontems, F.; Roumestand, C.; Gilquin, B.; Menez, A.; Toma, F. Refined structure of charybdotoxin: Common motifs in scorpion toxins and insect defensins. Science 1991, 254, 1521–1523. [Google Scholar] [CrossRef] [PubMed]
- Blanc, E.; Sabatier, J.M.; Kharrat, R.; Meunier, S.; el Ayeb, M.; Van Rietschoten, J.; Darbon, H. Solution structure of maurotoxin, a scorpion toxin from Scorpio maurus, with high affinity for voltage-gated potassium channels. Proteins 1997, 29, 321–333. [Google Scholar] [CrossRef]
- Johnson, B.A.; Stevens, S.P.; Williamson, J.M. Determination of the three-dimensional structure of margatoxin by 1H, 13C, 15N triple-resonance nuclear magnetic resonance spectroscopy. Biochemistry 1994, 33, 15061–15070. [Google Scholar] [CrossRef] [PubMed]
- Jaravine, V.A.; Nolde, D.E.; Reibarkh, M.J.; Korolkova, Y.V.; Kozlov, S.A.; Pluzhnikov, K.A.; Grishin, E.V.; Arseniev, A.S. Three-dimensional structure of toxin OSK1 from Orthochirus scrobiculosus scorpion venom. Biochemistry 1997, 36, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Cui, M.; Briggs, J.M.; Huang, X.; Xiong, B.; Zhang, Y.; Luo, X.; Shen, J.; Ji, R.; Jiang, H.; et al. Brownian dynamics simulations of the recognition of the scorpion toxin maurotoxin with the voltage-gated potassium ion channels. Biophys. J. 2002, 83, 2370–2385. [Google Scholar] [CrossRef]
- Jin, L.; Wu, Y. Molecular mechanism of the sea anemone toxin ShK recognizing the Kv1.3 channel explored by docking and molecular dynamic simulations. J. Chem. Inf. Model. 2007, 47, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wu, Y. Molecular mechanism of δ-dendrotoxin-potassium channel recognition explored by docking and molecular dynamic simulations. J. Mol. Recognit. 2011, 24, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Heinzelmann, G.; Huq, R.; Tajhya, R.B.; Chang, S.C.; Chhabra, S.; Pennington, M.W.; Beeton, C.; Norton, R.S.; Kuyucak, S. A potent and selective peptide blocker of the Kv1.3 channel: Prediction from free-energy simulations and experimental confirmation. PLoS ONE 2013, 8, e78712. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Kuyucak, S. Affinity and selectivity of ShK toxin for the Kv1 potassium channels from free energy simulations. J. Phys. Chem. B 2012, 116, 4812–4822. [Google Scholar] [CrossRef] [PubMed]
- Khabiri, M.; Nikouee, A.; Cwiklik, L.; Grissmer, S.; Ettrich, R. Charybdotoxin unbinding from the mKv1.3 potassium channel: A combined computational and experimental study. J. Phys. Chem. B 2011, 115, 11490–11500. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Qiu, S.; Cao, Z.; Wu, Y.; Li, W. Molecular basis of inhibitory peptide maurotoxin recognizing Kv1.2 channel explored by ZDOCK and molecular dynamic simulations. Proteins 2008, 70, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.H. Structural basis of the selective block of Kv1.2 by maurotoxin from computer simulations. PLoS ONE 2012, 7, e47253. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.H. Binding modes of two scorpion toxins to the voltage-gated potassium channel Kv1.3 revealed from molecular dynamics. Toxins 2014, 6, 2149–2161. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Robinson, A.; Gordon, D.; Chung, S.H. Modeling the binding of three toxins to the voltage-gated potassium channel (Kv1.3). Biophys. J. 2011, 101, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.H. Molecular dynamics simulations of scorpion toxin recognition by the Ca2+-activated potassium channel KCa3.1. Biophys. J. 2013, 105, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Q.; Ni, F.; Ma, J. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement. Proc. Natl. Acad. Sci. USA 2010, 107, 11352–11357. [Google Scholar] [CrossRef] [PubMed]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Ishida, Y.; Wakamatsu, K.; Kato, R.; Honda, H.; Ohizumi, Y.; Nakamura, H.; Ohya, M.; Lancelin, J.-M.; Kohda, D.; et al. Active site of μ-conotoxin GIIIA, a peptide blocker of muscle sodium channels. J. Biol. Chem. 1991, 266, 16989–16991. [Google Scholar] [PubMed]
- Reynolds, I.J.; Wagner, J.A.; Snyder, S.H.; Thayer, S.A.; Olivera, B.M.; Miller, R.J. Brain voltage-sensitive calcium channel subtypes differentiated by ω-conotoxin fraction GVIA. Proc. Natl. Acad. Sci. USA 1986, 83, 8804–8807. [Google Scholar] [CrossRef] [PubMed]
- Kauferstein, S.; Huys, I.; Lamthanh, H.; Stocklin, R.; Sotto, F.; Menez, A.; Tytgat, J.; Mebs, D. A novel conotoxin inhibiting vertebrate voltage-sensitive potassium channels. Toxicon 2003, 42, 43–52. [Google Scholar] [CrossRef]
- Shon, K.J.; Stocker, M.; Terlau, H.; Stühmer, W.; Jacobsen, R.; Walker, C.; Grilley, M.; Watkins, M.; Hillyard, D.R.; Gray, W.R.; et al. κ-Conotoxin PVIIA is a peptide inhibiting the Shaker K+ channel. J. Biol. Chem. 1998, 273, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Savarin, P.; Guenneugues, M.; Gilquin, B.; Lamthanh, H.; Gasparini, S.; Zinn-Justin, S.; Menez, A. Three-dimensional structure of κ-conotoxin PVIIA, a novel potassium channel-blocking toxin from cone snails. Biochemistry 1998, 37, 5407–5416. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, S.; Kuyucak, S. Why the Drosophila Shaker K+ channel is not a good model for ligand binding to voltage-gated Kv1 channels. Biochemistry 2013, 59, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dong, F.; Zhou, H.X. Electrostatic recognition and induced fit in the κ-PVIIA toxin binding to Shaker potassium channel. J. Am. Chem. Soc. 2005, 127, 6836–6849. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; Sollod, B.; King, G.F. Venom landscapes: Mining the complexity of spider venoms via a combined cDNA and mass spectrometric approach. Toxicon 2006, 47, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Swartz, K.J.; MacKinnon, R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron 1995, 15, 941–949. [Google Scholar] [CrossRef]
- Marvin, L.; De, E.; Cosette, P.; Gagnon, J.; Molle, G.; Lange, C. Isolation, amino acid sequence and functional assays of SGTx1. The first toxin purified from the venom of the spider scodra griseipes. Eur. J. Biochem. 1999, 265, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Johnson, J.H.; Hammerland, L.G.; Kelbaugh, P.R.; Volkmann, R.A.; Saccomano, N.A.; Mueller, A.L. Heteropodatoxins: Peptides isolated from spider venom that block Kv4.2 potassium channels. Mol. Pharmacol. 1997, 51, 491–498. [Google Scholar] [PubMed]
- Diochot, S.; Drici, M.D.; Moinier, D.; Fink, M.; Lazdunski, M. Effects of phrixotoxins on the Kv4 family of potassium channels and implications for the role of Ito1 in cardiac electrogenesis. Br. J. Pharmacol. 1999, 126, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Ruta, V.; Jiang, Y.; Lee, A.; Chen, J.; MacKinnon, R. Functional analysis of an archaebacterial voltage-dependent K+ channel. Nature 2003, 422, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Swartz, K.J. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon 2007, 49, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S.; Pallaghy, P.K. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 1998, 36, 1573–1583. [Google Scholar] [CrossRef]
- Takahashi, H.; Kim, J.I.; Min, H.J.; Sato, K.; Swartz, K.J.; Shimada, I. Solution structure of hanatoxin1, a gating modifier of voltage-dependent K+ channels: Common surface features of gating modifier toxins. J. Mol. Biol. 2000, 297, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Kim, S.; Roh, S.H.; Endoh, H.; Kodera, Y.; Maeda, T.; Kohno, T.; Wang, J.M.; Swartz, K.J.; Kim, J.I. Solution structure and functional characterization of SGTx1, a modifier of Kv2.1 channel gating. Biochemistry 2004, 43, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Lee, J.Y.; Kim, S.H.; Eu, Y.J.; Shin, S.Y.; Milescu, M.; Swartz, K.J.; Kim, J.I. Solution structure and lipid membrane partitioning of VSTx1, an inhibitor of the KvAP potassium channel. Biochemistry 2005, 44, 6015–6023. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Zamanian, M.; Bae, C.; Milescu, M.; Krepkiy, D.; Tilley, D.C.; Sack, J.T.; Yarov-Yarovoy, V.; Kim, J.I.; Swartz, K.J. Tarantula toxins use common surfaces for interacting with Kv and ASIC ion channels. eLife 2015, 4, e06774. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Roh, S.H.; Kim, S.; Lee, C.W.; Kim, J.I.; Swartz, K.J. Molecular surface of tarantula toxins interacting with voltage sensors in KV channels. J. Gen. Physiol. 2004, 123, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Milescu, M.; Vobecky, J.; Roh, S.H.; Kim, S.H.; Jung, H.J.; Kim, J.I.; Swartz, K.J. Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 2007, 130, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Nishizawa, K. Interaction between K+ channel gate modifier hanatoxin and lipid bilayer membranes analyzed by molecular dynamics simulation. Eur. Biophys. J. 2006, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Wee, C.L.; Bemporad, D.; Sands, Z.A.; Gavaghan, D.; Sansom, M.S.P. SGTx1, a Kv channel gating-modifier toxin, binds to the interfacial region of lipid bilayers. Biophys. J. 2007, 92, L07–L09. [Google Scholar] [CrossRef] [PubMed]
- Bemporad, D.; Sands, Z.A.; Wee, C.L.; Grottesi, A.; Sansom, M.S.P. Vstx1, a modifier of Kv channel gating, localizes to the interfacial region of lipid bilayers. Biochemistry 2006, 45, 11844–11855. [Google Scholar] [CrossRef] [PubMed]
- Wee, C.L.; Gavaghan, D.; Sansom, M.S.P. Interactions between a voltage sensor and a toxin via multiscale simulations. Biophys. J. 2010, 98, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Mihailescu, M.; Krepkiy, D.; Milescu, M.; Gawrisch, K.; Swartz, K.J.; White, S. Structural interactions of a voltage sensor toxin with lipid membranes. Proc. Natl. Acad. Sci. USA 2014, 111, E5463–E5470. [Google Scholar] [CrossRef]
- Chen, R.; Robinson, A.; Chung, S.H. Binding of hanatoxin to the voltage sensor of Kv2.1. Toxins 2012, 4, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; Vernon, R.; Thompson, J.; Tyka, M.; Sadreyev, R.; Pei, J.M.; Kim, D.; Kellogg, E.; DiMaio, F.; Lange, O.; et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins 2009, 77, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Suchyna, T.M.; Tape, S.E.; Koeppe, R.E.; Andersen, O.S.; Sachs, F.; Gottlieb, P.A. Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature 2004, 430, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Hurst, A.C.; Gottlieb, P.A.; Martinac, B. Concentration dependent effect of GsMTx4 on mechanosensitive channels of small conductance in E. coli spheroplasts. Eur. Biophys. J. 2009, 38, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Kamaraju, K.; Gottlieb, P.A.; Sachs, F.; Sukharev, S. Effects of GsMTx4 on bacterial mechanosensitive channels in inside-out patches from giant spheroplasts. Biophys. J. 2010, 99, 2870–2878. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.H. Effect of gating modifier toxins on membrane thickness: Implications for toxin effect on gramicidin and mechanosensitive channels. Toxins 2013, 5, 456–471. [Google Scholar] [CrossRef] [PubMed]
- Aneiros, A.; Garcia, I.; Martinez, J.R.; Harvey, A.L.; Anderson, A.J.; Marshall, D.L.; Engstrom, A.; Hellman, U.; Karlsson, E. A potassium channel toxin from the secretion of the sea anemone Bunodosoma granulifera. Isolation, amino acid sequence and biological activity. Biochim. Biophys. Acta 1993, 1157, 86–92. [Google Scholar] [CrossRef]
- Dauplais, M.; Lecoq, A.; Song, J.; Cotton, J.; Jamin, N.; Gilquin, B.; Roumestand, C.; Vita, C.; de Medeiros, C.L.; Rowan, E.G.; et al. On the convergent evolution of animal toxins. Conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 1997, 272, 4302–4309. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.W.; Byrnes, M.E.; Zaydenberg, I.; Khaytin, I.; de Chastonay, J.; Krafte, D.S.; Hill, R.; Mahnir, V.M.; Volberg, W.A.; Gorczyca, W.; et al. Chemical synthesis and characterization of ShK toxin: A potent potassium channel inhibitor from a sea anemone. Int. J. Peptide Protein Res. 1995, 46, 354–358. [Google Scholar] [CrossRef]
- Alessandri-Haber, N.; Lecoq, A.; Gasparini, S.; Grangier-Macmath, G.; Jacquet, G.; Harvey, A.L.; de Medeiros, C.; Rowan, E.G.; Gola, M.; Menez, A.; et al. Mapping the functional anatomy of BgK on Kv1.1, Kv1.2, and Kv1.3. Clues to design analogs with enhanced selectivity. J. Biol. Chem. 1999, 274, 35653–35661. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; Khoo, K.K.; Feng, Z.P.; Crossley, G.; Nugent, D.; Khaytin, I.; Chi, V.; Pham, C.; Calabresi, P.; Pennington, M.W.; et al. Potassium channel modulation by a toxin domain in matrix metalloprotease 23. J. Biol. Chem. 2010, 285, 9124–9136. [Google Scholar] [CrossRef] [PubMed]
- Tudor, J.E.; Pallaghy, P.K.; Pennington, M.W.; Norton, R.S. Solution structure of ShK toxin, a novel potassium channel inhibitor from a sea anemone. Nat. Struct. Biol. 1996, 3, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Gilquin, B.; Racape, J.; Wrisch, A.; Visan, V.; Lecoq, A.; Grissmer, S.; Menez, A.; Gasparini, S. Structure of the BgK-Kv1.1 complex based on distance restraints identified by double mutant cycles. Molecular basis for convergent evolution of Kv1 channel blockers. J. Biol. Chem. 2002, 277, 37406–37413. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.W.; Mahnir, V.M.; Khaytin, I.; Zaydenberg, I.; Byrnes, M.E.; Kem, W.R. An essential binding surface for ShK toxin interaction with rat brain potassium channels. Biochemistry 1996, 35, 16407–16411. [Google Scholar] [CrossRef] [PubMed]
- Dauplais, M.; Gilquin, B.; Possani, L.D.; Gurrola-Briones, G.; Roumestand, C.; Menez, A. Determination of the three-dimensional solution structure of noxiustoxin: Analysis of structural differences with related short-chain scorpion toxins. Biochemistry 1995, 34, 16563–16573. [Google Scholar] [CrossRef] [PubMed]
- Chi, V.; Pennington, M.W.; Norton, R.S.; Tarcha, E.J.; Londono, L.M.; Sims-Fahey, B.; Upadhyay, S.K.; Lakey, J.T.; Iadonato, S.; Wulff, H.; et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon 2012, 59, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Smith, B.J.; Sabo, J.K.; Crossley, G.; Nugent, D.; Khaytin, I.; Chi, V.; Chandy, K.G.; Pennington, M.W.; Norton, R.S. The D-diastereomer of ShK toxin selectively blocks voltage-gated K+ channels and inhibits T lymphocyte proliferation. J. Biol. Chem. 2008, 283, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Kalman, K.; Pennington, M.W.; Lanigan, M.D.; Nguyen, A.; Rauer, H.; Mahnir, V.; Paschetto, K.; Kem, W.R.; Grissmer, S.; Gutman, G.A.; et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J. Biol. Chem. 1998, 273, 32697–32707. [Google Scholar] [CrossRef] [PubMed]
- Lanigan, M.D.; Kalman, K.; Lefievre, Y.; Pennington, M.W.; Chandy, K.G.; Norton, R.S. Mutating a critical lysine in ShK toxin alters its binding configuration in the pore-vestibule region of the voltage-gated potassium channel, Kv1.3. Biochemistry 2002, 41, 11963–11971. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Qiu, S.; Yang, F.; Cao, Z.J.; Li, W.X.; Wu, Y.L. Unique mechanism of the interaction between honey bee toxin TPNQ and rKir1.1 potassium channel explored by computational simulations: Insights into the relative insensitivity of channel towards animal toxins. PLoS ONE 2013, 8, e67213. [Google Scholar] [CrossRef] [PubMed]
- Hilder, T.A.; Chung, S.H. Conductance properties of the inwardly rectifying channel, Kir3.2: Molecular and Brownian dynamics study. Biochim. Biophys. Acta 2013, 1828, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Hilder, T.A.; Chung, S.H. Conduction and block of inward rectifier K+ channels: Predicted structure of a potent blocker of Kir2.1. Biochemistry 2013, 52, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Doupnik, C.A.; Parra, K.C.; Guida, W.C. A computational design approach for virtual screening of peptide interactions across K+ channel families. Comput. Struct. Biotechnol. J. 2015, 13, 85–94. [Google Scholar] [PubMed]
- Chen, R.; Chung, S.H. Binding modes of μ-conotoxin to the bacterial sodium channel (NaVAb). Biophys. J. 2012, 102, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Pennington, M.W.; Wulff, H.; Singh, S.; Nugent, D.; Crossley, G.; Khaytin, I.; Calabresi, P.A.; Chen, C.Y.; Gutman, G.A.; et al. Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases. Mol. Pharmacol. 2005, 67, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Huq, R.; Tanner, M.R.; Chhabra, S.; Khoo, K.K.; Estrada, R.; Dhawan, V.; Chauhan, S.; Pennington, M.W.; Beeton, C.; et al. A potent and Kv1.3-selective analogue of the scorpion toxin HsTX1 as a potential therapeutic for autoimmune diseases. Sci. Rep. 2014, 4, 4509. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, Y.; Hong, J.; Hu, J.; Yang, W.; Xiang, F.; Yang, F.; Xie, Z.; Cao, Z.; Li, W.; et al. Toxin acidic residue evolutionary function-guided design of de novo peptide drugs for the immunotherapeutic target, the Kv1.3 channel. Sci. Rep. 2015, 5, 9881. [Google Scholar] [CrossRef] [PubMed]
- Finol-Urdaneta, R.K.; Glavica, R.; McArthur, J.R.; French, R.J. Polymodal, high affinity actions of μ-conotoxins on a bacterial voltage-gated sodium channel. Biophys. J. 2013, 104, 136a–137a. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, R.; Chung, S.-H. Computational Studies of Venom Peptides Targeting Potassium Channels. Toxins 2015, 7, 5194-5211. https://doi.org/10.3390/toxins7124877
Chen R, Chung S-H. Computational Studies of Venom Peptides Targeting Potassium Channels. Toxins. 2015; 7(12):5194-5211. https://doi.org/10.3390/toxins7124877
Chicago/Turabian StyleChen, Rong, and Shin-Ho Chung. 2015. "Computational Studies of Venom Peptides Targeting Potassium Channels" Toxins 7, no. 12: 5194-5211. https://doi.org/10.3390/toxins7124877
APA StyleChen, R., & Chung, S.-H. (2015). Computational Studies of Venom Peptides Targeting Potassium Channels. Toxins, 7(12), 5194-5211. https://doi.org/10.3390/toxins7124877